
ABSTRACT
New challenges and other topics in non-clinical safety testing of biotherapeutics were presented and discussed at the nineth European BioSafe Annual General Membership meeting in November 2019. The session topics were selected by European BioSafe organization committee members based on recent company achievements, agency interactions and new data obtained in the non-clinical safety testing of biotherapeutics, for which data sharing would be of interest and considered as valuable information. The presented session topics ranged from strategies of in vitro testing, immunogenicity prediction, bioimaging, and developmental and reproductive toxicology (DART) assessments to first-in-human (FIH) dose prediction and bioanalytical challenges, reflecting the entire space of different areas of expertise and different molecular modalities. During the 9th meeting of the European BioSafe members, the following topics were presented and discussed in 6 main sessions (with 3 or 4 presentations per session) and in three small group breakout sessions: 1) DART assessment with biotherapeutics: what did we learn and where to go?; 2) Non-animal testing strategies; 3) Seeing is believing: new frontiers in imaging; 4) Predicting immunogenicity during early drug development: hope or despair?; 5) Challenges in FIH dose projections; and 6) Non-canonical biologics formats: challenges in bioanalytics, PKPD and biotransformation for complex biologics formats. Small group breakout sessions were organized for team discussion about 3 specific topics: 1) Testing of cellular immune function in vitro and in vivo; 2) MABEL approach (toxicology and pharmacokinetic perspective); and 3) mRNA treatments. This workshop report presents the sessions and discussions at the meeting.
Introduction
BioSafe is the Non-clinical Safety expert group of the Biotechnology Innovation Organization (BIO), with the mission to identify and respond to key scientific and regulatory issues and challenges related to the non-clinical safety evaluation of biopharmaceuticals. In addition to the annual general membership meeting in the US, an annual BioSafe meeting is held in Europe for European BIO member companies.Citation1–5 The nineth Annual BioSafe European general membership meeting was hosted by Sanofi on November 5–6, 2019, in Chilly-Mazarin, south of Paris, France.
The meeting attendees were from the biopharmaceutical industry, small biotechnology companies and contract research organizations from Europe, but also from US, representing various disciplines, including pharmacology, toxicology and pathology, pharmacokinetics (PK) and bioanalytics. The objective of the meeting was to share experiences and insights into non-clinical safety assessments of biologics, covering various non-clinical safety topics, including developmental and reproductive toxicity, non-animal testing strategies, bioimaging, prediction of immunogenicity, first-in-human (FIH) dose projection and bioanalytics of complex biologic formats.
In each session, case examples were presented, and all sessions were followed by a podium discussion. Three hot topics, pre-selected by the organization committee, were discussed in small group breakout sessions. The breakout session leads then shared the consolidated discussion outcome with all attendees.
Session 1: developmental and reproductive toxicology assessment with biotherapeutics: what did we learn and where to go?
The first session, chaired by Guenter Blaich (AbbVie) and Adam Hey (AstraZeneca), focused on developmental and reproductive toxicology (DART) assessments beyond the historical approach of testing the potential risk of developmental toxicity of biotherapeutics in humans using the guinea pig or the rabbit instead of monkeys. In addition, alternative approaches, including thorough literature assessment, placental transfer, and information from genetically modified animals, that were used to request a waiver based on a weight-of-evidence (WoE) assessment were discussed. Finally, relevant changes from the revision of the International Conference on Harmonization (ICH) S5(R3) guideline were presented.
In the first talk, Andreas Hohlbaum (Ablynx/Sanofi) presented a case study using the guinea pig for DART assessment of caplacizumab, as the guinea pig is not the standard species used for DART evaluations, and published examples in particular for biologics are limited.
Caplacizumab is a humanized anti-von Willebrand Factor (vWF) Nanobody® for the treatment of acquired thrombotic thrombocytopenic purpura (aTTP). Caplacizumab targets the A1-domain of vWF, inhibiting the interaction between vWF and platelets. Clinical studies conducted in aTTP patients confirmed the rapid and sustained complete suppression of the vWF activity using an initial intravenous dose of 10 mg, and a maintenance subcutaneous (SC) 10 mg daily dosing regimen, with corresponding favorable efficacy and safety profiles.Citation6
General toxicity studies were performed in guinea pigs and cynomolgus monkeys, as caplacizumab is pharmacologically active in both species and does not bind to the A1-domain of vWF from mouse, rat, rabbit, hamster or dog. To support Phase 2 studies in women of childbearing potential, embryo-fetal development (EFD) studies were performed in guinea pigs.
DART assessment of caplacizumab included assessment of male and female fertility in a 13-week repeat-dose toxicity study in cynomolgus monkeys, according to ICHS6 (R1). No juvenile toxicity study was performed as aTTP only occurs in adulthood. The clinical experience with pregnant vWF1 disease patients exhibiting reduced levels of vWF indicated a low risk for caplacizumab as a highly targeted biologic. Therefore, a dedicated pre- and postnatal development (PPND) study was considered unnecessary, as it would not provide additional information. Nevertheless, Ablynx proposed to perform EFD studies in Dunkin Hartley guinea pigs rather than in cynomolgus monkeys, which was endorsed by the Paul Ehrlich Institute prior to the FIH study in healthy volunteers well in advance of regulatory interactions with the European Medicines Agency (EMA) and US Food and Drug Administration (FDA).
The choice of guinea pigs for EFD studies is unusual because there is limited background data and precedence for DART evaluations of biologics in this species. Exceptions are the fertility, early development and EFD studies that were performed with natalizumabCitation7,Citation8 and fertility studies with guselkumab.Citation9 Regarding reproduction, guinea pigs have a discoidal, hemomonochorial placental structure with a maternal/fetal transport barrier that is similar to the one in human, the endocrine control of pregnancy is similar to humans and the gestation period of around 68 days can be divided into trimesters, which resembles the human fetal development stages.Citation10,Citation11.
Caplacizumab was dosed in the EFD study daily from gestation day (GD) 6 to GD41 at 0, 0.05, 1 and 20 mg/kg via intramuscular (IM) injection with n = 18–20 pregnant females per group. IM was selected as route of administration as repeated intravenous (IV) dosing is not possible in guinea pig. Laparotomy was performed on GD61 and toxicokinetics (TK) and anti-drug antibodies (ADA) were assessed at 6 time points to confirm exposure throughout the treatment period. The no-observed-adverse-effect level (NOAEL) in this study was established at 20 mg/kg/day without a test item-related increase in the incidence of malformations, variations or retardations.
After discussion of these results with the Paul Ehrlich Institute, a placental transfer study was performed as no fetal drug concentrations were measured in the first study and PD assays (vWF, FVIII and ristocin cofactor activity (RICO)) were not available at the time. Caplacizumab was dosed from GD6 to GD41 or GD61 at 0, 20 and 40 mg/kg/day via IM injection to 5 animals/group. The NOAEL was at 40 mg/kg/day while active exposure of the dams was demonstrated by the TK data and full suppression of vWF induced platelet aggregation ex vivo in the RICO assay measuring the ability of vWF to interact with the platelet receptor GP1b-IX-V. Low fetal exposure was seen on GD41 and GD61. Active placental transfer of caplacizumab is not expected because high molecular weight proteins (> 5kD) inefficiently cross the placenta by diffusion, as the Nanobody does not interact with the FcRn receptor and as there is no literature evidence for target-mediated placental transfer.
Overall, the EFD studies in guinea pig provided conclusive results for caplacizumab risk assessment during the early clinical development stage of the project prior to recruiting women of childbearing potential in clinical trials and informed the respective sections of the EMA’s European public assessment report and FDA label. To our knowledge, this is the first and only example where an EFD in guinea pig did substitute for a study in cynomolgus monkey within the non-clinical development of a biologic.
General challenges with guinea pigs include the limited historic control data and smaller litter size (average 3–4) compared to rat and rabbits. In addition, there are no readily accessible peripheral veins for IV injections and blood collection and anesthesia is more challenging. Guinea pigs are more difficult to house and breed since they are stress sensitive, have a lower mating rate and longer gestation period and the assessment of successful pregnancy is more difficult. Therefore, selection of a contract research organization with relevant experience is critical. Artificial insemination at the breeder is recommended to assure a sufficient number of pregnant females. In addition, treated reserve animals should be included in the study design (e.g., 30 dams were treated per group to obtain 20 evaluable litters in the EFD study) to account for non-confirmed pregnancy, natural premature delivery, potential deaths during blood withdrawal and a background of spontaneous abortions.
We therefore recommend evaluating guinea pig cross-reactivity of novel biologics case by case, such as, for example, if an extensive DART evaluation is warranted due to a potentially high risk associated with the mode of action. Guinea pig studies can then be considered as alternative to studies in primates or in rodents with rodent specific surrogates.
In the next presentations, Peter Ulrich (Novartis) and Adam Hey (AstraZeneca) presented three case studies. The case study presentation by Peter Ulrich (Novartis) described the strategy of reproductive toxicity assessment of an immunosuppressive IgG1 antibody, NVS-0815. Comparison of amino acid sequences revealed identical sequence of the targeted epitope for the non-human primate (NHP) and the rabbit, but not for rodents. NVS-0815 is cross-reactive to the NHP and the rabbit target with binding affinity similar to human. In vitro pharmacology assays in all 3 species led to comparable results, indicating relevance of the rabbit as well. The safety profile in NHP was excellent, while the expected pharmacodynamic (PD) effects were observed. In compliance with ICH S6(R1), the rabbit was selected as a relevant species for the reproductive toxicity program for NVS-0815. To further confirm this, a dose range-finding (DRF) EFD study was conducted with additional investigation of PD effects in lymphatic tissues. Results from a previous study with a therapeutic antibody (IgG1) applied to pregnant rabbits revealed placental transfer at a rate of over 90% at GD29, whereas at GD19 a rate of only 1.5% was achieved. Based on these results, three dose levels were applied on GD7, 14, and 20. After Cesarian section on GD29, the main organs and lymphatic tissues were taken from the dams. Pups were externally examined and did not show any deviation or adverse effect. Lymphatic tissues of the dams showed the expected PD effects and no signs of general toxicity were observed. At the highest dose, a complete PD effect was observed, whereas incomplete effects were noted at the mid- and low dose. Immunogenicity was seen in low and mid-dose mother animals, which led to a disproportional exposure of the females after the second and third injection. No ADA were detected in high-dose animals, which is in line with the immunosuppressive mode of action. Based on the results of this preliminary study on rabbits, a full reproductive toxicity assessment will be conducted in parallel to future clinical development. In agreement with health authorities, these studies will be conducted with one high dose of NVS-0815 because at this dose full target receptor occupancy was demonstrated, whereas at lower doses confounding immunogenicity will lead to inconclusive results.
