
ABSTRACT
Bispecific antibodies (bsAbs) recognize and bind two different targets or two epitopes of the same antigen, making them an attractive diagnostic and treatment modality. Compared to the production of conventional bivalent monospecific antibodies, bsAbs require greater engineering and manufacturing. Therefore, bsAbs are more likely to differ from endogenous immunoglobulins and contain new epitopes that can increase immunogenic risk. Anti-A/B is a bsAb designed using a ‘knobs-into-holes’ (KIH) format. Anti-A/B exhibited an unexpectedly high immunogenicity in both preclinical and clinical studies, resulting in early termination of clinical development. Here, we used an integrated approach that combined in silico analysis, in vitro assays, and an in vivo study in non-human primates to characterize anti-A/B immunogenicity. Our findings indicated that the immunogenicity is associated with epitopes in the anti-B arm and not with mutations engineered through the KIH process. Our results showed the value of this integrated approach for performing immunogenicity risk assessment during clinical candidate selection to effectively mitigate risks during bsAb development.
Introduction
Bispecific antibodies (bsAbs) are engineered antibodies that bind two different antigens or two distinct epitopes of an antigen.Citation1 Given that some bsAbs have enhanced clinical efficacy compared to conventional monospecific monoclonal antibodies (mAbs), the use of bsAbs is emerging as an important component in cancer, autoimmune, and infectious disease treatment.Citation2 Over the past two decades, advances in protein engineering have led to a growing interest in the development of bsAbs for treatment of human diseases. As a result, more than 100 bsAbs are being evaluated in clinical trials for diagnostic and therapeutic applications.Citation3
The bsAb formats can be extensively engineered and contain various mutations such that these molecules differ from the sequences encoded by the human immunoglobulin genes. Therefore, bsAbs may have a higher immunogenic potential than conventional mAbs.Citation4–6 The development of immunogenicity and anti-drug antibodies (ADAs) can lead to reduced therapeutic efficacy, which limits the use of biotherapeutics in clinic. Indeed, 100% of patients treated with LY3415244, a bsAb against TIM-Citation3 and PD-LCitation1, developed ADAs, and hypersensitivity reactions were observed in 100% of patients treated with LY3415244, a bsAb against TIM-3 and PD-L1, developed ADAs, and hypersensitivity reactions were observed in 2 of the 12 (17%) patients, which led to early trial termination.Citation7 Despite these potential drawbacks, other bsAbs currently in the clinic were shown to be efficacious and exhibit acceptable low rates of immunogenicity.Citation8 In addition, several useful approaches have been proposed to manage the immunogenicity of bsAbs in patients.Citation9 Thus, bsAbs remain viable therapeutic candidates for clinical development.
Although the complete immunogenicity profile of a biotherapeutic can only be definitively characterized through costly and time-consuming clinical trials, various preclinical approaches can be used to assess T cell‐dependent immunogenicity. These include computational analysis to predict T cell epitopes, in vitro T cell assays and preclinical studies with animal models. Such methods can be informative for differentiating highly immunogenic candidates and improving overall safety by mitigating ADA generation.Citation10 Over the course of several optimization procedures to reduce immunogenicity risk by in silico T cell epitope prediction system, the number of T cell epitopes presented by emicizumab, a bsAb directed against FIXa and FX, were minimized, enabling the generation of a bsAb with low ADA incidence.Citation9
Most bsAbs are generated using one of two methods: chemical conjugation with crosslinkers, or genetic (protein or cell) engineering.Citation11,Citation12 Due to the homodimerization of heavy chains (HCs) and random pairing of light chains (LCs) to HCs, the co-expression of two different LCs and two different HCs in the same cell line can result in up to nine variants of undesirable mispaired species along with the target bsAbs.Citation13 One solution to overcome this pairing problem and increase the efficiency of recombinant bsAb production, is by using the ‘knobs-into-holes’ (KIH) technology. This is based on a combined protein engineering approach and assembly of the intact bsAb from two component half-antibodies. Mutations in the antibody CH3 domain promote selective HC heterodimerization while the expression of the half-antibodies circumvents the mispairing of an LC with a noncognate HC.Citation14–16
Anti-A/B is a full-length, humanized/human immunoglobulin G4 (IgG4) bsAb designed with the KIH technology. Administration of anti-A/B to healthy human subjects led to increasing levels of immunogenicity and an ADA positive incidence of 94% (16 of 17).Citation17 Despite the high ADA incidence, there was limited impact on anti-A/B exposure in the circulation and there were no apparent safety concerns. Moreover, robust immunogenicity responses against anti-A/B were observed in a multiple-dose toxicology study of cynomolgus monkeys. This study revealed an ADA-positive incidence of 97% (31 of 32) with a negative impact on the toxicokinetic (TK) and pharmacodynamics (PD) profile of anti-A/B in some animals.Citation17
In this study, we combined different approaches to characterize anti-A/B immunogenicity and evaluate whether KIH mutations contribute to the robust induction of ADAs. We used in silico and in vitro tools to rank immunogenicity of anti-A/B and three variant molecules anti-B/A, anti-B/B, and anti-A/A. The immunogenicity of these four molecules was also characterized and compared in vivo with cynomolgus monkeys. Our findings suggest that the observed immunogenicity is associated with epitopes in the anti-B arm and is not directly related to the mutations introduced during KIH engineering. Our results also highlight the value of using combined tools for evaluating immunogenicity risk early in development to reduce the likelihood of advancing a highly immunogenic candidate therapeutic and effectively mitigating financial and safety risks during the development phases.
