
ABSTRACT
The molecular interactions of mouse CD96 to CD155 ligand and to two surrogate antibodies have been investigated. Biophysical and structural studies demonstrate that CD96 forms a homodimer but assembles as 1:1 heterodimeric complexes with CD155 or with one of the surrogate antibodies, which compete for the same binding interface. In comparison, the other surrogate antibody binds across the mouse CD96 dimer and recognizes a quaternary epitope spanning both protomers to block exposure of the ligand-binding site. This study reveals different blocking mechanisms and modalities of these two antibodies and may provide insight into the functional effects of antibodies against CD96.
Introduction
Blockade of co-inhibitory immune checkpoint receptors or ligands has become a critical component of cancer therapy.Citation1 Numerous monoclonal antibodies targeting CTLA-4, PD-1, and PD-L1 are already approved for marketing, and a new wave of therapeutics and combinations targeting other checkpoints is on the horizon.Citation2,Citation3 Among them, TIGIT (WUCAM, VSIG9, VSTM3), CD96 (TACTILE), and CD112R (PVRIG) are immunoreceptors in the nectin family that have been identified as important regulators of immune responses.Citation4–6 These co-inhibitory receptors are expressed on T and natural killer cells, and their blockade has shown enhanced anti-tumor immune responses.Citation7–9 However, it has also been reported that CD96 has co-stimulatory rather than co-inhibitory activity.Citation10 Nonetheless, monoclonal antibodies against these receptors entered, and advanced in, clinical development.Citation11
The receptor–ligand interactions between the nectin family members are expansive. Binding of CD155 (PVR, nectin-like molecule 5, necl5) and CD112 (PVRL2, nectin-2) ligands are mutually exclusive for CD96 and CD112R, respectively, but they both interact with TIGIT and the co-stimulatory receptor CD226. Additionally, CD111 (PVRL1, nectin-1) and CD113 (PVRL3, nectin-3) bind to CD96 and TIGIT, respectively.Citation12,Citation13 Structural characterizations of these receptors, ligands, and their complexes have provided insight into their binding epitopes and molecular assemblies. Homophilic interactions of the ligands, albeit weak, are formed with their membrane distal immunoglobulin (Ig) domain,Citation14–16 but dissociate to form receptor-ligand heterodimers with CD96 and CD226 at a 1:1 binding stoichiometry.Citation17,Citation18 This same ligand interface is also used to bind TIGIT, revealing a common solution for recognition to either the co-inhibitory or co-stimulatory receptors. These dimeric interactions are mediated through a paired “lock-and-key” binding mode between each Ig domain formed by an AX6G motif of one subunit and a T(Y/F)P motif on the neighboring molecule. In contrast to CD96-CD155 or CD226-CD155 complex crystal structures, TIGIT can form homodimers on the protein face opposite of the lock-and-key, suggesting a 2:2 heterotetrameric arrangement.Citation19,Citation20 These structural studies have elucidated the epitopes and mechanisms of these immunoreceptors that would be desirable to target for direct blockade of ligand interactions.
For therapeutic antibody development, pharmacology and toxicology studies are required, but therapeutic candidates can have poor mouse or cynomolgus (cyno) monkey cross-reactivity, which is required for animal studies, due to low sequence identity differences between orthologs. For instance, the ectodomains of human and mouse CD96 have a sequence identity of 59%, and thus the creation of human/mouse/cyno cross-reactive antibodies is challenging. In this study, we shed light on the structural and functional properties of surrogate anti-mouse CD96 antibodies and mouse CD96 receptors that may influence their activity and provide implications for the human setting.
Results
Mouse and human CD96 form dimers in solution that dissociate to form complexes with CD155 ligand
Size exclusion chromatography with multi-angle light scattering (SEC-MALS) was used to investigate the molecular states of the CD96 and CD155 proteins, as well as their protein–protein complex assemblies. The theoretical monomeric molecular weight of the full ectodomain (three Ig domains) of human and mouse CD96 is ~42 kDa, but they both had an observed protein mass of ~86–90 kDa (). Furthermore, the first Ig domain (D1) of human and mouse CD96 also had an observed mass twice that of their theoretical monomeric molar mass, indicating that CD96 forms a homodimer in solution using its membrane distal D1 domain.