Adam Hey (AstraZeneca) first presented a case with NVS148, a combination of two antibodies directed against cytomegalovirus (CMV). One antibody targets the surface proteins gB and the other targets the 5-member complex (gH, gL, UL128, UL130 and UL131). Both targets are important for viral infection, cell to cell spread and cell to cell fusion. Both IgG1 antibodies have an unaltered Fc part and are developed for the prevention of intrauterine human CMV transmission in pregnant women. In accordance with ICH S6 R1, when targeting a foreign target and in the absence of a pharmacologically relevant species, a short-term 4-week study in rat was the only in vivo study conducted with weekly IV doses of 500 and 50 mg/kg as the highest doses of the two antibodies. No toxicological or safety issues were observed in the study. An additional in vitro assessment of antibody-dependent cell-mediated cytotoxicity (ADCC) potency was done in parallel with and using the anti-CMV IVIG compound Cytotect®CP as a “safe reference”, since this had previously been given safely to pregnant women. NVS148 showed similar to or lower potency for eliciting ADCC compared to Cytotect®CP. In consultancy with FDA and EMEA, waivers for conduct of chronic toxicity studies, reproductive toxicity studies and carcinogenicity studies were obtained. Regarding the assessment of tissue cross-reactivity, however, specific additional requests were received from FDA. When assessing binding to the full panel of tissues from adult humans, rats and cynomolgus monkeys, rat and monkey were negative with no staining. In human tissues staining was observed in multiple tissues to scattered cells. Using in-situ hybridization, these cells were subsequently confirmed to be positive for CMV and thus represented on-target binding. At the pre-IND meeting FDA, based on the indication in pregnant women, requested additional assessment of binding to human, rat and monkey fetal tissues and also additional assessment of binding to human placenta and inclusion of confirmed CMV-negative tissues to assess any binding in the absence of the virus. For fetal tissues only major organs and in humans only tissues from a single donor were available. Again, rat and monkey were negative, and human placental and fetal tissues from a CMV-positive donor showed similar scattered staining of confirmed CMV positive cells as observed with adult tissues. Finally, testing CMV-negative tissues, which were extremely difficult to obtain but stained negative in the single donor available, confirmed the absence of off-target staining in CMV-negative tissues.
In conclusion, in accordance with ICH S6 R1, a waiver for conduct of reproductive studies was obtained for NVS148. However, based on the indication in pregnant women, additional work on tissues cross-reactivity was requested by FDA, resulting in a total of 5 such studies, including assessment in fetal human tissues.
In his second case study, Adam Hey presented a case on NVS031 where assessment of juvenile toxicity was done as part of the enhanced PPND (ePPND) study in cynomolgus monkeys. NVS031 is a follow-up candidate to NVS030; both antibodies are IgG1 monoclonal antibodies (mAbs) against IgE, with indications of asthma and urticaria. In juvenile studies with NVS030, weekly doses of up to 250 mg/kg had resulted in thrombocytopenia, which had not been observed in any of the previous repeated dose (up to 26 weeks) studies in adolescent/adult monkeys. Therefore, for NVS031 there was a desire to test if any similar effect on platelets and thrombocytopenia would be observed. After consultation with FDA it was decided to perform this assessment as part of the planned ePPND study where weekly SC doses of 0, 15 or 150 mg/kg were applied. The juvenile assessment would then be done by using the offspring from the low dose (15 mg/kg) group and dose these for 4 weeks starting at the end of the planned 6 months post-natal follow-up period. This resulted in a design with 5 control animals and 10 treated animals dosed 150 mg/kg SC on a weekly basis with assessments of clinical signs, body weight and clinical pathology (platelet counts), macro and microscopic findings, computerized tomography (CT) scans and echocardiography and complete end of study necropsies. No mortalities, clinical signs or changes in body weight were observed. Following CT scans, normal anatomy of visceral organs and appearance of skeleton was observed and no echocardiography changes, no effects on platelets or signs of thrombocytopenia were seen. All treated animals were exposed to NVS031, and no ADAs were observed in any of the animals.
In conclusion, no test item-related changes in platelet counts or in bleeding time were observed with NVS031. These results were repeatedly and successfully used in multiple health authority interactions with FDA, Health Canada and EMA where the NVS030 juvenile data was repeatedly being brought up in relation to NVS031 also targeting IgE.
Benno Rattel (Amgen) then discussed the challenges of assessing the potential developmental toxicity of drugs developed for the treatment of advanced cancer patients. Oncology molecules are regulated by the ICH S9 guidance “Nonclinical Evaluation for Anticancer Pharmaceuticals”,Citation12 which states that toxicology studies of anticancer pharmaceuticals assessing the EFD should be available when the marketing application is submitted and are not considered essential to support clinical trials in patients with advanced cancer. EFD studies can, however, be omitted altogether from the marketing application dossier in cases of anticancer drugs that are genotoxic and target rapidly dividing cells or belong to a class that has already been well characterized as causing developmental toxicity. Additionally, in cases where the NHP is the only pharmacologically relevant species and the mechanism of action is expected to yield a reproductive toxicity risk, and/or use of knock-out (KO) animals or of surrogate biologics in rodents have demonstrated a reproductive risk, a WoE assessment of the reproductive risk can be provided instead of conducting reproductive studies. Indeed, an NHP study to assess a hazard to EFD should not be considered a default approach and an EFD study in NHP is not always warranted if the WoE clearly outlines the risk(s).Citation13 Reproductive risk assessment using WoE would comprise a thorough review of public databases and literature, an assessment of the potential effects of target biology, expression, and pharmacology on EFD, a review of existing repeat-dose toxicity data of the respective or related molecule(s) and an assessment of established direct or indirect effects of the therapeutic in nonpregnant animals and patients, as well as on potential placental transfer. The reproductive risk assessment of daratumumab (Darzalex®)Citation14 was presented in detail as an example of acceptable WoE assessment, based mainly on scientific literature without performing reproductive and developmental toxicology studies. There are several recent FDA approvals of biologics with no EFD studies, as listed by Rocca et al.Citation15 These authors have also proposed a conceptual decision tree for evaluating whether an in vivo study in pregnant animals is warranted or not.
Three case studies of bispecific T-cell engager (BiTE®) molecules, which are only cross reactive to NHPs, were presented to illustrate the WoE approach. Case Study 1 was a BiTE® molecule for which published studies have characterized potential off-tumor target expression during EFD. Consistent with the more extensive expression pattern during development, genetic mutations in the target have been associated with malformations in mice. Thus, Amgen concluded that the published studies clearly indicated that the target is expressed during EFD and is involved in normal skeleton formation, suggesting that embryonic exposure to the BiTE® molecules would likely result in embryofetal toxicity. Therefore, an EFD study in NHPs was considered unnecessary or unable to provide additional data that would significantly change the WoE-based human risk assessment. However, the FDA did not agree, concluding that, while it acknowledged the importance of the target in EFD, the data on expression of the target and the data from KO mice on its own were insufficient to inform on the risk of the BiTE® molecules during development.
In Case Study 2, the tumor antigen targeted by the BiTE® molecules was restricted to a specific hematologic cell type or population, and no off-target binding was detected in normal human tissues. All the effects observed in the repeat-dose toxicity studies represented the expected PD effects and were reversible. The target did not seem to be essential for EFD, as shown in target KO mice, which were outwardly normal, survived with no unusual morbidity and all major organs were present. This specific BiTE® molecules lacked an Fc portion, so substantial fetal exposure was not expected, as high molecular weight proteins (>5 kDa) do not cross the placenta by simple diffusion.Citation16 Even if very low amounts of BiTE® molecules were to be transported by passive diffusion, exposure of developing embryo/fetus was not expected to cause any effects besides the known PD effects, i.e., target cell depletion with repopulation from bone marrow stem cells following cessation of treatment. Of all cancer patients with the disease, the population of women of child-bearing potential was estimated to be only ~3%. Furthermore, the pharmaceutical was very likely to be administered in combination with different teratogenic and embryo-fetally toxic drugs. Based on this WoE, the FDA and EMA granted a waiver allowing Amgen not to conduct an EFD study in NHPs.
The cancer targeted by the BiTE® molecules of Case Study 3 only occurs in adult populations, except in very rare circumstances. Repeat-dose data suggested that administering the BiTE® molecule to NHPs for longer than 3 weeks will result in a high incidence of ADA formation with a subsequent loss of exposure. Amgen submitted a Pediatric Investigation Plan with justification for a product-specific waiver based on the disease not occurring in any specified pediatric subset and several products having previously been granted product-specific waivers for the treatment of this cancer in all subsets of the pediatric population on the grounds that the condition occurs only in adult populations. However, the EMA did not agree in this case, and Amgen was asked for more comprehensive information concerning the potential use of the product in the pediatric population given its mechanism of action and expanding on its potential significant therapeutic benefit in other indications, with a focus on other pediatric malignancies overexpressing the target.
In the last talk of the DART session, Paul Barrow (Roche) gave an update on the revision of the ICH S5(R3) guideline and how it will affect the DART testing of biopharmaceuticals. The revised guideline “Detection of toxicity to reproduction for human pharmaceuticals” is currently at ICH Step 3 and is expected to be adopted in early 2020. This will be the first major revision of the first safety guideline issued by the ICH, back in 1993.Citation17 The ICH S5(R3) Expert Working Group is composed of 31 individuals representing 15 organizations from 11 regions or countries.Citation18 The new guideline is expanded in scope to include biopharmaceuticals and vaccines. A new section on DART risk assessment will also be included. While the guideline brings about no radical changes to the DART testing of biopharmaceuticals, it is updated to incorporate the relevant recommendations on DART testing from more recent ICH guidelines, such as M3, S6 and S9. It should be determined if pregnancy alters the toxicokinetic properties of the pharmaceutical. The determination of drug levels in the conceptus or neonate remains optional, though the determination of infant levels in an NHP ePPND study is highly advisable. Evidence of excretion in milk can be obtained, when warranted, by sampling milk or by demonstrating exposure in offspring during the pre-weaning period. It is not essential, therefore, to develop and validate a bioanalytical method in milk specifically for this purpose. It is accepted that mating studies are not feasible in NHPs. If a fertility evaluation is based on the histopathological examination of the reproductive organs in a repeated dose study, it is important to allocate sexually mature animals to the study. It is not essential, however, to use sexually mature non-rodents when testing biopharmaceuticals intended for the treatment of advanced cancer. A dedicated fertility study is required for biopharmaceuticals that are pharmacologically active in rodents.
For biopharmaceuticals that only have pharmacological activity in NHPs, DART testing in NHPs should only be performed as a last resort. Rodent DART studies using surrogate molecules or genetically modified animal models are encouraged. If consistent with the pharmacological activity of the biopharmaceutical, EFD studies are performed in both a rodent (rat or mouse) and a non-rodent (rabbit or minipig) species, along with a PPND study in a rodent. An EFD study in a single species, without a PPND study, is sufficient for biopharmaceuticals intended for the treatment of advanced cancer. An ePPND study can be used in the place of the EFD and PPND studies for biopharmaceuticals with no pharmacological activity in non-primate species, but should only be undertaken if there is a potential impact on hazard identification.Citation15 Determination of exposure and duration of exposure and the inclusion of juvenile toxicity endpoints in an ePPND study can potentially avoid the need for a dedicated juvenile study.Citation19 All of the recommendations on the testing of vaccines comply with the FDA guidance on “Considerations for developmental toxicity studies for preventive and therapeutic vaccines for infectious disease indications”,Citation20 which has become the de-facto standard for this purpose. The high dose level for the DART testing of biopharmaceuticals can be limited to a dose that results in at least 10-times the human exposure, based on AUC or Cmax, at the maximum recommended dose (MRD), provided that it elicits the maximum biological effect. The risk assessment section states that there is less concern for developmental findings occurring at more than 10-times the human exposure at the MRD and that transient findings, such as fetal variations, are of less concern than fetal death or malformations.
Three small group breakout sessions
Breakout #1: testing of cellular immune function in vitro and in vivo
Led by Ulrike Hopfer (Roche), this breakout session discussed in vitro and in vivo testing strategies of cellular immune functions with a focus on immune-modulatory molecules. There was an agreement that the key trigger of pro-active studies is a good target assessment. This guides follow-up activities and should include an assessment of the biology, type of modality, targeted disease, dose, route, contamination, or redundant structures that can result in a scoring system. Immunogenicity testing, assessing a potential immune response against self as well as different T-cell-dependent antibody response (TDAR) setups were discussed in more detail.