Results
In silico immunogenicity analysis of anti-A/B bsAb variants
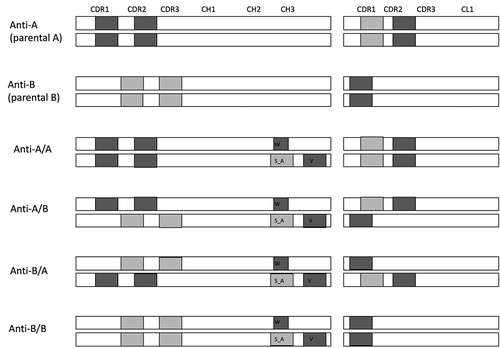
To characterize the source of immunogenicity in anti-A/B and identify the potential T cell epitopes in the LC and HC portions of anti-A/B, six sequence variants were analyzed with two in-silico applications: NetMHCIIpan 3.2,Citation18 and EpiMatrix. We tested the two parental monoclonal antibodies, anti-A monospecific (anti-A) and anti-B monospecific (anti-B), along with four antibodies carrying KIH mutations, anti-A/A, anti-B/B, anti-A/B, and anti-B/A (Supplementary Table 1). The parental molecules are humanized or human antibodies engineered in the antigen-binding fragment (Fab) to generate high-affinity binding to the target antigens, even though they lack the KIH mutations. The complete sequences of the antibodies (Fab + Fc portions) were examined. Sequences common to the human endogenous antibodies, based on alignment with human proteomic data, were excluded. To assess the potential immunogenicity risk of each molecule, we used NetMHCIIpan 3.2 to examine potential epitopes recognized by the 26 most common HLA-II (human leukocyte antigen class II) moleculesCitation19 (), or EpiMatrix with the nine common HLA-DR molecules (Supplementary Table 2). Both in silico analyses showed similar results. New epitopes were predicted for each of the molecules tested and these included those predicted to have strong binding to a single HLA-II or to several HLA-II molecules (promiscuous epitopes) ( and ). The predicted new epitopes were in the complementarity-determining region (CDR)1 and CDR2 of the HC and LC of anti-A, CDR2 and CDR3 of anti-B HC and CDR1 of anti-B LC, as well as in the KIH mutations regions (). Overall, the parental anti-A had more predicted new epitopes than did the parental anti-B, and the KIH mutations added ten strong binding epitopes three of which were promiscuous epitopes (). In addition, the anti-A/B and anti-B/A sequences had higher numbers of potentially strong binding epitopes and promiscuous epitopes in comparison to the parental anti-A and anti-B mAbs and anti-A/A and anti-B/B antibodies ( and Supplementary Table 2).
Table 1. In silico immunogenicity analysis of the parental monoclonal antibodies, anti-A/B, and its variant molecules based on NetMHCIIpan 3.2. T cell epitopes were identified based on their predicted binding affinity (percentile rank ≤10%) to 26 common HLA-II. Nine amino acid peptides that are identical to common endogenous human antibodies were excluded from the analysis
Figure 1. Schematic illustration of the areas predicted to have new T cell epitopes. New epitopes may be single or clusters (more than 1 epitope in close proximity). New epitopes were predicted by in silico immunogenicity analysis. Dark gray, promiscuous epitopes; light gray strong binding epitopes; knob mutation, T336W (w); hole mutations, T366S and L368A (S_A) and Y407V (v)
In vitro immunogenicity risk analysis of anti A/B bsAb variants
We assessed the immunogenicity risk of anti-A/B and the variants with KIH mutations in vitro using a T cell proliferation assay as previously described.Citation20 We examined enriched CD8−CD25low PBMC cells from 40 healthy donors in this in vitro assay. The cells were challenged with biotherapeutics and proliferating cells were evaluated by flow cytometry by quantifying the BrdU+CD4+CD3+CD14− cells. As controls and reference for biotherapeutics with high and low immunogenicity risk, we included HuA33, with a 73% ADA incidence,Citation21 and bevacizumab, with a 0.6% ADA incidence (USPI, 2020). Keyhole limpet hemocyanin (KLH) was used as a positive control. To quantify differences in immunogenicity risk, we calculated a stimulation index (SI) by dividing the maximum percentage of CD4+ proliferating cells for each treatment by the maximum percentage of proliferating CD4+ cells for unstimulated control cells. A positive donor response was determined based on SI greater than or equal to 2.0 (SI ≥ 2.0). Based on this threshold, the percentage of donors reacting to the bsAbs anti-A/B or anti-B/A was similar to the percentage of donors that reacted to HuA33 (). In addition, we performed a standard receiver-operated curve (ROC) analysis, which is a threshold-independent measurement (see Methods) and compared the T cell proliferation of the tested molecules to bevacizumab and HuA33 (Supplementary Table 3). Based on both SI ≥ 2.0 analysis and ROC analysis, anti-A/A led to T cell proliferation in a similar percentage of donors as bevacizumab, and had a lower immunogenicity in this assay than anti-A/B, anti-B/A and anti-B/B. These findings indicated that anti-A/B and anti-B/A may have an increased immunogenic risk compared to anti-A/A, even though this antibody carried the KIH mutations.