Table 1. SEC-MALS of CD96 and CD155 proteins and complexes
In contrast, both human and mouse CD155 are monomeric in solution (), which was also previously reported from analytical ultracentrifugation studies with human CD155.Citation15 The observed masses of the human and mouse CD96-CD155 complexes correspond to one CD96 protomer bound to one CD155 monomer (), consistent with the 1:1 binding heterodimeric complex observed in the human CD96-CD155 crystal structure.Citation17 These studies suggest that CD96 exists as a homodimer, but separates so that each protomer, using the same interface that forms the homophilic interactions, individually binds CD155 ligand as a 1:1 heterodimeric complex. Additionally, this binding arrangement is conserved between the orthologous human and mouse proteins. These results also indicate that the dissociation constant of the CD96 homodimer is weaker than the CD96–CD155 interactions (KD 10 µM).Citation17
While TIGIT has been reported to be a homodimer in crystal structures at an interface that is not shared by the ligands, resulting in a heterotetrameric complex with CD155 or CD112, it is monomeric in solution by SEC-MALS but may self-associate at high concentrations (KD > 1 mM).Citation19,Citation20 The putative TIGIT dimer could form at the high local concentrations required for crystallization or on the cell surface. Notwithstanding, CD96 likely cannot accommodate a similar dimeric arrangement as seen for TIGIT due to potential steric clashes from predicted N-linked glycosylation sites (Figure S1), but instead forms a homodimer using its lock-and-key interface.
Binding and blocking properties of surrogate antibodies to mouse CD96
The binding affinities between the surrogate antibodies mCD96-A and mCD96-B to mouse CD96 D1 domain were measured by surface plasmon resonance (SPR). While mCD96-A had moderate affinity to mouse CD96 (KD 47 nM), mCD96-B has very high affinity and binds with a KD <0.01 nM ( and Figure S2). In comparison to mCD96-A, mCD96-B has >5000-fold greater affinity with a 10-fold faster association rate and a nearly immeasurable dissociation rate. Since mCD96-A had considerably weaker affinity than mCD96-B, affinity maturation was performed on mCD96-A to improve its binding strength to mouse CD96. A single positional NNK scanning library in each of the mCD96-A complementarity-determining regions (CDRs) of the heavy and light chain was synthesized and cloned into a phage vector for scFv display. Affinity maturing variants were selected over rounds of semi-automated phage selections. Three individual mutations were enriched at heavy-chain G56K and F101M and light-chain G50H (Kabat numbering), and each of these positions moderately improved affinity to mouse CD96 (Table S1). Combinations of these three substitutions were assessed by SPR and the triple mutant mCD96-A-7 had the highest affinity, with a ~ 6-fold improved affinity over the mCD96-A parent as a result of its slower dissociation rate ( and Table S1).
Table 2. SPR-binding kinetics and ligand blocking activity of anti-mouse CD96 antibodies
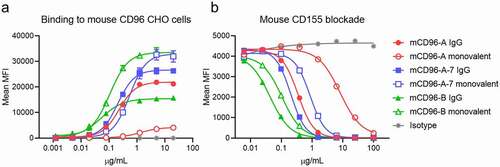
In vitro binding and blocking assays were performed using mouse CD96-expressing Chinese hamster ovary (CHO) cell lines to assess activity of mCD96-A, higher affinity mCD96-A-7 and mCD96-B antibodies. The antibodies were generated as standard bivalent IgGs or as monovalent IgGs, where only one arm can bind to mouse CD96. By direct binding, mCD96-B showed stronger affinity compared to mCD96-A or mCD96-A-7 and, as expected, each of the IgGs had enhanced affinity over their monovalent counterparts due to avidity (). The antibodies exhibited a range of maximum mean fluorescence intensity (MFI) values at the highest antibody concentrations. For instance, mCD96-A-7 had nearly double the signal as mCD96-B despite being a weaker binder, hinting that these antibodies may have distinct epitopes. Interestingly, each of the monovalent antibodies also had a higher MFI than the bivalent IgGs, which could be a result of their binding valency to CD96. The antibodies have varying levels of mouse CD155 ligand blocking activity proportionate to their affinities (). Furthermore, mCD96-A had greater differences in blocking activity between the monovalent and bivalent forms than the other antibodies and avidity for mCD96-A likely contributes more to compensate for its weak monovalent binding (Table S2). In all cases, the monovalent antibodies had less potent activity than the bivalent IgGs and the blocking IC50 values were consistent with the monovalent antigen-binding fragment (Fab) SPR affinities with mCD96-B > mCD96-A-7 > mCD96-A ().