More frequently used methods to investigate immunogenicity across companies are in silico analyses, MHC-associated peptide proteomics (MAPPS) and dendritic cell (DC)/T-cell assays that are primarily used for immune modulators. Challenges include availability of good quality material, sufficient ranking candidates and databases. There is a need for high throughput systems. These strategies provide rather a ranking than a no-go signal across companies; one challenge is to get suitable controls, as they may become less positive than the candidate may. One should also consider that ADAs elicited by many ADA-positive molecules on the market have no effect on PK, PD or safety.
Few methods exist to investigate a potential effect of the immune system against self-antigens and primarily focus on the target biology and the safety assessment. Challenges of non-clinical models include the assessment of inducible targets, the animal model itself (young, healthy animals), the limited number of study animals, a potential xenogeneic immune response, species-specific differences or the predictability in a non-disease model. As resident T-cells are also important, there is a need to investigate the right-cell type and to expand future assays to more differentiated cell types. Furthermore, there is a significant association between immune-related adverse events in cancer patients and the likelihood of objective antitumor response for some immune modulators in oncology.
Participants in this breakout session also discussed the use of TDAR assays, which is initiated at various timepoints across companies from DRF studies and IND-enabling packages up to 13-week toxicology studies or even ePPND studies (depending on the question). Health authorities tend to increase questions for immune stimulators in this regard. While the standard TDAR assay with keyhole limpet hemocyanin (KLH) is more frequently used, alternative (CD8 T-cell) stimulated TDAR models using, for example, tetanus toxins are being explored for oncology products. Most companies embed this TDAR into pharmacology or PK/PD studies, primarily if required by heath authorities. Clinical trial designs also imbedded a TDAR assay for some compounds.
Taken together, the testing of cellular immune function should be triggered by a robust target assessment taking the respective cell types, mode of action, modality, and targeted disease into consideration. Further work is necessary to develop new strategies to assess inducible targets, develop various TDAR assays and investigate more differentiated cell types.
Breakout #2: MABEL approach (toxicology and PK)
The Minimum Anticipated Biological Effect Level (MABEL) workshop, which was chaired by Benno Rattel (Amgen), started by reviewing the EMA guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products issued in 2007 (see also the MABEL presentation in Session 5).Citation21,Citation22 Participants reported case examples of how they selected the most appropriate test system for the definition of a minimum biological effect level. In addition to in vitro pharmacology data, these examples comprised data derived from in vivo disease models, findings from repeat-dose toxicity studies in NHPs including challenges with bell-shaped dose–response curves and PK/PD data from NHPs titrated to the low end of the dose–response curve. There was a general agreement that PK/PD modeling should be improved to better describe and predict clinically relevant doses. For example, this could be helped by taking receptor expression as well as the presence of effector cells in the targeted tissue into account or by considering drug concentrations at the local level in the compartment(s) of the targeted site of action. The break-out group concluded that a case-by-case selection of the best in vitro or in vivo pharmacologically relevant parameters and test systems for modeling of the safe FIH starting dose as well as subsequent dose escalations should be followed based on the totality of available data.
Breakout #3: mRNA treatments
mRNA was first discovered in 1961 and was successfully transfected into cells in vitro for the first time in 1989.Citation23 In 1990, Wolff et al.Citation24 successfully injected mRNA into mouse skeletal muscle resulting in expression of the encoded protein. Although this was the first demonstration that mRNA could be used as a therapeutic, the instability and immunogenicity potential of mRNA made this seem unattainable.
Many challenges existed for mRNA to become a therapeutic modality. The first is the delivery of the mRNA, which requires it to pass through the extracellular matrix, cross the cell membrane, and escape the endosome to take residence in the cytosol. The mRNA that is delivered must be engineered to ensure the appropriate sequences are included to allow for successful and efficient translation by the cellular machinery. Therefore, translatability and stability are key factors required to optimize mRNA as a viable therapeutic modality.
Many of the attendees of this session, which was chaired by Jennifer Fretland (Sanofi) were there to learn about mRNA therapies and only a small percentage had experience working with mRNA as a therapeutic modality. Some high-level challenges discussed were related to safety, and the key challenge agreed upon was that mRNA therapies are not yet classified by the regulators and therefore no regulatory guidelines have been published. There are translational challenges that exist since the mRNA is not the pharmacologically active substance and there is no demonstration of controlling the “bioavailability” of the pharmacologically active encoded protein(s). Therefore, how to identify a safe therapeutic window poses a continual challenge. Drug–drug interaction (DDI) potential of the encoded proteins must also be considered and a dosing strategy to mitigate should be included in the dose escalation planning. Understanding the distribution of the mRNA was explored by most companies that were represented in this session or were requested by regulatory agencies. Although there is no evidence that mRNA is immunogenic, there are data that suggest it could be; therefore, the immunogenicity potential of this modality is still evolving.
Session 2: non-animal testing strategies
The second session of the meeting was co-chaired by Andrea Kiessling (Novartis) and Petra Schmitt (AbbVie). With animal studies coming increasingly under scrutiny, the evolving field in drug development of non-animal testing strategies is becoming attractive to the safety assessment of biologics. The session’s focus was on how such approaches could be applied to biologics. Examples of non-traditional techniques are provided to assess on- and off-target toxicity.
Petra Schmitt (AbbVie) asked in her introduction whether the time has come for non-traditional safety assessment approaches of biologics. In the “Replacement, Reduction, and Refinement” (3 R) regulatory age, health authorities have become more willing to consider use of alternative ways of safety assessment, such as translational pharmacological and toxicological databases, complex in vitro models and other non-traditional methods. In the European Union, many guidelines include 3Rs as a requirement, and some make direct reference to translational databases like eTox (Draft Reflection paper on the qualification of non-genotoxic impurities, EMA/CHMP/SWP/545,588/2017).Citation25 In addition, non-animal testing methods are under review by the Joint Research Center, the European Commission’s science and knowledge service, informing evidence-based policy making, with the review expected by 2019/2020.Citation26–28 The Head of Medical Agencies and the EMA also formed a Joint Big Data Taskforce to discuss key criteria and requirements for large database systems and data use.Citation29 While chemicals are more commonly the focus of these efforts, reference is made to biologics, and thus the groundwork is laid for a generally applicable principle. In the US, the Critical Path Institute’s Predictive Safety Testing ConsortiumCitation30 shares internally developed new and improved safety testing methods among 250 participating scientists, which include FDA, EMA and Pharmaceuticals and Medical Devices Agency scientists, with the aim to submit them for regulatory qualification. The six working groups have a strong translational focus and evaluate non-clinical and clinical biomarkers in the areas of kidney, liver, testicular toxicity, pancreatic, skeletal muscle as well as vascular injury.
In drug development, generally the aim of safety evaluation is to gain reliable information that can be used for decision-making and that will achieve regulatory acceptance. Not only availability of in vitro and in vivo data, but also a good understanding of the toxic mechanism and the organ or tissue biology increase the credibility of the assessment. In this context, non-animal testing strategies could be useful through a combination of various in vitro techniques, which complement each other, and work in combination with in silico knowledge management and predictive modeling. This way, early on in drug development quantitative structure–activity relationship models, -omics and translational database mining may serve to inform and mature a heuristic computational model and the simplistic understanding of the potential safety profile of the drug. This initial model could then inform and refine in vitro assays, which focus on the identified key concerns by using new stem cell technologies, label-free cell assays, micro-scale systems, new safety biomarker like circulating omics, or other alternative approaches. These data in turn will again mature predictive modeling before studies in humans are started. Some of these techniques are the topic of the further presentations of this session. So far, the closest to regulatory acceptance of such a combined approach is the field of physiology-based kinetics modeling.Citation31
Overall, the area of non-animal testing strategies has the potential to streamline safety assessment and to address all three of the 3Rs, which makes it valuable. It is in flux, and as it is at its early stage, current efforts to evolve it need to be seen as an investment in the future.
Mohan Rao (AbbVie) introduced computational strategies to predict potential safety-related endpoints for therapeutic biologics, including small, medium and large molecules. Details on computational methods for predicting immunogenicity, potential off-target binding sites (Fab- and Fc-mediated), aggregation sites, iso-electric point (i.e., computational pH titration), protease cleavage sites, and antigen–antibody interactions were presented, including the potential role of mannose receptor 2 (MR2) in nonspecific uptake of antibody–drug conjugates (ADC). The utility of computational off-target framework called Off-Target Safety Assessment tool (OTSA)Citation32 to predict potential off-targets and associated outcomes for drug molecules that are attached as payload to a mAb was discussed. The OTSA computational process rapidly screens compounds in silico beyond the current in vitro off-target screening panels. Additionally, a structural approach was presented that exploits antigen–antibody interactions to predict potential Fab-mediated weak off-target effects.
Two examples were shared with the audience:
Case study 1: Here, a computational approach of combining transcriptomics and structural bioinformatics was applied to identify potential antigens for the ADC platform. A novel strategy was used to focus on conformational epitopes as mAb targets, which also grants cross-reactivity with preclinical species. In the example presented, the initial mAb had been designed to target the antigen (called Target 1), its selection was based on transcriptomics analysis. However, the designed mAb showed specific binding to the human target only and no binding was observed to the cynomolgus analogue of target 1. A multiple sequence alignment of target 1 with preclinical species suggested cynomolgus is closest to human, with a computed sequence identity of 85%. Further structural analysis on the computed models of human and cynomolgus target 1 revealed the presence of a conserved 25 amino-acid epitope on the target 1 surface. A new set of mAbs was therefore designed for this conserved region of target 1. Some of these mAbs targeting this region provided cynomolgus cross-reactivity.
Case study 2: To minimize nonspecific uptake of ADCs, binding of various carbohydrate moieties to MR2 were investigated by computational modeling. For this, a three-dimensional (3D) model of the lectin domain of human MR2 was constructed. The carbohydrate binding site of human MR2 is highly conserved and aligns with the computed structural model of cynomolgus MR2. Mono, di, tri and oligosaccharides of mannose were computationally docked onto the models of MR2 to identify optimal saccharide length needed to completely inhibit MR2 activity. The computational study suggested mannose trisaccharide is structurally compatible to the binding site of MR2. Thus, mannose trisaccharide (or its analogs) can potentially minimize MR2-mediated nonspecific uptake and its related adverse outcomes, which is in agreement with in vitro findings.
The presentation of Reiner Class (UCB) focused on the assessment of drug hepatotoxicity in 3D Liver Microtissues with an expanded panel of cytotoxicity markers (aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and ATP). Human 3D Liver Microtissues (hLiMT) consisting of primary liver cells are an attractive tool for in vitro drug safety assessment due to preservation of liver-specific functions and metabolic activity over a long culturing time. It was previously shown that hLiMT outperform 2D primary hepatocytes in prediction of hepatotoxicants causing drug-induced liver injury (DILI). However, biologics have never been tested in this system.
The capability of the in vitro system to discriminate between interferon (IFN)-lambda (clinically associated with hepatotoxicity) and IFN-alpha 2a (approved, no hepatotoxicity) was assessed. AST, a clinically relevant marker for liver damage, was combined with LDH and intracellular ATP, both established in vitro toxicity markers. Individual hLiMT were exposed to IFN-lambda and IFN-alpha 2a for 7 days with three compound additions. Measurement of all 3 markers was performed for a single microtissue, with LDH and AST measurements multiplexed in the same microtissue supernatant.