Figure 2. In vitro immunogenicity analysis of anti-A/B and three variant molecules. Peripheral blood mononuclear cells (PBMCs) from 40 donors were collected from human blood and incubated with the 4 test molecules and 3 control molecules (bevacizumab, HuA33, and keyhole limpet hemocyanin [KLH]) or tissue culture medium. The fraction of BrdU+CD4+ CD3+ CD14- was measured by flow cytometry. Percentage of donors positive for each testing molecule is calculated based on stimulation index (SI) ≥ 2 with a reference to KLH signals
![Figure 2. In vitro immunogenicity analysis of anti-A/B and three variant molecules. Peripheral blood mononuclear cells (PBMCs) from 40 donors were collected from human blood and incubated with the 4 test molecules and 3 control molecules (bevacizumab, HuA33, and keyhole limpet hemocyanin [KLH]) or tissue culture medium. The fraction of BrdU+CD4+ CD3+ CD14- was measured by flow cytometry. Percentage of donors positive for each testing molecule is calculated based on stimulation index (SI) ≥ 2 with a reference to KLH signals](/cms/asset/3507dc2e-7d1d-4f67-8dd1-0b02a96131f3/kmab_a_1944017_f0002_b.gif)
Cynomolgus monkey immunogenicity results and analysis
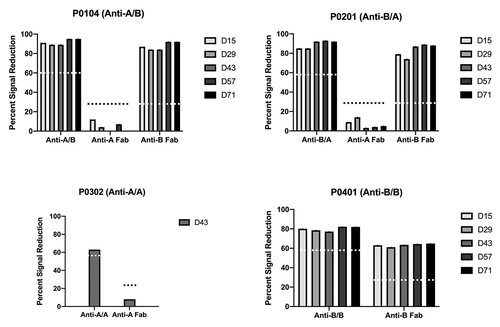
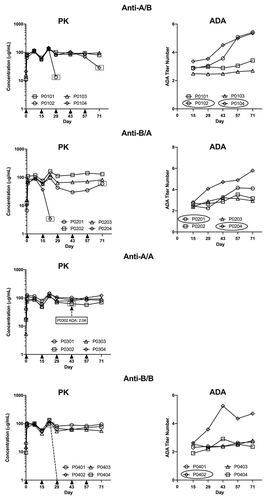
To assess immunogenicity in a preclinical in vivo experiment, we subcutaneously (SC) injected four male cynomolgus monkeys each once every 2 weeks for a total of 5 doses with 10 mg/kg anti-A/B or the variants or with the vehicle control article as the placebo. We monitored for the development of ADA by taking blood samples once at baseline; on days 15, 29, 43 and 57 prior to administration of the antibody; and on day 71. All of the animals receiving any antibody that contained the B portion anti-A/B, anti-B/A, or anti-B/B developed ADAs (). In contrast, only one (animal P0302) of the four animals receiving anti-A/A developed ADAs with a titer of 2.04; the ADAs were only detected on day 43. For the other antibodies, we observed a trend of increasing titer values throughout the study. Following the first dose on Day 15, titer values ranged from 1.90 to 3.36. In comparison, after treatment on Day 71, titer values ranged from 2.70 to 5.79 (). Titer values are reported in log scale, therefore the observed range represents 10- to 100-fold difference in ADA production.
Table 2. Immunogenicity of anti-A/B and its variant molecules in cynomolgus monkeys. Male monkeys received an injection of the indicated testing molecule subcutaneously once every 2 weeks for a total of 5 doses
To determine the target of the ADAs, we performed competition enzyme-linked immunosorbent assays (ELISAs) with the injected test molecule or the anti-A or anti-B Fab portions. The ADAs in all animals injected with molecules containing the anti-B portion were effectively competed by the injected molecule and the anti-B Fab, but were not competed by the anti-A Fab, as shown in the representative data (, ). These results suggested that ADAs were predominantly against the anti-B Fab portion. The only sample that was competed by anti-A Fab was from the one animal (P0302) that developed ADAs upon administration of anti-A/A ().
Figure 3. Representative characterization results of ADAs in animals treated with anti-A/B, anti-B/A, anti-A/A or anti-B/B. ADAs of each testing molecule were characterized using the corresponding competitive binding enzyme-linked immunosorbent assay (ELISA). Percent of signal reduction by pre-incubating the sample with the indicated molecule (anti-A/B, anti-B/A, anti-A/A, anti-B/B or the indicated Fab molecules) is shown. The dotted lines indicated the assay threshold for each test molecule
Of note, an earlier ADA competing experiment with the two parental whole molecules (anti-A and anti-B) and the two Fabs (anti-A Fab and anti-B Fab) from an original anti-A/B cynomolgus monkey toxicology study showed similar competing ability whether the whole parental molecules or the corresponding Fabs were used to perform the competition ELISA (Supplementary Figure 1)
Toxicokinetics in cynomolgus monkeys
In the same study, TKs of anti-A/B and its three variant molecules (anti-B/A, anti-A/A and anti-B/B) were characterized following five SC doses of 10 mg/kg once every 2 weeks (Q2W) in cynomolgus monkeys (). For each animal, we determined the maximum observed concentration after antibody administration (Cmax), time of the maximum observed concentration (Tmax), the area under the serum concentration curve from time 0 immediately before the first dose through the first 14 days (area under the curve (AUC)0–14), from the time of the last dose administered through the last 14 days (AUC56-70), and from time 0 to the last measurable time point (AUC0-last). Cmax peaked on day 21, after the second dose, for all antibodies in all animals except for 2 animals administered anti-B/A. In those two animals, anti-B/A concentrations peaked after the first dose (day 14). AUC0-last values across the four animals in each group with values ranging from a low of 4490 ± 2020 µg•day/mL for anti-B/B to a high of 6210 ± 996 µg•day/mL for anti-A/A. There was no evident accumulation observed after biweekly SC administrations as indicated by the similar AUC values after the last dose compared to the first dose.