Figure 1. Binding and ligand blocking of anti-mouse CD96 bivalent (filled symbols) and monovalent (open symbols) IgGs on cells. (a) Binding of antibodies to mouse CD96-expressing CHO cells (mean ± SD, n = 3; independent experiments performed three times). (b) Antibody blocking of mouse CD155 tetramers (mean ± SD, n = 2, independent experiments performed three times). All antibodies were expressed in the mIgG1-D265A isotype
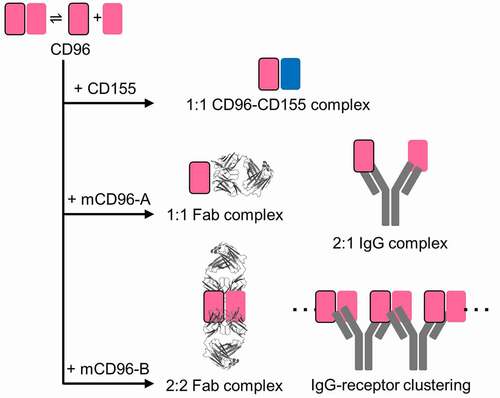
Different binding stoichiometries of surrogate antibodies to mouse CD96
The molar mass of mCD96-A and mCD96-B in complex with the mouse CD96 D1 domain were determined by SEC-MALS. The mCD96-A and the affinity-matured mCD96-A-7 Fab complexes had an observed molar mass of ~65 kDa (). This result suggests that these variants form a 1:1 complex with mouse CD96. In contrast, the mCD96-B Fab complex had an observed mass of ~132 kDa corresponding to two Fabs and two mouse CD96 protomers (), indicating that two mCD96-B Fabs bind to a mouse CD96 homodimer and assemble with a 2:2 binding stoichiometry. While mCD96-A and mCD96-A-7 IgG complexes had a molar mass of ~167 kDa, which is indicative of an antibody bound to two mouse CD96 protomers, the mCD96-B IgG complex was >2 MDa (). These results demonstrate differences in binding and blocking mechanism between these antibodies where mCD96-B IgG may bridge mouse CD96 homodimers to create large oligomers, while each antibody arm of the mCD96-A and mCD96-A-7 IgGs bind to individual mouse CD96 protomers ().
Table 3. SEC-MALS of antibody-CD96 complexes. The individual Fab and IgG molecular weights are approximately 48 kDa and 143 kDa, respectively
Figure 2. Cartoon schematic of binding assemblies of CD155 ligand, Fabs, and IgGs to mouse CD96 reveals two distinct antibody blocking mechanisms
Crystal structures of surrogate antibody complexes reveal distinct epitopes on CD96 monomers or dimers
To understand the molecular basis of antibody recognition to mouse CD96, the structures of the mCD96-A and mCD96-B complexes were determined by x-ray crystallography at 3.12 Å and 2.45 Å resolution, respectively (Table S3 and Figure S3). The mCD96-A Fab binds a similar epitope as the CD155 ligand on CD96, confirming that it is a direct blocker of CD155 interactions (). The mCD96-B complex has two Fabs bound to a mouse CD96 D1 homodimer and each Fab makes interactions across a quaternary epitope composed of both CD96 protomers, thus stabilizing the CD96 homodimer (). Although the mCD96-B Fabs come in close proximity, they do not form any Fab–Fab interactions when bound to CD96 (Figure S4). Furthermore, the mouse CD96 dimer has a similar orientation and interface residues as the human CD96-CD155 complex at their respective D1 domains (). Altogether, these structures confirm the observed masses of the complexes calculated by SEC-MALS and illustrate distinct molecular recognition and blocking mechanisms of mCD96-A and mCD96-B.