Cell viability profiles were able to show hepatotoxic effects of IFN-lambda, while IFN-alpha 2a had no effect. The decrease in ATP levels correlated well with an increase of AST and LDH signal, while a decrease in ATP levels correlated with a mild or no release of AST and LDH. These results suggest that hLiMT are a suitable tool for an in vitro assessment of hepatotoxic biologics and their potential mechanism of toxicity, and could be incorporated into the routine preclinical safety assessment.
Lauriane Cabon (Roche) gave an overview on 3D in vitro models currently developed and used to evaluate toxicities of immunotherapies. The field of cancer has recently seen outstanding progress, with the widespread adoption of immunotherapy leading to durable tumor regression in some patients.Citation33 However, these drugs can cause serious immune adverse events such as autoimmunity and tissue inflammation, some of which are life-threatening or irreversible.Citation34 Therefore, there is a great need for better understanding and prediction of responses to immunotherapies so that more patients can benefit from these breakthrough medicines. Because our immune system is the result of a long co-evolution with microbes, human immunity has specific features that are not captured in preclinical models such as mice or non-human primates.Citation35,Citation36 To bridge the gap between preclinical tools and clinical outcomes, the field has turned toward advanced, physiological human-relevant in vitro models of tissues and organs rather than standard cell cultures. Indeed, the latter cultures have previously failed to predict acute cardiac failure induced by T cell receptor-engineered T cells, whereas induced pluripotent stem cell-derived cardiomyocytes revealed the clinically observed off-target toxicity.Citation37,Citation38
3D cell cultures are assembled manually, using microfluidic devices or manufactured by bioprinting. Cells can be cultured in suspension, embedded in extracellular matrix (ECM)/hydrogel (as single cells, spheroids or organoids), cultured on top of the ECM/scaffold, or on tissue culture inserts for mechanical support and creating an air–liquid interface (ALI).Citation39 One major technical issue inherent to the field is the sourcing of fresh tissue material matched with peripheral blood mononuclear cells (PBMCs) or tissue-resident immune cells. Of course, the addition of relevant immune cell subsets increases the complexity of these assays. Several complex multicellular models are currently used at Roche to address various questions ranging from cytokines-mediated toxicities, on-target off-tumor toxicities, off-target cross-reactivity and autoimmunity, infections, inflammation risks. The multidisciplinary team of experts is focusing on clinically relevant readouts and use of marketed molecules to validate models, in partnership with external stakeholders.
Examples of 3D lung models applied to evaluate immunotherapy-induced pulmonary toxicities were then presented. Mentioned cases involved submerged and ALI cultures, airway organoids, primary cell alveolus-on-chip model and microfluidic lung-on-chip, all cocultured with PBMCs in order to recapitulate clinical toxicities such as interleukin (IL)-2-induced capillary leak syndrome. Dr. Cabon concluded with discussion on the way forward to advance these human in vitro tools enough to replace in vivo models in non-clinical studies and broaden their application to personalized safety.
Session 3: seeing is believing: new frontiers in imaging
The third session, chaired by Wouter Driessen (Roche) and Chantal Carrez (Sanofi), highlighted key case examples about how imaging can make an impact on drug development and aid in decision-making. The focus was on how quantitative imaging applied in preclinical studies can add value and novel insights from the discovery phase to the clinical stage. The session illustrated that by pushing the frontiers, imaging can become a cornerstone technology in drug development.
Iina Laitinen (Sanofi) presented on quantitative magnetic resonance imaging (MRI) of liver function providing a noninvasive safety biomarker.
THE TRISTAN APPROACH: Imaging biomarkers (IB) play an important role in drug development and healthcare.Citation40 When developed and validated, IBs can be invaluable in preclinical safety assessment, early-phase drug development, regulatory drug development, labeling, and managing adverse effects of drugs in clinical practice. One well-known example of important IBs is ultrasound-derived left ventricular ejection fraction in cancer therapeutics-related cardiotoxicity, which is used to manage safety issues all the way from preclinical to marketing, and they are frequently used by regulators in drug labeling.
A different roadmap is required for IB validation than for validation of conventional “biospecimen” biomarkers, and in particular parallel rather than sequential approaches to technical (or assay) validation, standardization, biological validation, and clinical validation are needed. It is also critically important to establish the benefit of any new IB over existing non-imaging assays, otherwise it is unlikely to be adopted.
The focus for this consortium is in imaging-based approaches for detection and characterization of drug-induced liver injury (DILI) and drug-induced interstitial lung disease (DIILD), as well as metabolic therapies that are included in our approach to better address bio-distribution of biologics, recognizing that drug safety is crucial in all disease areas.Citation41
TRISTAN LIVER IMAGING PROJECT: Inhibition of hepatobiliary transporters (particularly bile salt export pump) has been identified as a key initiating mechanism by which drugs may cause DILI. Furthermore, inhibition of hepatic uptake transporters may cause elevated drug concentrations in plasma, while inhibition of biliary efflux transporters can cause intra-hepatocyte drug accumulation; both of these processes could result in DDIs. Noninvasive whole-body imaging enables the direct investigation of hepatobiliary function in vivo, and quantification of effects of drugs on hepatobiliary transporter function.
The aim of the work package is to develop a standardized gadoxetate dynamic contrast–enhanced-MRI-based imaging biomarker, and demonstrate that this IB is valid for the assessment of liver transporter inhibition in rats and humans in vivo.
Michael Niehues (Bayer) presented on visualizing drug and drug effects in tissue. The investigation of tissue distribution, biotransformation and potential accumulation of drugs or drug candidates in the body is key to pharmaceutical research and development. In recent years, mass spectrometry imaging (MSI) has become an increasingly relevant label-free tool to study and image ex vivo the spatial distribution of xenobiotics and endogenous molecules in tissue sections or even small clinical biopsies.
Various ionization techniques are available to generate MSI data. However, the most commonly applied is matrix-assisted laser desorption/ionization (MALDI) MSI, often coupled to high-resolution mass spectrometers. Various practical examples were shown that illustrate the capabilities of MALDI-MSI to tackle questions in the field of non-clinical as well as clinical pharmacology, drug metabolism and pharmacokinetics (DMPK) and toxicology. One key feature of this technique is the capability to detect and visualize simultaneously dozens, if not hundreds of molecules from a single measurement. Hence, if detectable by MALDI-MS, such experiments generate knowledge on the spatial distribution of drug(s), metabolites and the effect of a drug on endogenous marker molecules (i.e., drug impact on neurotransmitters). Does a pharmacologically active compound reach the target region and at which concentration? What is the influence of formulation or administration route on drug distribution? Can MALDI-MSI data lead to the identification of biomarkers or help in tumor and tissue classification? What is the composition of drug-induced deposits in kidney and what is the mechanistic reason for such finding? These and other questions are illustrated of how MALDI-MSI can deliver meaningful answers to support the understanding of drug properties.
In conclusion, the broader applicability of MALDI-MSI is demonstrated with small molecules, but several reports also show that it is suitable for proteins and new modalities (i.e., oligonucleotides). As a label-independent approach, MALDI-MSI experiments allow multiplexed spatial analysis, which can be of great value for questions in early research, as well as for late development of drugs and beyond.
Erwan Jouannot (Sanofi) discussed imaging of biologics to support drug development. Over the past decade, mAbs and other targeted biologic therapies have emerged as transformative treatments for many diseases. Targeted biologicals have shown particular clinical benefits in oncology, with therapies inducing favorable tumor response and reducing severe adverse effects. Some biological properties of these agents can be investigated with nuclear imaging. ImmunoPET using labeled biologics as probes for positron emission tomography (PET) represents an exciting imaging option to noninvasively assess antigen expression in tumors and demonstrate tumor targeting of biological drugs. It further allows quantification of selective tumor uptake and highlights potential sinks in normal organs.
First, the use of immunoPET to characterize the in vivo fate of a series of trispecific mAb candidates in various preclinical models was described. The accumulation of the radioactive mAbs in normal organs was monitored noninvasively by quantification of the PET signal on whole-body scans. The PK profile of the radiolabelled mAbs was assessed by sampling 5 µl blood at regular time points post-probe administration. Final tissue biodistribution of labeled mAbs was assessed ex vivo by scintillation counting. The results highlighted the influence of the mouse model on biodistribution in normal organs and were useful to rank constructs/models for development.
The second example explored the tissue biodistribution in a rodent tumor model of an antibody recognizing a murine target and its human counterpart. The limited tumor uptake observed for the murine mAb even when increasing the dose was likely due to a high partition to the liver, which expressed the target. In contrast, no liver uptake was seen for the human antibody due to the lack of cross reactivity with the murine antigen.
Finally, the translational potential of an ADC that stimulates the immune system in rodents and NHP was explored. Results obtained on immunocompetent mice were nicely translatable to NHPs with liver and spleen uptake limiting blood exposure of the ADC compared to the naked antibody.
In conclusion, 89Zr-labeled mAbs are unique tools to noninvasively monitor tissue/tumor biodistribution and enable specific visualization of antigen-positive tumors in vivo. These preclinical experiments pave the way toward clinical application of immunoPET as a support to drug clinical development.
Finally, Pierre Maliver (Roche) discussed digital pathology in drug development. We generally distinguish classical from digital and computational pathology. Digital pathology encompasses not only the basic slide digitization, but also all the tools that allow the digital analysis of the slides, thanks to algorithms and/or artificial intelligence tools. Digital images are used especially in macroscopy or microscopy (remote peer reviews, slide annotation and image analysis). The pathologist is instrumental in all stages of the digital pathology workflow, and not only in the interpretation of image analysis results, in early development as well as in preclinical safety drug development.
Digital Pathology also provides the unique ability to perform a global study data evaluation digitally, associating digital pathology (like image analysis) with in life (clinical signs), clinical pathology and PKPD data. Therefore, digital pathology is part of a comprehensive, integrative preclinical study data evaluation in drug development.
Session 4: predicting immunogenicity during early drug development: hope or despair?
The fourth session of the meeting was chaired by Sven Kronenberg (Roche) and Daniel Kramer (Sanofi).
In the first presentation, Sebastian Spindeldreher (Integrated Biologix) shared some of the key results of the European Innovative Medicines Initiative project Anti-Biopharmaceutical Immunization: prediction and analysis of clinical relevance to minimize RISK (ABIRISK) to introduce to the topic. ABIRISK was a large consortium consisting of 38 partners from academia (26), pharma industry (9) and small- to medium-sized enterprises (3). The project started in March 2012 and ended in February 2018. The research in ABIRISK was divided into 4 work packages that were interlinked. One focused on the conduct of clinical studies, including cohort management, PK and ADA assay development and validation as well as sample analysis. The second one explored mechanisms of the anti-drug immune response in patient-derived samples aiming at increasing the general understand of immunogenicity and identifying markers that may predict an immune repose at a patient level. The third work package addressed the evaluation and development of technologies for predicting immunogenicity. The fourth work package ensured the connection of all data through a database and integration and evaluation of the data to enable identification of potential predictive patterns. In his presentation, Dr. Spindeldreher provided insights into the work performed under work package 3.