Table 3. Toxicokinetic parameters of anti-A/B and its variant molecules in cynomolgus monkeys. Data were determined by non-compartmental analysis and are presented as mean ± standard deviation (N = 4)
Effect of ADAs on antibody concentration and safety
To evaluate the effect of ADAs on the exposure of anti-A/B, anti-B/A, and anti-B/B, we compared ADA titer and administered antibody concentration in matched animals (). Animals with the highest ADA titers had reduced concentrations of administered antibodies, indicating that the presence of ADAs negatively affected the exposure of the administered antibodies. In the anti-A/B group, animals P0102 and P0104 had significantly decreased concentrations of anti-A/B by day 29 and day 71, respectively, compared with the values in the preceding samples (, top). These animals also had the highest ADA titers, although the timing of ADA titer increase did not correlate precisely with the reduction in anti-A/B concentration. This may reflect the timing of the development of ADAs with higher binding affinities. A similar correlation between ADA titer and reduction in dosed antibody concentration was noted for animal P0204 administered anti-B/A and animal P0402 administered anti-B/B (). Animals with lower ADA titers had little or no change in the concentration of the administered antibody. Anti-A/A concentration was maintained throughout the dosing period in all animals, including animal P0302, which had a moderate ADA titer of 2.04 that was only detected on day 43 ().
Figure 4. Negative impact of the ADA response on exposure of the administrated antibodies. Administered antibody concentrations (left graphs) and ADA titers (right graphs) in animals administered anti-A/B, anti-B/A, or anti-B/B treated animals. For animals administered anti-A/A, the single animal with a detectable ADA titer at one time point only is indicated on the antibody concentration PK graph. Arrows on x axes of each PK plot indicated antibody injections. A dotted line indicates that the concentration of injected antibody was undetectable in the next sample. Animals with reduced injected antibody concentration are indicated with ovals in the ADA titer graphs and the symbols are similarly highlighted in the concentration graphs
No test item-related clinical observations were noted in any group during the study. Test item-related changes were limited to minor clinical pathology changes consisting of transient elevation in circulating eosinophils in all groups, but predominantly in animals administered anti-A/A and anti-A/B, and evidence of inflammation in animals P0104 and P0301 (mildly decreased albumin and increased globulins). These changes were not correlated with the presence of ADA. Macroscopic and microscopic pathology was limited to assessment of the injection site. Minimal, mononuclear, perivascular infiltrates were present in all groups, consistent with a normal reaction to injected heterologous protein material ().
Table 4. Microscopic findings at the injection sites of cynomolgus monkeys administered anti- A/B and its variant molecules
Discussion
The past decade has seen increased interest in bsAbs as a therapeutic modality. Currently, more than 100 bsAbs are in various clinical trials for the treatment of cancer and autoimmune or infectious diseases.Citation3 We observed a high immunogenicity with a bsAb, anti-A/B, following its administration in cynomolgus monkeys and humans.Citation17 We applied an integrated approach that combined in silico analysis, an in vitro T cell assay with human peripheral blood mononuclear cells (PBMCs), and an in vivo study in cynomolgus monkeys to assess the immunogenic responses to anti-A/B.
In silico methods are helpful for analysis of antibody properties and for predicting potential immunodominant epitopes within biotherapeutics.Citation22 Such computational analyses have been used to design biotherapeutics with less immunogenic risk by removing potential T cell epitopes.Citation9,Citation23 We used two predictive algorithms to identify peptides that can bind to HLA-II within the linear sequence of anti-A/B, its three variant molecules, and the two parental molecules. Our analyses indicated a higher number of potential T cell epitopes in two bsAbs, anti-A/B and anti-B/A, in comparison to the four monospecific antibodies with or without KIHs. This is presumably because the two bsAbs contain sequences of two different Fabs (each one corresponding to the parental antibody). Consequently, the repertoire of epitopes presented by the bsAbs were increased in comparison to those in the monospecific antibodies. This may contribute to the greater risk of immunogenicity observed in bsAbs. In addition, our computational analysis predicted that bsAbs, anti-A/B and anti-B/A, contain more promiscuous T cell epitopes than the four monospecific antibodies.
Consistent with the in silico analysis, anti-A/B and anti-B/A had a high immunogenic risk in both the in vitro T cell assay and the in vivo study with cynomolgus monkeys. The in vitro T cell assay indicated a relatively lower immunogenicity risk for anti-A/A than for anti-A/B, anti-B/A and anti-B/B. The in vivo study showed robust immunogenic response in animals treated with the two bsAbs and anti-B/B, but not anti-A/A. Additionally, in the cynomolgus monkeys the ADAs were predominantly against the anti-B Fab of anti-A/B, anti-B/A and anti-B/B, suggesting that high ADA incidence was restricted to the three molecules containing anti-B arm(s). Animals treated with anti-A/B versus anti-B/A had similar immunogenicity outcomes, indicating no difference in anti-B positioning relative to the arm containing the knob or the hole mutations. Taken together, these studies strongly suggested the anti-B Fab sequence is a key contributing factor to high immunogenicity.
Another interesting observation is that the anti-B arm of the bsAb is highly immunogenic, but not in the context of the anti-B parental molecule. Anti-B parental molecule, an IgG1 mAb without KIH modifications, had an ADA incidence of 21% (7 of 34), as determined in a previous anti-B cynomolgus monkey toxicology study. Although a low number of animals (n = 4) were tested in the current in vivo study, animals treated with anti-B/B (with KIH) had notably higher ADA incidence (100%, 4 of 4) and ADA responses compared to the anti-B parental molecule. For example, ADA titer, which was calculated in decimal logarithm, ranged from 1.90 to 5.24 for anti-B/B (with KIH) and 1.46 to 2.48 for anti-B parental molecule. The precise mechanism for this outcome is not fully understood. Overall, the product quality of the four KIH molecules was consistent with each other, and was similar to the two parental molecules produced in Chinese hamster ovary (CHO) cells as well. Nevertheless, a few notable factors, including difference in isotype (IgG1 vs IgG4), CHO vs E. coli expression (leading to differences in potential host cell protein impurities and the absence of glycosylation), as well as the antibody engineering work and production processes (standard mAb bioprocess vs. assembly from 2-cell KIH), might contribute to the different immunogenicity for anti-B and anti-B/B.