Figure 3. Antibodies mCD96-A and mCD96-B bind distinct epitopes on monomeric or dimeric mouse CD96 to block CD155. The crystal structures of CD96 in complex with (a) CD155 ligand, (b) mCD96-A Fab, and (c) mCD96-B Fab. The complexes are aligned onto CD96, represented as a light gray surface. In the mCD96-B structure, the other protomer of CD96 that forms the dimer is shown as a dark gray surface. CD155, mCD96-A, and mCD96-B are colored as blue, red, or green cartoons, respectively. The CD96-CD155 complex is of the human orthologs from PDB ID 6ARQ. (d) Epitopes of CD155, mCD96-A, and the CD96 dimer interface are colored blue, red, or dark gray on the surface representation of CD96. Each CD96 protomer is aligned in the same orientation. Regions contacted by the mCD96-A heavy chain or light chain are colored red or light red, respectively. (e) Epitope of mCD96-B on the surface of the CD96 homodimer, with each protomer colored light or dark gray. Regions contacted by the mCD96-B heavy chain or light chain are colored green or light green, respectively
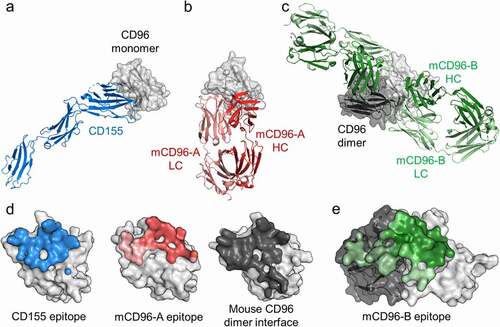
Figure 4. Comparisons of the human and mouse CD96 D1 domains. (a) Structural alignment of the D1 domains of the human CD96-CD155 heterodimeric complex (PDB ID 6ARQ) and the mouse CD96 homodimer from the mCD96-B complex. (b) Sequence alignment of the human and mouse CD96 D1 domains. Interacting residues to human CD96 by CD155 or of the mouse CD96 homodimer conserved across both protomers were calculated by Areaimol and are highlighted blue or gray, respectively

The CD155 ligand and mCD96-A bind an overlapping footprint also shared by the CD96 homodimer, showing that the homodimer must dissociate to expose the ligand or mCD96-A binding site (). On average, 1,320 ÅCitation2 is buried in the mCD96-A complex (650 ÅCitation2 on mouse CD96 and 670 ÅCitation2 on mCD96-A), where the heavy and light chains contribute to 77% and 23% of the Fab buried surface area, respectively, by all six CDRs. In contrast, the mCD96-B antibody has a distinct CD155 blocking mechanism compared to mCD96-A as it holds the mouse CD96 homodimer together to prevent exposure of the ligand-binding site rather than directly competing for the same epitope (). A total of 2,220 ÅCitation2 is buried at the mCD96-B interface on average, which is considerably larger than the footprint of mCD96-A. Approximately 1,150 ÅCitation2 is buried on each mouse CD96 epitope split roughly 63% and 37% on each protomer and 1,070 ÅCitation2 is buried on each Fab contributed equally by both the heavy chain and light chain through interactions with all six CDRs. Each mouse CD96 protomer and the mCD96-B antibody variable domains are positioned toward one another where the VH primarily interacts with one mouse CD96 protomer and the VL binds the other protomer.
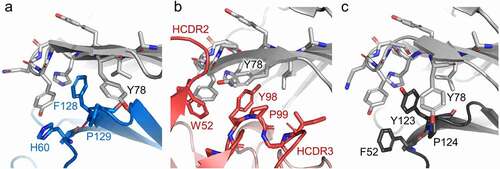
While CD155 and mCD96-A have a different approach angle to engage CD96 (), there are some similarities in their molecular recognition to the receptor. The mCD96-A Fab has a hydrophobic HCDR3 that contains a PYP amino acid stretch, similar in sequence to the signature T(Y/F)P of the key motif of the nectin receptors and ligands. Structural comparisons reveal that the tyrosine and proline residues from all three structures overlay to interact with the AX6G lock of CD96, demonstrating that mCD96-A mimics the lock-and-key interactions (). The large aromatic HCDR2 W52 residue of mCD96-A is also buttressed against the lock, resembling the ancillary key described for CD96–CD155 interactions.Citation17 In addition, the CD96 Y78 residue adjacent to the lock adopts different rotamer conformations in each of the complexes, suggesting that it has an induced fit orientation.