In the course of ABIRISK, several mouse models were developed and evaluated for utility in the assessment of antigenic potential of biotherapeutics. The so-called BRGSF mouse modelCitation42 was found to be the most promising one giving rise to a KLH-specific human IgM, IgG and CD4 T cell response that was human leukocyte antigen (HLA), and not mouse MHC, restricted (unpublished data). However, no responses were found to etanercept, Factor VIII, infliximab, or even to aggregated infliximab. Nevertheless, this is the first humanized mouse model for which a human IgG response was demonstrated, indicating that this model provides a good basis for further optimization.
Another model assessed was the human artificial lymph node assay.Citation43 In this assay some responses to KLH were observed, but, since these could not be consistently reproduced, the assay was not further evaluated in the context of ABIRISK.
An early event during an ADA response is the activation of the innate immune system. Therefore, ABIRISK researchers evaluated monocyte-derived DC activation assays for their capacity to respond to protein aggregates by looking at changes in mRNA expression, cytokine release, intracellular signaling, and changes in surface maturation maker expression. In this study, aggregated infliximab induced a marked activation of DCs as measured by an increase in CD83 and CD86 surface expression, IL-1β, IL-6, IL-8, IL-12, tumor necrosis factor (TNF), CCL3, and CCL4 mRNA upregulation and release of respective proteins, and phosphorylation of the intracellular signaling proteins Syk, ERK1/2, and Akt.Citation44 Adalimumab and rituximab aggregates induced only slight changes in these parameters and a challenge with natalizumab did not lead to any changes under these stress conditions.Citation44 This assay turned out to be quite robust since the source of monocytes and their differentiation into DCs did not affect results. Also, results from the different readouts were well aligned.
This was not the case for T cell assays. An objective of ABIRISK was the evaluation of the robustness of T cell assay applied for antigenicity assessment (unpublished data). Four different T cell assay protocols were applied to rank the biotherapeutics infliximab, rituximab, adalimumab, natalizumab, IFN beta-1a (Rebif®) and IFN beta-1b (Betaferon®), which show quite different immunogenicity incidence rates in the clinic. The four T cell assays followed very different approaches with regard to the cells used for the assay, the stimulation of the antigen presenting cells, the number of T cell (re-) stimulations and the readouts for T cell activation. The results of these studies indicated very different rank orders for the biotherapeutics. Of note, the labs running the T-cell assays were blinded, and thus could not adjust their assays to the mode of action of the biotherapeutics. However, some of the biotherapeutics, such as the IFNs and the TNF-blockers, have direct effects on the cells used in these in vitro assays. This confirms therefore that T-cell assays cannot be conducted blindly and without proper adaptation. Consequently, knowledge on the biology and mechanism of action of the drug is essential, as these can interfere with assays.
One of the major and most important outcomes of ABRISK for antigenicity assessment approaches was the demonstration of the biological relevance of the applied in vitro concepts and their translatability to patients. This “biological validation” was conducted by comparing T cell epitopes in patients and healthy volunteers. In this study, T cell epitopes were discovered which gave rise to recall responses of memory T cell derived from patients who had been treated with rituximab and infliximab and who developed immunogenicity during the course of the treatment.Citation45 The very same T cell epitopes were also recognized by naïve CD4 T cells derived from healthy, drug-naïve blood donors after in vitro stimulation with the same drugs. In addition, there was a substantial overlap of the identified T cell epitopes and the peptides presented in vitro on HLA class II by dendritic cells that were identified by MAPPs. These data indeed confirm that in vitro T cell epitope mapping is highly relevant for and translates into clinical T cell responses in patients who develop immunogenicity.
This approach was recently also applied to identify T cell epitopes in secukinumab and ixekizumab, two anti-IL-17 antibodies that are used to treat autoimmune diseases such as psoriasis and psoriatic arthritis.Citation46 Consistent with previous data, the frequency of preexisting T cells to secukinumab was significantly lower as compared with ixekizumab. In this study, the difference in the T cell precursor frequency could be attributed to a number of mutations in the complementarity-determining regions (CDRs) of ixekizumab that are not found in any of the germline family members.
In conclusion, it is still not possible to predict immunogenicity incidence or consequences, and there is still a need for better understanding of immunological events that trigger ADA and for better and novel technologies, such as appropriate animal models and improved in vitro models that better mimic in vivo and systems biology-based mathematical models. Nevertheless, quite some progress was made over the past 10 years and available assays can now be applied with much more confidence than in the past. ABIRISK increased the overall knowledge in the field and boosted interest in the academic environment, which is leading to an increase in research activities in the field of immunogenicity.
The goal of the second lecture by Ernesto Luna (Sanofi-Pasteur) was to discuss the utility of coupling in vitro biological assays of human immune function (the MIMIC system) with in silico (computational) prediction approaches to provide insights into the potential immunogenicity of drugs/vaccines intended for human use. The MIMIC® system is composed of a series of sensitive and reliable culture assays for the interrogation of human innate (short-term inflammation) and adaptive (long-term memory) immunity.Citation47,Citation48 Relying on the modular nature of the platform where various facets of immune function are evaluated in distinct assay systems, a 4-gate strategy () was devised to define how the MIMIC platform can be used to examine the sequential steps leading to an immune reaction: Gate 1 represents the analysis of whether a therapeutic agent is immunoreactogenic/immune-cytotoxic via induction of cytokines/chemokines associated with inflammation and lymphocyte stimulation; gate 2 reflects the ability of the drug compound to induce DC activation, maturation and cytokine secretion; gate 3 highlights whether it is antigenic/immunogenic via activation of autologous CD4 + T lymphocytes; and gate 4 represents the ability of biologics to drive B cell activation (antibody generation). Focusing on gate 3 (induction of CD4 + T cells response), datasets showing the utility (capacity to generate responses to naïve and recall antigens), sensitivity (capacity to recognize one or more antigenic peptides in a pool of non-antigenic peptides) and specificity (HLA-matched responses) of the MIMIC CD4 + T cell assay were highlighted. As a case study of coupling in silico and biological assays to examine CD4 T cell immunogenicity, published results were shown illustrating how in silico predictions that the newly emergent pandemic A/California/07/2009 (H1N1) virus had cross-reactive T cell epitopes with prior circulating influenza strains could be verified in the MIMIC CD4 T cell assay.Citation49 Through this analysis, it was shown that the computational tools used were 80–90% accurate in predicting CD4 + T cell epitopes and their HLA-DRB1-dependent response profiles. Combined, these results suggest coupling immuno-informatics to define CD4 + T cell epitopes with highly sensitive in vitro biological assays to verify these in silico predictions to understand human cellular immunity, including cross-protective responses, and to define CD4 + T cell epitopes. Based on the previous data using vaccines and certain immuno-therapeutics, MIMIC can be a useful translational platform to serve as a powerful tool to evaluate CD4 T cell immunogenicity and de-immunization strategies of immuno-therapeutics intended for human use.
Figure 1. Schematic of the 4-gate strategy of MIMIC immunogenicity evaluations
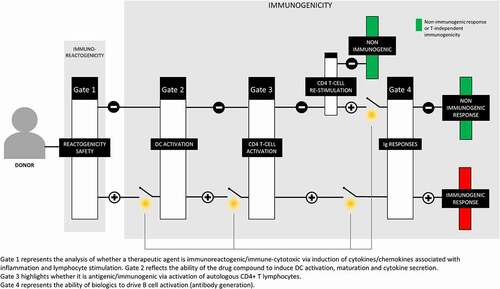
In the third presentation, Daniel Kramer (Sanofi) shed light on Sanofi´s strategy of how to best use available immunogenicity prediction tools during the development of biotherapeutics.
Although immunogenicity represents a substantial hurdle for the development of biologics, the biopharmaceutical industry will only learn about immunogenicity of their products once they have completed repeated dose clinical trials. For that reason, many companies are trying to “predict” human immunogenicity already early on, i.e., during the drug discovery phase. Unfortunately, immunogenicity in wild-type animals is not predictive for the human situation and genetically engineered animal models (e.g., humanized mice) are not yet considered mature enough for this purpose. Therefore, most companies currently focus on in silico T-cell epitope prediction and in vitro prediction tools such as DC/T-cell assays.
Both approaches, in silico and in vitro, have their intrinsic advantages, but also shortcomings. In silico algorithms provide a fast, cost-effective way to identify potential T-cell epitopes in biotherapeutics. In turn, they are usually over-predictive as binding of a peptide to the MHC-II is necessary, but not sufficient for a human ADA response. The major advantage of in vitro assays is that they do mimic a T cell-dependent immune response most closely, i.e., uptake of the therapeutic protein by antigen-presenting cells followed by digestion in their endosome, and presentation of fragments on the surface of MHC-II, but also activation of T-cells. However, they do require large numbers of donors reflecting the world´s MHC-II population and are labor intensive and quite variable. Their major downside though is that they are prone to react to several extrinsic (non-sequence derived) factors, such as aggregates or impurities. This limits their use in drug discovery, as early drug product batches are usually not well purified, bearing the risk of false positive results.
Currently no tool can predict the ADA incidence in humans. Because of that, their main application is limited to the assessment of relative immunogenicity. During drug discovery they should be used to select the leads/development candidates with the least number of predicted T-cell epitopes amongst equally scoring candidates in a multiparametric setting (potency, manufacturability, stability, PK, etc.). Sanofi uses in silico prediction algorithms for this purpose, as they offer high throughput and do not suffer from the presence of extrinsic immunogenicity factors in early drug product batches. Results at this stage might also guide further de-immunization during lead optimization if sequence liabilities (non-human T-cell epitopes) are identified in otherwise promising leads. However, due to the over-predictiveness of in silico tools, identified T-cell epitopes should always be confirmed via in vitro assays before embarking in de-immunization. This can be limited to peptides (instead of whole proteins) and reactive HLA alleles identified by the in silico tools.
During clinical development in vitro tools can support a comparability exercise in respect to critical quality attributes for immunogenicity (comparing pre-change to post change material). In vitro tools might also help during biosimilar development to address remaining concerns in respect to non-sequence-derived differences to the originator.
In summary, currently no immunogenicity prediction tool can predict the ADA incidence in humans. Consequently, prediction results for a single development candidate are not at all useful. Prediction tools find application when the assessment of relative immunogenicity is important. In drug discovery this is mainly the selection of the least immunogenic leads and development candidates in a multiparametric setting. During clinical development they are best suited to support a comparability exercise or the development of biosimilars in respect to extrinsic immunogenicity.
In the final talk, Céline Marban-Doran (Roche) provided the Roche perspective and insights on predicting immunogenicity. As more complex biotherapeutics become available on the market, the occurrence of immunogenicity is rising. The ADAs formed against a biotherapeutic can have dramatic consequences, such as loss of efficacy and neutralization of the drug or adverse events. Unwanted immunogenicity to biotherapeutics is a complex and multifactorial challenge, which makes it very difficult to predict with a single assay.
At Roche, a preclinical immunogenicity categorization for the clinical candidates has been implemented based on the outcome of preclinical in vivo and in vitro assays, but also other risk considerations such as ADA-related safety signals in toxicology.
The preclinical in vitro assays (DC-T cell assay, and MAPPs if a signal is observed in the DC-T cell assay) are performed during lead optimization with up to 5–6 candidates to address their sequence-based immunogenicity and to contribute to candidate lead selection. The selected candidate (or a mouse surrogate) is then tested in the in vivo human IgG1 transgenic mouse model to identify potential mechanism-related immunogenicity in vivo. The huIgG1 mouse model bears a mini-repertoire of human IgG1 proteins that makes it tolerant to human IgG1 antibodies.Citation50 Several Roche mAbs have been tested in this model and those with clinical immunogenicity showed high ADA response in the mice (unpublished data).