Many factors contribute to the generation of ADAs in patients, including route of administration, dosing regimen, and interaction of T cells and B cells in the follicular and germinal centers.Citation24,Citation25 These factors may explain the similar high immunogenicity risk observed with anti-A/B, anti-B/A and anti-B/B in our animal model and not with anti-A/A, despite anti-A/A having a higher number of predicted epitopes than anti-B/B. These discrepancies emphasize the need for multiple and combined tools to assess immunogenicity risk of biotherapeutics. Furthermore, our data indicate that anti-A/A with KIH mutations has a low immunogenicity risk, suggesting that KIH platform itself is likely not responsible for higher immunogenicity. This is supported by the low ADA associated with vanucizumab, an IgG1 bsAb that contains the KIH mutations.Citation26
Despite the advancements in tools for assessing immunogenicity of biotherapeutics and identification of T cell epitopes, limitations exist. One limitation for computationally predicting the binding affinity of epitopes to HLA-II is the tendency to over-predict the number of true epitopes that will be presented by HLA-II, be recognized by the T cell receptor (TCR), and lead to T cell activation. Additional experiments evaluating epitopes by proteomic analysis of major histocompatibility complex-associated peptides proteomics (MAPPs) might verify the number of potential epitopes that are truly presented by HLA-II molecules. With the advancement of artificial intelligence-based algorithms to recognize epitope presentation, as well as TCR-peptide HLA-II recognition, the immunogenicity predictions are expected to improve.Citation27,Citation28 In addition, experiments are warranted to evaluate whether the increased immunogenicity is caused by novel structural epitopes that are present in the anti-B/B and not in the parental anti-B antibody. Due to the low prediction power of computational B cell epitopes,Citation29 we focused on in silico examination of the potential T cell epitopes based on linear sequence. Thus, increased immunogenicity due to the conformational structure of anti-B/B or B cell immunodominant epitopes could not be ruled out. Those potential B cell epitopes could bind to antigen-specific surface B cell receptor and lead to activation of naïve B cells and a more robust ADA generation than was observed for anti-B.
The current in vitro assays can help assess the presence of T cell epitopes, but cannot account for the presence of B cell epitopes, patient genetics, or treatment-related factors. Those factors are crucial to regulating the immune response within the patient. B cell cloning work was planned to characterize immunodominant B cell epitopes of anti-A/B and its variants in the animal study. However, clone sorting of blood samples failed due to lack of probe specificity. Since they are closely related to humans, cynomolgus monkeys have considerable immunological similarities with humans that make them a good model for nonclinical safety assessment of novel biotherapeutics.Citation30 However, the genetic difference of HLA-II between humans and monkeys limits the use of monkeys as precise predictors of clinical ADA. Indeed, in a study evaluating 33 mAbs, the incidence of formation of ADAs in non-human primates and patients was comparable in only 59% of the antibodies tested.Citation31 In addition, animal studies can be costly and time consuming, therefore may not be a practical tool for routine use in immunogenicity ranking tests.
Considering that bsAbs hold greater risk to be immunogenic than monospecific antibodies, our findings highlight the need for early and comprehensive immunogenicity risk assessment of bsAbs prior to clinical candidate selection. This holistic approach can be extremely valuable to reduce immunogenicity associated with novel and complex molecules like bsAbs and to mitigate program development risks.
Materials and methods
Testing molecules
Anti-A/B is a humanized/human IgG4 bsAb therapeutic that binds to two soluble targets, A and B. The KIH mutations are introduced to the CH3 domain of the anti-A/B CH3 region, with the knob (K) mutation (T336W) in the anti-A CH3 region and the hole (H) mutations (T366S, L368A, or Y407V) in the anti-B CH3 region. The three variant molecules anti-B/A, anti-A/A, and anti B/B were generated following a similar process as anti-A/B with a few exceptions. Anti-B/A has the knob mutation in the anti-B CH3 and the hole mutations in the anti-A CH3; anti-A/A and anti-B/B have the same KIH containing CH3, but with a single target A or B, respectively. Anti-A/B and its three variant molecules were produced at Genentech (South San Francisco, CA) in E. coli. Briefly, two anti-A half-antibodies, carrying either knob or hole mutations, and two anti-B half-antibodies, carrying either knob or hole mutations, were expressed in E. coli and purified separately using Protein A affinity chromatography. Four bsAb molecules were then assembled in vitro, purified downstream, formulated in 20 mM histidine acetate, 240 mM sucrose, 0.03% (w/v) polysorbate 20, pH 6.0 ± 0.3, and filled into sterile vials and stored at 2–8 C. Testing of the four KIH products showed that they were suitable for use in animal studies and that they were of comparable quality. Size exclusion-HPLC performed according to industry standards was used to determine the level of HMW forms, or aggregates. The four KIH products were of similar purity and quality as the parental anti-B and anti-A monoclonal antibodies. Parental anti-B and anti-A/A had 1.1% high molecular weight (HMW). Parental anti-B had 0.9% HWM, Anti-A/B had 0.4% HMW, anti-B/A had 0.6% HWM and anti-B/B had 0.7% HWM.
Additional test antibodies
mcKLH (ImjectTM mcKLH; Thermo Fisher Scientific) was used as positive control and inclusion criteria for in vitro T cell immunogenicity assay. HuA33 was produced based on the published patent sequence. Both HuA33 and bevacizumab were produced in CHO cells.