Figure 5. CD96 molecular recognition similarities at the lock-and-key interface. The (a) CD155, (b) mCD96-A, (c) and mouse CD96 dimer complexes are represented as cartoons and the residues of the ligand, mCD96-A, and opposing CD96 protomer that form the key are shown as sticks and are colored blue, red, or dark gray, respectively. The CD96 lock and Y78 residues in each of the complexes are shown as sticks and are colored light gray
The structural basis of mCD96-A-7 affinity maturation was modeled onto the mCD96-A complex, which generally substituted for residues with much larger side chains that may contribute to additional buried surface area contacts to CD96. The HCDR3 F101 residue is buried and a flexible glycine amino acid precedes both HCDR2 G56K and LCDR2 G50H, suggesting that there may potentially be conformational rearrangements in the CDR loops from these substitutions (Figure S5).
Discussion
The immuno-oncology field has seen much success from checkpoint inhibitors that provide anti-tumor efficacy against human cancers. Following on this success, there has been an emergence of other targets, such as TIGIT, LAG-3, Tim-3, and CD96, with growing interest in bringing novel therapeutics to patients resistant to existing therapies. In the absence of species cross-reactive antibodies, surrogate antibodies are used to understand the blockade of these pathways. In this work, the distinct binding and blocking mechanisms of anti-mouse mCD96-A and mCD96-B surrogate antibodies have been characterized and their mechanisms for blocking CD155 ligand engagement have been revealed.
By SEC-MALS and crystallographic studies, mouse CD96 was discovered to be a homodimer in solution using a similar lock-and-key interface as other members of the nectin family. The homodimer dissociates to expose the interface for CD155 ligand engagement. The surrogate antibody mCD96-A also binds an epitope of the CD96 monomer similar to CD155, revealing that this interface can be commonly used for homodimeric interactions, binding of ligand, or blocking antibody mCD96-A. The mCD96-B antibody, however, holds together the mouse CD96 homodimer, effectively preventing the CD155 binding interface from revealing itself, and can form large clusters of receptors upon binding. Furthermore, mCD96-B binds a quaternary epitope on CD96 where each Fab simultaneously engages each protomer, as has been seen for antibodies targeting multimeric antigens on influenza and HIV.Citation21,Citation22
For the ligand and antibodies to bind mouse CD96 with different binding stoichiometries suggests that CD96 is in equilibrium between its dimeric and monomeric forms. The mCD96-A antibody has a slower association rate to CD96 compared to mCD96-B, suggesting a kinetic component for the mouse CD96 dimer to open to reveal the binding epitope on each protomer, albeit on SPR sensors. In contrast, mCD96-B does not require this open conformation, but can bind to the preformed CD96 dimer. These recognition differences are also reflected in the cell-binding experiments where mCD96-A and mCD96-A-7 have a higher maximum MFI signal than mCD96-B as IgGs, likely due to individual protomer engagement by mCD96-A and mCD96-A-7, while mCD96-B can avidly bind CD96 with both antibody arms. In addition, the monovalent mCD96-B antibody has double the signal than as a bivalent IgG, which is reflective of two single-arm antibodies capable of binding a CD96 dimer compared to a mCD96-B IgG spanning across neighboring CD96 molecules.
The different binding and blocking mechanisms of mCD96-A and mCD96-B shed light on considerations for desired properties of antibody-based therapeutics against CD96. Despite having much lower affinity and blocking activity, mCD96-A directly blocks ligand interactions but would not cluster mouse CD96 on the surface of cells. In contrast, mCD96-B can connect multiple CD96 receptors to potentially form large networks. Head-to-head comparisons of these antibodies with matched isotypes, and perhaps affinity, will be needed to investigate the relationship between anti-tumor activity and epitope. Altogether, this study underscores the need to understand the molecular mechanisms of surrogate antibodies and their relevance to functionally equivalent therapeutic antibodies.