Based on the results of the preclinical immunogenicity testing, but also other risk considerations, clinical candidates fall into one of the following:
Category 1: no signal identified in any of the preclinical assay and no other risk considerations.
Category 2: signal detected in at least one preclinical assay or evidence of preexisting ADA but no other risk considerations.
Category 3: as per category 2, but with ADA-related safety signals in toxicology studies and/or other risk considerations, such as potential for neutralization of endogenous targets or ADA-mediated enhancement of mode of action.
The categorization of the clinical candidates is done between Clinical Candidate Selection and Entry-into-Human and is designed to guide the Entry-into-Human trial protocols (e.g., for bioanalytical assays on ADA measurement, frequency of sampling for ADA testing) and clinical ADA mitigation strategies.
Session 5: challenges in FIH dose projections
The fifth session was chaired by Andreas Baumann (Bayer) and Jennifer Fretland (Sanofi) and covered case examples for FIH dose projections for T-cell engagers, MABEL determination for agonists and other antibody-based therapies, and finally using optimal FIH dose and regimen at target identification to influence the design of multi-specific antibodies.
Wolfgang Richter (Roche) provided an overview of the entry-into-human (EIH) dose selection for a CD20-CD3-T-cell bispecific antibody based on predicted human cytokine release derived from PKPD data for cytokine release in cynomolgus monkeys. The selection of a safe, but as high as possible, EIH starting dose is a common challenge for immunomodulatory compounds. EIH starting doses for CD3-bispecific compounds are usually selected using the MABEL approach based on in vitro data. This approach often leads to very low starting doses. For the present CD20-CD3-T-cell bispecific antibody, alternative options were explored using PKPD data from cynomolgus monkeys. Cytokine release was selected as the PD endpoint, as it is related to both the mode of action and safety of T-cell bispecific antibodies. A PKPD model for cytokine release (IL-6, TNF and IFNγ release) was developed using monkey data obtained from toxicity studies. This PKPD model comprised a two-compartment PK model linked to an indirect response PD model. In the PD model, cytokine formation is a function of both drug and B cell levels in the circulation. Scaling to humans was done for the PK part using conventional allometric approaches, while for the PD part a correction was included for in vitro potency differences in cytokine release as determined in cynomolgus and human whole blood assays. The starting dose was selected using the scaled PKPD model to keep the projected cytokine level below a clinically acceptable threshold. IL-6 release was found to determine the starting dose. As the data in healthy cynomolgus monkey do not reflect potential cytokine release from tumor, a 10-fold safety factor was added for selection of the starting dose. This approach allowed EIH at a starting dose markedly higher than the one based on in vitro MABEL assessment. The starting dose was well tolerated. Comparison of projected and observed IL-6 release in patients indicated a tendency of the model to overestimate IL-6 release in patients.
Benno Rattel (Amgen) started his presentation by giving the regulatory history behind the generation of the EMA guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products issued in 2007 following the TGN1412 incident.Citation21,Citation22,Citation51 This guideline introduced the concept of using the MABEL to determine the Maximum Recommended Safe Starting Dose (MRSD) for FIH trials. MABEL is the dose/exposure that results in minimal PD effects in humans, which could be either a biological effect (e.g., cytotoxicity, T-cell activation) or expressed in terms of receptor occupancy of blood-based cell surface targets.Citation22 It was coined to understand the lowest animal dose, systemic exposure or concentration required to produce pharmacological activity in in vivo and/or in vitro in animal/human test systems. Consideration of a Pharmacologically Active Dose, however, had already been initially proposed in the FDA´s 2005 guideline about the MRSD.Citation52 As described in the above guideline the following modes of action and factors might require special attention: (1) A mode of action that involves a target which is connected to multiple signaling pathways (target with pleiotropic effects)for example, leading to various physiological effects, or targets that are ubiquitously expressed, as often seen in the immune system; (2) A biological cascade or cytokine release including those leading to an amplification of an effect that might not be sufficiently controlled by a physiological feedback mechanism (e.g., in the immune system or blood coagulation system); (3) The novelty of the agent, its biological potency and its mechanism of action; (4) The degree of species-specificity of the agent; and (5) The steepness of dose–response curves of biological effects in human and animal cells, dose–response data from in vivo animal studies, PK and PD modeling, the calculation of target occupancy versus concentration and the calculated exposure of targets or target cells in humans in vivo.
Initially, the minimum biological effect level was defined as an EC/ED20 in the most sensitive in vitro or in vivo test system. Though this approach guaranteed low and safe clinical starting doses, it has also resulted in multiple-dose steps/cohorts in FIH trials with anticancer drugs at subtherapeutic dosages, bringing no benefit to severely or terminally ill patients. To reduce the number of cohorts needed to reach clinically relevant doses with expected clinical PD effects, MABEL should be scrutinized to reflect the respective patient situation as closely as possible. Potential approaches include using ex vivo patient tumor material instead of very sensitive tumor cell lines, optimizing incubation times mimicking exposure duration in patients, simulating in vivo drug concentration profiles, reflecting the relevant hot or cold tumor effector to target cell ratios for T-cell engaging molecules or adding constituents to the buffer that play a critical role in vivo as they interfere with activity, e.g., shed target. These topics were also discussed in the MABEL Breakout#2.
Celine Amara (Sanofi) presented a case study regarding a stepwise approach to estimate FIH dose for an mAb used for the treatment of an autoimmune disease. She described the first step of calculating the MRSD, per the FDA Guidance. This calculation was based on the dose at the NOAEL from the repeated dose monkey GLP toxicology study. The second step was to use allometry to scale human PK from NHP targeting the NOAEL exposure observed in the GLP toxicology study. The last step was to incorporate PK/PD modeling using the TDAR-KLH inhibition observed in the preclinical TDAR study in nonhuman primates. The feedback from the regulators was supportive of the various approaches, but they challenged the use of the TDAR PKPD modeling approach due to the high variability in the data, which may lead to a poor translatability from monkey to human.
Finally, Lindsay Avery (Sanofi), described how the complexity of biotherapeutics has increased with an increase in modality diversity, including multispecific targeting. The addition of multispecific targeting increases the need for understanding how to best cover multiple targets with a fixed stoichiometry. An overview of model-based approaches used in early discovery was provided to guide the design of bispecific antibodies at the target identification stage. Each antigen may exhibit similar or different kinetic behaviors like half-life, internalization rates, and/or expression rates, which affect the amount of target coverage for each antigen with the use of a single bispecific biotherapeutic. A tiered model-based approach was presented to determine the amount of target coverage at a competitive dose and regimen. Once feasibility was assessed, further modeling was performed to determine ideal affinity ranges for each target to appropriately bind at the site of action. Sensitivity analysis was performed to understand each drug-related parameter and its impact on predicted target coverage. This approach guided teams for informed bispecific antibody design, prioritized experiments that would inform on the most sensitive parameters, and triaged the most challenging multispecific pairs.
Session 6: non-canonical biologics formats: challenges in bioanalytics, PKPD and biotransformation for complex biologics formats
The sixth session was co-chaired by Wolfgang Richter (Roche) and Tobias Paehler (Sanofi). This session addressed points to consider and some of the key challenges that arise with these non-canonical formats for bioanalysis, PK and biotransformation assessments. In addition, the PKPD assessment of multi-specific biologics were discussed.
Rita Martello and Olivier Pasquier (Sanofi) presented on bioanalytical tool to unravel the complexity of biologics. Measurements of biotherapeutics in biological matrix are essential during the drug development process in order to assess their PK, efficacy and safety. Traditionally, this has been carried out by using ligand-binding assays (LBA), but currently liquid chromatography coupled to mass spectrometry (LC-MS) is emerging as a promising technique.Citation53,Citation54
LC-MS assays are based on two approaches: bottom-up and intact analysis. In the bottom-up approach, biotherapeutics are subjected to tryptic digestion and the deriving peptides are then analyzed and quantified via LC-MS. In the intact analysis, biotherapeutics are analyzed and quantified based on the total molecular weight, without previous reduction to peptides.Citation55
LC-MS is gaining popularity for being highly selective and possessing multiplexing ability, whereas LBA performs better in terms of throughput and sensitivity.Citation56–59 A decision-tree that helps to select the right assay and three case studies using this approach were presented. Some factors to considers are: limit of quantification to be achieved, moieties to be quantified (total drug vs free drug) and whether in vivo integrity of the compound must be confirmed.Citation60
In the first case, the performance of bottom-up LC-MS and LBA was compared: both assays measured the same moiety of the drug (total) and showed similar results confirming the expectations. In the second case, three different assays were compared (bottom-up LC-MS, top-down LC-MS and LBA). Bottom-up LC-MS has been selected for an early screening, as it is not affected by ADA interference. The top-down approach has confirmed the in vivo integrity of the construct and LBA has been preferred as late-stage assay because of higher throughput. The third was a case study where two assays (LC-MS and LBA) that did not deliver the same results were compared; the reason might be due to the two methods measuring different moieties of the drug.
In conclusion, given the complexity of biotherapeutics analysis, a clear bioanalytical strategy should be established in order to successfully deliver reliable data. LBA and LC-MS can be considered either equivalent or complementary techniques: decisions must be driven by different factors such as the stage for the project and scientific questions to answer. Also, in order to correctly interpret the data, efforts should be invested in identifying the moiety of the biotherapeutics measured.
Another advantage of LC-MS is the ability to deeply characterize biotransformations and glycosylation heterogenicity, which might impact the efficacy and safety of biotherapeutics.Citation61–63
In a case example referring to a therapeutic IgG1 mAb, a deamidation of an asparagine (N) residue localized in the CDR1 of the heavy chain was observed within the drug substance batches to a limited degree of occurrence. This deamidation could potentially decrease the binding affinity toward its target antigen and the main concern was to know if this deamidation process could take place during the in vivo phase.Citation64,Citation65 Plasma samples from a PK study in NHP were subjected to a tryptic digestion and the specific tryptic peptide carrying the specific deamidation site was analyzed by LC – high-resolution MS to determine the ratio between its deamidated and non-deamidated forms over the PK profile, which was ultimately deemed to remain within acceptable limits despite its slight increase over time in vivo.
Furthermore, the glycan analysis of this mAb revealed the presence of Mannose-5 (Man 5) glycoform at low levels in addition to the main G0F and G1F glycoforms. The percentage of Man 5 species varied from batch to batch (from few % to about 15%) depending on the batch production yield, the longer incubation periods in the bioreactor, resulting in higher percentages of Man 5 glycoform. It is reported that Man five glycoform is subjected to a higher in vivo clearance due to specific Man five receptors as compared to the other glyco-variants.Citation66,Citation67 Concerning this mAb, it became necessary to define the maximum percentage of Man 5 acceptable in the different batches that would not significantly affect the overall in vivo clearance of the drug. Plasma samples from a specific PK study in NHP treated with two different batches of the mAb (e.g., low and high Man five species) were subjected to a tryptic digestion. The tryptic peptides EEQYNSTYR carrying the different N-glycans were monitored by LC – high-resolution MS to determine the PK profiles of each glycoforms. As anticipated, a higher overall clearance of the batch containing the higher percentage of Man 5 species was observed, and these results contributed to establish a specification regarding the maximum percentage of Man 5 species acceptable in further batches.
Overall, the use of LC-MS can allow the monitoring of targeted biotransformation evolution during the PK profile of biotherapeutics, as well as monitoring several co-administered forms of biologics in order to better investigate their safety and efficacy.