In silico epitope prediction
The IEDB prediction tool for HLA‐II was used to predict potential T cell epitopes by applying the NetMHCIIpan 3.2 method.Citation18 For each sequence, we evaluated the potential binding affinity of the core peptide to the 26 most common HLA-DR, HLA-DP, and HLA-DQ alleles.Citation19 Based on the algorithm ranking, 15-mer peptides were considered potential T cell epitopes if the relative binding affinity ranking was within the top 10%. Epitopes predicted to bind to equal or more than five different HLA-II molecules were defined as potential promiscuous epitopes. Additionally, EpiMatrix (Epivax) algorithm was used to evaluate binding of epitopes to HLA-DR.Citation32 Sequences of nine amino acid peptides that were identical to sequences that exist in endogenous human antibody sequences were excluded from the analysis by performing blastp alignment to the sequence tested.
In vitro T cell immunogenicity assay
PBMCs were collected from anonymous healthy volunteers participating in the Genentech blood donor program, following written, informed consent from the Western Institutional Review Board. PBMCs were isolated by density gradient centrifugation using Uni-Sep blood separation tubes (Accurate Chemical & Scientific; Westbury, NY). Blood was obtained from Genentech blood donation program. BrdU assay to follow T cell proliferation in response to exposure to biotherapeutics was performed as previously described.Citation20 For each donor, responses were compared to a negative control, consisting of cells exposed to medium only (unstimulated cells).
T cell data analysis
The stimulation index (SI) was calculated by dividing the maximum percentage of CD4+ CD3+ CD14-BrdU+ cells of each treatment by the maximum percentage of CD4+ CD3+ CD14-BrdU+ cells of unstimulated cells. For determining the positive donors in the assay, a threshold of SI ≥2 was used. This threshold was determined based on previous experiments to allow maximum sensitivity without detecting large numbers of false-positive responses and provided the optimal signal-to-noise ratio in this assay for bevacizumab and HuA33. The percentage of donors that responded to a treatment (% positive donors) was calculated by dividing the number of donors that had a positive response (SI ≥ 2.0) by the total number of donors examined.
ROC analysis was done by generating ROCs and comparing the AUC of the tested molecules to bevacizumab and HuA33. A value of 0.5 for AUC indicates that there is no discriminatory ability between reference (HuA33 or bevacizumab) and molecule tested. ROC curves above 0.5 show a discriminating ability between reference and molecule tested.
Immunogenicity study of anti-A/B and variant molecules in cynomolgus monkeys
The in vivo study was conducted at Covance Laboratories, Madison, in accordance with the IACUC, the USDA Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals. Anti-A/B, anti-B/A, anti-A/A, or anti-B/B (10 mg/kg) were administered Q2W by SC injection to male cynomolgus monkeys for 8 weeks (5 total doses on days 1, 15, 29, 43, and 57). The study included a vehicle control (n = 1 animal) and four treatment groups with four animals in each group. Blood for TK analyses was collected at baseline, approximately 2 hours and 24 hours post dose on Day 1, once on days 8 and 22, and 71, and prior to the administration of the dose (predose) on days 15, 29, 43, and 57 and routinely processed to serum. Blood for ADA analyses was collected at baseline, once on day 71, and predose on days 15, 29, 43 and 57 and routinely processed to serum.
Animals were observed daily and subject to detailed clinical observations and body weight measurement on days of dosing and once on Study Days 8, 22, 36, 50, 64, and 71. Blood samples were collected for hematology, clinical chemistry, and coagulation parameter assessment twice at baseline and on Study Days 29, 57, and 71. Terminal assessments were limited to examination of the injection site.
ADA measurement in cynomolgus monkeys
ADAs for the four testing molecules were screened using four homogeneous bridging ELISAs as described previouslyCitation33 with a few modifications. The anti-A/B ADA assay is described in detail here and the anti-B/A, anti-A/A, and anti-B/B ADA assays were performed identically using cognate reagents (biotin- and digoxin [DIG] – conjugated testing molecules).
In the anti-A/B ADA assay, study serum samples were prepared at a minimum dilution of 1:20 in assay diluent [1× phosphate-buffered saline (PBS), 0.5% bovine serum albumin (BSA), 0.05% Tween 20%, and 0.05% ProClin 300, pH 7.4]; biotin- and DIG-conjugated anti-A/B (2 µg/mL) were co-incubated with pre-diluted controls or samples in a round-bottom polypropylene Costar plate (Cat#3365, Corning, Glendale, AZ) overnight at room temperature (RT) with agitation. Signal was detected with horseradish peroxidase-conjugated IgG fraction mouse anti-DIG mAb (40 ng/mL) (Cat# 200–032-156, Jackson ImmunoResearch, West Grove, PA).
Anti-B/A, anti-A/A and anti-B/B ADA assays had the same assay conditions as the anti-A/B ADA assay except the corresponding biotin- and DIG-conjugated testing molecule was used for each assay. The assay parameters, such as screening cutpoint factor (CPF), relative sensitivity, and drug tolerance and specificity were evaluated according to previously described recommendations.Citation34,Citation35 A sample with an assay signal equal or above the cutpoint was considered screening positive for the corresponding testing molecule. The qualified anti-A/B ADA assay had a screening CPF of 1.78 and relative assay sensitivity of 38 ng/mL based on a surrogate positive control (sheep anti-human IgG polyclonal antibody, Cat#AU003CUS01, Binding Site, San Diego, CA). The anti-B/A ADA assay had a screening CPF of 1.75 and a relative sensitivity of 45 ng/mL; the anti-A/A ADA assay had a CPF of 1.51 and sensitivity of 23 ng/mL; and the anti-B/B ADA assay had a CPF of 1.54 and sensitivity of 47 ng/mL. All four ADA assays detected 1 μg/mL of the surrogate positive control in the presence of 100 μg/mL of anti-A/B or the three variant antibodies.
ADA specificity measurement
Samples that tested positive in the screening assay were subjected to competitive ELISA assay to evaluate the immunogenic domain(s) targeted by the ADAs. Samples were pre-incubated in the presence (spiked) or absence (unspiked) of the corresponding testing molecule, anti-A Fab or anti-B Fab or the injected antibody, at RT for 1 hour, and then subjected to the screening homogeneous bridging ELISA described above. The percentage of signal reduction relative to that in the unspiked samples was calculated as follows: [(signals of unspiked samples – signals of spiked samples)/(signals of unspiked samples)] × 100.
The confirmatory threshold for each antibody or Fab was determined based on the percentage of signal reduction observed in serum samples from 25 drug‑naïve cynomolgus monkeys with a target untreated positive rate (UTPR) of 1%. A sample with a signal reduction equal or above the confirmatory threshold for the target molecule was considered positive for that molecule. The confirmatory thresholds for each target molecule are as follows: 45% for anti-A/B, 56% for anti-B/A, 59% for anti-A/A, 59% for anti-B/B, 26% for anti-A Fab, and 25% for anti-B Fab.
Toxicokinetic analysis in cynomolgus monkeys
A Gyros-based generic assay was developed to quantify serum concentrations of the four antibody molecules. Antibody molecules were captured with biotin-labeled sheep anti-human IgG (Binding Site Product Code AU003.M) diluted to 100 µg/mL in wash buffer 1 (1× PBS, 0.01% Tween 20, pH 7.4). Antibodies were detected with Alexa647-labeled sheep anti-human IgG diluted to 25 nM in Rexxip F buffer (Gyros Protein Technologies Product Number P0004825). Both capture and detection antibodies were conjugated using a 10:1 ratio of label:antibody. An 8-point standard curve encompassed the range 1.83 to 4000 ng/mL with a 1:3 serial dilution prepared in assay diluent (PBS, 0.5% BSA, 0.25% CHAPS, 5 mM EDTA, 0.35 M NaCl, 0.05% Tween 20, 0.05% ProClin300, pH 7.4 ± 0.1) containing 10% pooled serum from drug-naïve cynomolgus monkeys.
Samples were prepared at a minimum dilution of 1:10 in assay diluent, followed by four 1:2 serial dilutions. Capture reagent; standards, controls, and samples; and detection reagents were uploaded sequentially to Bioaffy 200 CDs (Gyros Protein Technologies Product Number P0004180), according to the vendor’s manual for analysis on the Gyros xP workstation.
The qualified generic Gyros pharmacokinetic assay has a standard curve ranging from 1.83 to 4000 ng/mL (in-well) at 1:3 serial dilutions. The lower limit of quantification of the assay is 300 ng/mL (neat concentration). Accuracy of the assay was assessed using controls prepared by spiking each testing molecule in pooled cynomolgus monkey serum at five concentrations representing the low, mid, and high portions of the assay standard curve. Assay accuracy ranged from 12.3% to 18.1%. The intra-assay coefficient of variation (CV) ranged from 4.6% to 9.1%.
The serum concentration versus time data were used to calculate clearance parameters in cynomolgus monkeys using noncompartmental analysis Phoenix® WinNonlin® version 6.4 (Certara USA, Inc., NJ). Nominal sample collection times and nominal dosing solution concentrations were used in the data analysis. All analyses were based on individual animal data. Maximum observed concentration after drug administration (Cmax), time of maximum concentration observed (Tmax), area under the serum concentration-time curve after first (AUC0-14) and last doses (AUC56-70), and area under the serum concentration-time curve from time 0 to the last measurable time point (AUC0-last) were reported as mean ± standard deviation.
List of abbreviation
Supplemental Material
Download Zip (457.4 KB)Acknowledgments
The authors would like to thank Patricia Y. Siguenza and Paul Carter for their support of the project and colleagues at the Department of Protein Analytical Chemistry, the Core Biophysical Characterization and Reagent Facility, and the Critical Reagents System for providing critical reagents. The authors would like to thank Richard Vandlen for providing critical assay reagents. Additionally, we thank Nancy R. Gough (BioSerendipity, LLC) and Anshin Biosolutions for editorial assistance.
Disclosure statement
All authors are Genentech employees and stockholders of the Roche Group. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–10. doi:10.1016/j.molimm.2015.01.003.
- Li H, Saw PE, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. 2020;17:451–61. doi:10.1038/s41423-020-0417-8.
- Huang S, van Duijnhoven SM, Sijts AJ, van Elsas A. Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. J Cancer Res Clin Oncol. 2020;146:1–12. doi:10.1007/s00432-019-03068-x.
- Nie S, Wang Z, Moscoso-Castro M, D’Souza P, Lei C, Xu J, Gu J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antibody Ther. 2020;3:18–62. doi:10.1093/abt/tbaa003.
- Talotta R, Rucci F, Canti G, Scaglione F. Pros and cons of the immunogenicity of monoclonal antibodies in cancer treatment: a lesson from autoimmune diseases. Immunotherapy. 2019;11(3):241–54. doi:10.2217/imt-2018-0081.
- Staton TL, Peng K, Owen R, Choy DF, Cabanski CR, Fong A, Brunstein F, Alatsis KR, Chen H. A phase I, randomized, observer-blinded, single and multiple ascending-dose study to investigate the safety, pharmacokinetics, and immunogenicity of BITS7201A, a bispecific antibody targeting IL-13 and IL-17, in healthy volunteers. BMC Pulm Med. 2019;19:1–16. doi:10.1186/s12890-018-0763-9.
- Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT, Szpurka AM, Copeland V, Hodi FS, Rottey S, et al. Safety and Immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res. 2021;27(10):2773–81. doi:10.1158/1078-0432.CCR-20-3716.
- Paz-Priel I, Chang T, Asikanius E, Chebon S, Emrich T, Fernandez E, Kuebler P, Schmitt C. Immunogenicity of emicizumab in people with hemophilia A (PwHA): results from the HAVEN 1-4 studies. Blood. 2018;132(Supplement 1):633. doi:10.1182/blood-2018-99-118492.
- Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PloS One. 2013;8(2):e57479. doi:10.1371/journal.pone.0057479.
- De Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol. 2008;8:620–26. doi:10.1016/j.coph.2008.08.002.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs: Taylor & Francis. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, Umaña P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019;154:21–31. doi:10.1016/j.ymeth.2018.11.008.
- Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther. 2018;12:195. doi:10.2147/DDDT.S151282.
- Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270:26–35. doi:10.1006/jmbi.1997.1116.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng Des Sel. 1996;9:617–21. doi:10.1093/protein/9.7.617.
- Spiess C, Merchant M, Huang A, Zheng Z, Yang N-Y, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31:753–58. doi:10.1038/nbt.2621.
- Peng K, Siradze K, Fischer SK. Characterization of robust immune responses to a bispecific antibody, a novel class of antibody therapeutics. Bioanalysis. 2021;13:239–52. doi:10.4155/bio-2020-0281.
- Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406. doi:10.1111/imm.12889.
- Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. 2011;63:325–35. doi:10.1007/s00251-011-0513-0.
- Ito S, Ikuno T, Mishima M, Yano M, Hara T, Kuramochi T, Sampei Z, Wakabayashi T, Tabo M, Chiba S, et al. In vitro human helper T-cell assay to screen antibody drug candidates for immunogenicity. J Immunotoxicol. 2019;16:125–32. doi:10.1080/1547691X.2019.1604586.
- Welt S, Ritter G, Williams C, Cohen LS, John M, Jungbluth A, Richards EA, Old LJ, Kemeny NE. Phase I study of anticolon cancer humanized antibody A33. Clin Cancer Res. 2003;9:1338–46.
- Ulitzka M, Carrara S, Grzeschik J, Kornmann H, Hock B, Kolmar H. Engineering therapeutic antibodies for patient safety: tackling the immunogenicity problem. Protein Eng Des Sel. 2020;33. doi:10.1093/protein/gzaa025.
- Dingman R, Balu-Iyer SV. Immunogenicity of protein pharmaceuticals. J Pharm Sci. 2019;108:1637–54.
- Fu K, March K, Alexaki A, Fabozzi G, Moysi E, Petrovas C. Immunogenicity of Protein Therapeutics: a Lymph Node Perspective. Front Immunol. 2020;11:11. doi:10.3389/fimmu.2020.00791.
- Lu Y, Khawli LA, Purushothama S, Theil F-P, Partridge MA. Recent advances in assessing immunogenicity of therapeutic proteins: impact on biotherapeutic development. J Immunol Res. 2016;2016.
- Hidalgo M, Martinez-Garcia M, Le Tourneau C, Massard C, Garralda E, Boni V, Taus A, Albanell J, Sablin M-P, Alt M, et al. First-in-human phase I study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin Cancer Res. 2018;24(7):1536–45. doi:10.1158/1078-0432.CCR-17-1588.
- Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–W54. doi:10.1093/nar/gkaa379.
- Schneidman-Duhovny D, Khuri N, Dong GQ, Winter MB, Shifrut E, et al. Predicting CD4 T-cell epitopes based on antigen cleavage, MHCII presentation, and TCR recognition. PloS One. 2018;13:e0206654. doi:10.1371/journal.pone.0206654.
- Reche P, Flower DR, Fridkis-Hareli M, Hoshino Y. Peptide-based immunotherapeutics and vaccines 2017. J Immunol Res. 2018 Jul 15;2018:4568239. doi: 10.1155/2018/4568239. PMID: 30116752; PMCID: PMC6079504.
- Kovalova N, Knierman MD, Brown-Augsburger PL, Wroblewski VJ, Chlewicki LK. Correlation between antidrug antibodies, pre-existing antidrug reactivity, and immunogenetics (MHC class II alleles) in cynomolgus macaque. Immunogenetics. 2019;71:605–15. doi:10.1007/s00251-019-01136-7.
- van Meer PJ, Kooijman M, Brinks V, Gispen-de Wied CC, Silva-Lima B, et al. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. MAbs: Taylor & Francis. pp.810–16. 2013. doi:10.4161/mabs.25234
- De Groot AS, Moise L, Terry F, Gutierrez AH, Hindocha P, Richard G, Hoft DF, Ross TM, Noe AR, Takahashi Y, et al. Better epitope discovery, precision immune engineering, and accelerated vaccine design using immunoinformatics tools. Front Immunol. 2020;11:442. doi:10.3389/fimmu.2020.00442.
- Peng K, Siradze K, Quarmby V, Fischer SK. Clinical immunogenicity specificity assessments: a platform evaluation. J Pharm Biomed Anal. 2011;54:629–35. doi:10.1016/j.jpba.2010.09.035.
- FDA.gov. Center for drug evaluation and research. Immunogenicity testing of therapeutic protein products —Developing and validating assays for anti-drug antibody detection. 2019. accessed 2021 Mar 8. https://wwwfdagov/regulatory-information/search-fda-guidance-documents/immunogenicity-testing-therapeutic-protein-products-developing-and-validating-assays-anti-drug
- Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, Fiscella M, Gorovits B, Kirschner S, Moxness M, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81. doi:10.1016/j.jpba.2008.09.020.