Armin Sepp (GSK) discussed going from mice to men and from in vivo to in silico.
Two-pore physiologically based PK for biologics (PBPK) describes the tissue distribution and elimination kinetics of soluble proteins as a function of their hydrodynamic radius and the physiological properties of the organs. Whilst rodent data allow the critically important organ-specific lymph flow rates to be estimated, the validation of the resulting model in humans remains largely confined to the plasma compartment only, as no tissue distribution data are available that would not be distorted by target-mediated binding of the molecules tracked in the studies. This limitation has been overcome with the data set from ThorneloeCitation68 where the tissue distribution time course of a high-affinity 89Zr-labeled albumin-binding domain antibody (AlbudAb™) GSK3128349 in healthy human volunteers was followed using positron emission tomography (PET/CT). The rodent data parameterized biologics PBPK model was found to provide the correct prediction of albumin-like plasma half-life for GSK3128349 AlbudAb, its volume of distribution and distribution-phase kinetics in humans. The model was subsequently adapted to evaluate the safety and DMPK properties of a 89R-labeled human domain antibody with no albumin-binding activity. The results obtained identified the risk that even a small overall administered dose of 15 MBq can result in high exposure in the kidneys due to rapid renal secretion of the molecule. As a result, the proposed study has not progressed.
Finally, Simone Schadt (Roche) presented about the increasing relevance of biotransformation studies for complex therapeutic proteins.Citation69 Biotransformation studies for therapeutic proteins are conducted in an issue-driven approach for compound optimization (stability, bioavailability), PK/PD characterization and to identify biotransformation products contributing to efficacy or safety. A typical trigger to embark on biotransformation studies is a mismatch between different bioanalytical assays (e.g., total versus target-binding competent), and biotransformation products may or may not be pharmacologically active. The toolbox for studying the biotransformation of therapeutic proteins includes affinity purification from biological matrices, separation by size exclusion, reverse phase or ion-exchange HPLC combined with radio detection and high-resolution MS. Two case examples of the biotransformation of immune cytokines were shared. Finally, the need for new tools that allow human in vitro assessment of large molecules ADME early on was discussed.
Conclusions
The nonclinical safety assessment of biopharmaceuticals is a multidisciplinary and integrated approach focusing on the specific characteristics related to the dedicated product in focus.
To meet the challenges posed by these novel modalities, an integrated nonclinical development approach is required for biopharmaceuticals, with close co-operation and alignment between pharmacologists, non-clinical safety scientists (toxicologists, pathologists, safety pharmacologists), as well as DMPK and bioanalytical scientists.
The use of relevant new technologies, development strategies and scientific information as well as a clear understanding of the current regulatory requirements with careful use of animals are essential components for building a solid development strategy.
The nineth annual BioSafe meeting in Europe served as a scientific platform for exchange and discussion, with an agenda allowing a focus on specific safety aspects (e.g., DART), but also on general regulatory challenges (e.g., non-animal testing strategies or FIH dose projection).
Through presentations, podium discussions and breakout sessions, attendees shared innovative ideas, development strategies and case studies, including overcoming non-clinical challenges (both scientific and regulatory). Such knowledge exchange promotes the use of optimized and streamlined non-clinical development programs for biopharmaceuticals (highest quality science applied to the minimum number of studies and animals used) that improves human risk assessment and facilitates early clinical and market entry of these innovative products.
Abbreviations
ABIRISK, Anti-Biopharmaceutical Immunization: prediction and analysis of clinical relevance to minimize RISK; ADA, anti-drug antibody; aTTP, acquired thrombotic thrombocytopenic purpura; BiTE®, bi-specific T-cell engager; CMV, cytomegalovirus; DART, Developmental And Reproductive Toxicology; EFD, embryo-fetal development; FIH, First-In-Human; GD, gestation day; KO, knock-out; LBA, ligand-binding assay; LC-MS, liquid chromatography – mass spectrometry; MSI, mass spectrometry imaging; NOAEL, no-observed-adverse-effect level; NHP, non-human primate; PET, positron emission tomography; PPND, pre-and postnatal development; TDAR, T-cell-dependent antibody response; TK, toxicokinetics; vWF, von Willebrand Factor; WoE, weight-of-evidence.
Disclosure statement
Thomas Kissner, Chantal Carrez, Daniel Kramer, Jennifer Fretland and Tobias Paehler are employees of Sanofi; Guenter Blaich and Petra Schmitt are employees of AbbVie; Andreas Baumann is an employee of Bayer AG; Sven Kronenberg, Ulrike Hopfer, Wouter Driessen and Wolfgang Richter are employees of Roche; Adam Hey is an employee of AstraZeneca; Andrea Kiessling is an employee of Novartis Pharma; Benno Rattel is an employee of Amgen.
References
- Kronenberg S, Baumann A, de Haan L, Hinton HJ, Moggs J, Theil FP, Wakefield I, Singer T. Current challenges and opportunities in nonclinical safety testing of biologics. Drug Discov Today. 2013;18(23–24):1138–17. doi:10.1016/j.drudis.2013.08.003.
- Baumann A, Flagella K, Forster R, de Haan L, Kronenberg S, Locher M, Richter WF, Theil FP, Todd M. New challenges and opportunities in nonclinical safety testing of biologics. Reg Tox Pharm. 2015;69(2):226–33. doi:10.1016/j.yrtph.2014.04.005.
- Brennan FR, Baumann A, Blaich G, de Haan L, Fagg R, Kiessling A, Kronenberg S, Locher M, Milton M, Tibbits J. Nonclinical safety testing of biopharmaceuticals – addressing current challenges of these novel and emerging therapies. Reg Tox Pharm. 2015;73(1):265–75. doi:10.1016/j.yrtph.2015.07.019.
- Blaich G, Baumann A, Kronenberg S, de Haan L, Ulrich P, Richter WF, Tibbits J, Chivers S, Tarcsa E, Caldwell R. Non-clinical safety evaluation of biotherapeutics – challenges, opportunities and new insights. Reg Tox Pharm. 2016;80:1–14. doi:10.1016/j.yrtph.2016.08.012.
- Ulrich P, Blaich G, Baumann A, Fagg R, Hey A, Kiessling A, Kronenberg S, Hvid Lindecrona R, Mohl S, Richter WF. Biotherapeutics in non-clinical development: strengthening the interface between safety, pharmacokinetics-pharmacodynamics and manufacturing. Reg Tox Pharm. 2018;94:91–100. doi:10.1016/j.yrtph.2018.01.013.
- Sargentini-Maier ML. Clinical pharmacology of caplacizumab for the treatment of patients with acquired thrombotic thrombocytopenic purpura. Expert Rev Clin Pharmacol. 2019;12(6):537–45. doi:10.1080/17512433.2019.1607293.
- Wehner NG, Shopp G, Rocca MS, Clarke J. Effects of natalizumab, an alpha4 integrin inhibitor, on the development of hartley guinea pigs. Birth Defects Res B Dev Reprod Toxico. 2009;86(2):98–107. doi:10.1002/bdrb.20189.
- Wehner NG, Skov M, Shopp G, Rocca MS, Clarke J. Effects of natalizumab, an alpha4 integrin inhibitor, on fertility in male and female guinea pigs. Birth Defects Res B Dev Reprod Toxicol. 2009;86(2):108–16. doi:10.1002/bdrb.20191.
- Krayer JA Unique Toxicology development program: tremfya an anti-IL23 inhibitor for the treatment of psoriasis and other indications. Presented at: Charles River 24th Annual Biotech symposium: Biotechnology-derived therapeutics. Perspectives on non-clinical development; 2018 Sep; La Jolla, CA.
- Suckow M, Stevens K, Wilson R, The laboratory rabbit, guinea pig, hamster, and other rodents. American College of Laboratory Animal Medicine. 2012;1288. Elsevier Inc. doi:10.1016/C2009-0-30495-X.
- Pentsuk N, van der Laan JW. An interspecies comparison of placental antibody transfer: new insights into developmental toxicity testing of monoclonal antibodies. Birth Defects Res B Dev Reprod Toxicol. 2009;86(4):328–44. doi:10.1002/bdrb.20201.
- Nonclinical Evaluation for Anticancer Pharmaceuticals. ICH guideline S9. International Council for Harmonisation; 2009. https://database.ich.org/sites/default/files/S9_Guideline.pdf.
- Questions and Answers Nonclinical Evaluation for Anticancer Pharmaceuticals. ICH document S9 Q&A. International Council for Harmonisation; 2018. https://database.ich.org/sites/default/files/S9_Q%26As_Q%26As.pdf.
- CDER Pharmacology Review BLA 761036. U.S. food and drug administration. 2015. [accessed 2019 Nov 8]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/761036Orig1s000PharmR.pdf.
- Rocca M, Morford LL, Blanset D, Halpern WG, Cavagnao J, Bowman CJ. Applying a weight of evidence approach to the evaluation of developmental toxicity of biopharmaceuticals. Regulatory Toxicology and Pharmacology. 2018;98:69–79. doi:10.1016/j.yrtph.2018.07.006.
- Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. ICH guideline S6(R1). International Council for Harmonisation; 2011. https://database.ich.org/sites/default/files/S6_R1_Guideline_0.pdf.
- Detection of toxicity to reproduction for medicinal products & toxicity to male fertility. ICH guideline S5(R2). European Medicines Agency; 1993. [accessed 2019 Nov 8]. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s-5-r2-detection-toxicity-reproduction-medicinal-products-toxicity-male-fertility-step-5_en.pdf.
- Detection of toxicity to reproduction for human pharmaceuticals. ICH guideline S5(R3). International Council for Harmonisation; 2019. Accessed 2019 Nov 11. https://www.ich.org/page/safety-guidelines.
- Nonclinical safety testing in support of development of paediatric medicines. ICH draft guideline S11 step 2b. European Medicines Agency; 2018. [accessed 2019 Nov 9]. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-s11-nonclinical-safety-testing-support-development-paediatric-medicines-step-2b-draft_.pdf.
- Guidance for industry: considerations for developmental toxicity studies for preventive and therapeutic vaccines for infectious disease indications. FDA. 2006. [accessed 2019 Nov 11]. http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/ucm092170.pdf.
- Guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products. European medicines agency. 2007. [accessed 2019 Nov 12]. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-mitigate-risks-first-human-clinical-trials-investigational-medicinal_en.pdf.
- Guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products. European medicines agency. Revision 1, 2017 [accessed 2019 Nov 8]. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational_en.pdf.
- Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci USA. 1989;86(16):6077–81. doi:10.1073/pnas.86.16.6077.
- Wolff JA, Malone RW, Williams P, Chong W, Ascadi G, Jani A. Direct gene transfer into mouse muscle in vivo. Science. 1990;247(4949):1465–68. doi:10.1126/science.1690918.
- Reflection paper on the qualification of non-genotoxic impurities (Draft). European medicines agency. 2018. [accessed 2019 Nov 11]. EMA/CHMP/SWP/545588/2017, https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-qualification-non-genotoxic-impurities_en.pdf.
- Reviewing the use of alternative methods in biomedical research. The JRC’s EU reference laboratory for alternatives to animal testing (EURL ECVAM). European Commission; 2018 [accessed 2019 Nov 12]. https://ec.europa.eu/jrc/en/science-update/alternative-methods-biomedical-research.
- Zuang V, Viegas Barroso JF, Belz S, Berggren E, Bernasconi C, Bopp S, Bouhifd M, Bowe G, Campia I, Casati S. EURL ECVAM Status Report on the Development, Validation and Regulatory Acceptance of Alternative Methods and Approaches (2017), EUR 28823, Luxembourg (LUX/Luxembourg): Publications Office of the European Union. 2017. http://publications.jrc.ec.europa.eu/repository/handle/JRC108831.
- Zuang V, Dura A, Asturiol BD, Viegas Barroso FF, Batista Leite S, Belz S, Berggren E, Bernasconi C, Bopp S, Bouhifd M. EURL ECVAM Status Report on the Development, Validation and Regulatory Acceptance of Alternative Methods and Approaches (2018), EUR 29455. Luxembourg (LUX/Luxembourg): Publications Office of the European Union, 2018. http://publications.jrc.ec.europa.eu/repository/handle/JRC113594.
- Summary Report of the Heads of Medicines Agencies (HMA) - EMA Joint Big Data task force. European Medicines Agency; 2019 Feb 13. https://www.ema.europa.eu/en/documents/minutes/hma/ema-joint-task-force-big-data-summary-report_en.pdf.
- Predictive safety testing consortium. Homepage. Critical Path Institute; 2006 Mar 16. https://c-path.org/programs/pstc/.
- Paini A, Leonard JA, Joossens E, Bessems JGM, Desalegn A, Dorne JL, Gosling JP, Heringa M, Klaric M, Kliment T. Next generation physiologically based kinetic (NG-PBK) models in support of regulatory decision making. Computational Toxicology. 2019;9:61–72. doi:10.1016/j.comtox.2018.11.002.
- Rao M, Gupta R, Liguori M, Hu M, Huang X, Mantena S, Mittelstadt SW, Blomme EAG, Van Vleet TR. Novel computational approach to predict off-target interactions for small molecules. Front Big Data. 2019;2:25. doi:10.3389/fdata.
- Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–50. doi:10.1038/s41568-019-0116-x.
- Martins F, Sofiya L, Sykiotis G, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–80. doi:10.1038/s41571-019-0218-0.
- Saber H, Gudi R, Manning M, Wearne E, Leighton JK. An FDA oncology analysis of immune activating products and first-in-human dose selection. Regul Toxicol Pharmacol. 2016;81:448–56. doi:10.1016/j.yrtph.2016.10.002.
- Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20(2):273–86. doi:10.1093/biostatistics/kxx069.
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra103. doi:10.1126/scitranslmed.3006034.
- Sanderson JP, Crowley DJ, Wiedermann GE, Quinn LL, Crossland KL, Tunbridge HM, Cornforth TV, Barnes CS, Ahmed T, Howe K. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. OncoImmunology. 2019 Nov;9(1):1682381. doi:10.1080/2162402X.2019.1682381
- Koledova Z. 3D cell culture: an introduction. Methods Mol Biol. 2017;1612:1–11. doi:10.1007/978-1-4939-7021-6_1.
- IMI-TRISTAN. Innovative medicines initiative - translational imaging in drug safety assessment homepage. Bayer AG; [accessed 2019 Nov 12]. https://www.imi-tristan.eu/.
- Kenna JG, Waterton JC, Baudy A, Galetin A, Hines CDG, Hockings P, Patel M, Scotcher D, Sourbon S, Ziemian S. Methods in pharmacology and toxicology, New York, NY, humana press, noninvasive preclinical and clinical imaging of liver transporter function relevant to drug-induced liver injury. Drug-Induced Liver Toxicity. 2018:125–50. doi:10.1007/978-1-4939-7677-5_30.
- Li Y, Mention JJ, Court N, Masse-Ranson G, Toubert A, Spits H, Legrand N, Corcuff E, Strick-Marchand H, Di Santo JP. A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur J Immunol. 2016. doi:10.1002/eji.201546132.
- Giese C, Demmler CD, Ammer R, Hartmann S, Lubitz A, Miller L, Müller R, Marx U. A human lymph node in vitro - challenges and progress. Artif Organs. 2006;30(10):803–08. doi:10.1111/j.1525-1594.2006.00303.x.
- Morgan H, Tseng SY, Gallais Y, Leineweber M, Buchmann P, Riccardi S, Nabhan M, Lo J, Gani Z, Szely N. Evaluation of in vitro assays to assess the modulation of dendritic cells functions by therapeutic antibodies and aggregates. Front Immunol. 2019;10:1–14. doi:10.3389/fimmu.2019.00601.
- Hamze M, Meunier S, Karle A, Gdoura A, Goudet A, Szely N, Pallardy M, Carbonnel F, Spindeldreher S, Mariette X. Characterization of CD4 T Cell epitopes of infliximab and rituximab identified from healthy donors. Front Immunol. 2017 May 05;8. doi:10.3389/fimmu.2017.00500.
- Spindeldreher S, Karle A, Correia E, Tenon M, Gottlieb S, Huber T, Maillere B, Kolbinger F. T Cell epitope mapping of Secukinumab and Ixekizumab in healthy donors. MAbs. 2020;12(1):1707418. doi:10.1080/19420862.2019.1707418.
- Drake III DR, Nguyen MN, Kachurin A, Wittman V, Parkhill R, Moser JM, Burdin N, Moreau M, Mistretta N, Byers AM. Abstract. In vitro biomimetic model of the human immune system for predictive vaccine assessments. DISRUPTIVE SCIENCE AND TECHNOLOGY. 2012;1:28–40. doi:10.1089/dst.2012.0006.
- Higbee RG, Byers AM, Dhir V, Drake D, Fahlenkamp HG, Gangur J, Kachurin A, Kachurina O, Leistritz D, Ma Y. An immunologic model for rapid vaccine assessment - a clinical trial in a test tube. Altern Lab Anim. 2009;37(Suppl 1_suppl):19–27. doi:10.1177/026119290903701S05.
- Schanen BC, De Groot AS, Moise L, Ardito M, McClaine E, Martin W, Wittman V, Warren WL, DrakeIII DR. Coupling sensitive in vitro and in silico techniques to assess cross-reactive CD4(+) T cells against the swine-origin H1N1 influenza virus. Vaccine. 2011;29(17):3299–309. doi:10.1016/j.vaccine.2011.02.019.
- Bessa J, Boeckle S, Beck H, Buckel T, Schlicht S, Ebeling M, Kiialainen A, Koulov A, Boll B, Weiser T. The Immunogenicity of antibody aggregates in a novel transgenic mouse model. Pharm Res. 2015;32(7):2344–59. doi:10.1007/s11095-015-1627-0.
- Expert Scientific Group on Phase One Clinical Trials. Expert Scientific Group on Phase One Clinical Trials: Final Report. London (UK): The Stationery Office; 2006.
- FDA: guidance for industry estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. 2005 Jul. https://www.fda.gov/media/72309/download.
- An B, Zhang M, Qu J. Toward sensitive and accurate analysis of antibody biotherapeutics by liquid chromatography coupled with mass spectrometry. Drug Metabolism and Disposition. 2014;42(11):1858–66. http://dmd.aspetjournals.org/content/42/11/1858.pdf. doi:10.1124/dmd.114.058917.
- Law WS, Genin JC, Miess C, Treton G, Warren AP, Lloyd P, Krantz C. Use of generic LC–MS/MS assays to characterize atypical PK profile of a biotherapeutic monoclonal antibody. Bioanalysis. 2014;6(23):3225–35. https://future-science.com/doi/ 10.4155/bio.14.167.pdf.
- Boyne M, Bose R Target Proteins: bottom‐up and top‐down proteomics. In: Gross ML, Chen G, Pramanik BN, editors. Protein and Peptide Mass Spectrometry in Drug Discovery. 2011. p. 89–100. https://onlinelibrary.wiley.com/doi/pdf/ 10.1002/9781118116555.ch3.
- Ramagiri S, Hybridizing MI. LBA with LC-MS/MS: the new norm for biologics quantification. Bioanalysis. 2016;8(6):483–86. doi:10.4155/bio.16.9.
- An B, Zhang M, Qu J. LC/MS versus immune‐based bioanalytical methods in quantitation of Therapeutic Biologics in biological matrices. In: Gad SC, editor. Pharmaceutical Sciences Encyclopedia. 2015. p. 313–329. doi:10.1002/9780470571224.pse556.
- Zheng J, Mehl JT, Zhu Y, Xin B, Olah TV. Application and challenges in using LC-MS assays for absolute quantitative analysis of therapeutic proteins in drug discovery. Bioanalysis. 2014;6(6):859–79. doi:10.4155/bio.14.36.
- Xu W, Jiang H, Titsch C, Gadkari S, Batog A, Wang B, Pillutla R. Concerted application of LC–MS and ligand binding assays to better understand exposure of a large molecule drug. Bioanalysis. 2018;10(16):1261–72. https://ncbi.nlm.nih.gov/pubmed/29923414.pdf. doi:10.4155/bio-2018-0108.
- Lee JW, Kelley M, King L, Yang J, Salimi-Moosavi H, Tang MT, Rogers C. Bioanalytical approaches to quantify “Total” and “Free” therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. Aaps Journal. 2011;13(1):99–110. https://ncbi.nlm.nih.gov/pmc/articles/pmc3032085.pdf. doi:10.1208/s12248-011-9251-3.
- Haberger MB. Assessment of chemical modifications of sites in the CDRs of recombinant antibodies Susceptibility vs. functionality of critical quality attributes. mAbs. 2014;6(2):327–39. doi:10.4161/mabs.27876.
- Yu MB. Production, characterization and pharmacokinetic properties of antibodies with N-linked Mannose-5 glycans. mAbs. 2012;4(4):475–87. doi:10.4161/mabs.20737.
- Xu X, Qiu H, Li N. LC-MS multi-attribute method for characterization of biologics. Journal of Applied Bioanalysis. 2017;3(2):21–25. http://betasciencepress.com/index.php/jab17003.pdf. doi:10.17145/jab.17.003.
- Sydow JL. Structure-based prediction of Asparagine and Aspartate degradation sites in antibody Variable Regions. PLOS ONE. 2014;9(6):e100736. doi:10.1371/journal.pone.0100736.
- Bults PB. LC-MS/MS-based monitoring of in vivo Protein biotransformation: quantitative determination of Trastuzumab and its deamidation products in human plasma. Anal Chem. 2016;88(3):1871–77. doi:10.1021/acs.analchem.5b04276.
- Higel FS. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. European Journal of Pharmaceutics and Biopharmaceutics. 2016;100:94–100. doi:10.1016/j.ejpb.20.
- Zhou QQ. The mechanistic impact of N-Glycosylation on stability, pharmacokinetics, and immunogenicity of therapeutic Proteins. J Pharm Sci. 2019;108(4):1366–77. doi:10.1016/j.xphs.2018.11.029.
- Thorneloe KS, Sepp A, Zhang S, Galinanes-Garcia L, Galette P, Al-Azzam W, Vugts DJ, van Dongen G, Elsinga P, Wiegers J. The biodistribution and clearance of AlbudAb, a novel biopharmaceutical medicine platform, assessed via PET imaging in humans. EJNMMI Res. 2019;9(1):45. doi:10.1186/s13550-019-0514-9.
- Schadt S, Hauri S, Lopes F, Edelmann MR, Staack RF, Villaseñor R. Are biotransformation studies of therapeutic Proteins needed? scientific considerations and technical challenges. Drug Metabolism and Disposition. 2019;47(12):1443–56. doi:10.1124/dmd.119.088997.