Materials and methods
Protein expression and purification
Human or mouse chimeric Fab and IgG constructs, using the variable domains of the mouse CD96 antibodies that originated from rat immunizations with mouse CD96 antigen and were derived using proprietary technology, were individually cloned into the pTT5 expression plasmids (GenScript) with N-terminal osteonectin signal peptides. The Fabs have a poly-histidine tag fused to the C-terminus of the heavy chain. Mouse CD96 and mouse CD155 were similarly cloned into the pTT5 expression plasmid with a C-terminal polyhistidine and BirA biotinylation tag. Mouse CD96 was also cloned into another pTT5 plasmid with a C-terminal polyhistidine tag for crystallography studies. Monovalent antibodies were cloned using mutations as previously described.Citation23 Briefly, the monovalent antibodies contained K439E/K409D heavy-chain mutations on the CD96 binding arm and Y122C/C209V/E356 K/D399K and S121C/C214V mutations on the heavy and light chains on the other arm that is unable to bind CD96, respectively, and were transfected at a 1:1:1:1 (HC1:LC1:HC2:LC2) ratio. The Fabs, CD96, and CD155 supernatant were purified using Ni Sepharose excel (GE Healthcare Life Sciences) and buffer exchanged into 1x phosphate-buffered saline (PBS). Both monovalent and bivalent IgG supernatants were purified using MAbSelect PrismA (GE Healthcare Life Sciences) and buffer exchanged into 1x PBS. The monovalent antibodies were further purified by cation exchange. All proteins passed standard quality checks such as purity (>95%), aggregation (<5%), endotoxin (<0.1 EU/mg), and mass spectrometry to confirm identity.
Surface plasmon resonance spectroscopic determination of binding parameters
SPR was used to determine binding parameters for the Fabs to mouse CD96 with a BIACORE® T200 SPR spectrometer (Biacore AB, Uppsala, Sweden). Three surface capacities (34–50 response units) of mouse CD96 were captured on a CM4 chip with amine-coupled anti-Avi antibody (Genscript, catalog number A00674) and were then bound by a concentration series of the Fabs in HBS pH 7.4 running buffer supplemented with 0.05% Tween-20 and 1 g/L bovine serum albumin at 37°C. All data were double-referenced and fitted to a 1:1 Langmuir binding model with mass transport to determine equilibrium dissociation constants (KD), as well as association (ka) and dissociation (kd) rate constants.
Affinity maturation of mCD96-A
Phage selections were performed to select mCD96-A variants with improved affinity. A positional NNK scanning library in the mCD96-A CDRs was synthesized by GeneWiz and had a theoretical diversity of 2,272 members. Four rounds of automated phage selections were performed using a KingFisher Flex Purification System (Thermo Fisher Scientific). Increased stringency was applied in each round by decreasing antigen concentration and increasing wash cycles and lengths (round 1: 500 nM mouse CD96, 1 × 1 minute wash; round 2: 100 nM mouse CD96, 1x 2-min wash; round 3: 20 nM mouse CD96, 2x 2-min washes; round 4: 4 nM mouse CD96 2x 4-min washes). Streptavidin-coated magnetic beads (Promega) were used to capture the CD96-scFv-phage complexes. Phage was eluted with 0.1 N HCl, neutralized, and amplified overnight in XL1-blue cells (Agilent). Single colonies were plated from each round and phage was cultured for enzyme-linked immunosorbent assay-binding evaluation to mouse CD96. The scFv from the phage clones were also amplified by PCR for Sanger sequencing. Positions G56K in HCDR2, F101M in HCDR3, and G50H in LCDR2 were enriched. These substitutions were combined to create variant mCD96-A-7 and its improved affinity was confirmed by SPR.
Size exclusion chromatography with multi-angle light scattering (SEC-MALS)
Human and mouse CD96 complexes were formed with 2x excess CD96 antigen and then normalized to 2 mg/mL. Protein samples (40 μg) were injected onto Acquity UPLC Protein BEH SEC columns (200 Å, 1.7 μm, 4.6 mm x 300 mm and 125 Å, 1.7 μm, 4.6 mm x 300 mm, Waters) attached to an Acquity UPLC H-Class system (Waters) at an isocratic flow rate of 0.3 mL/min in 1x PBS, 0.05% Na azide. The eluted peaks were analyzed using a UV detector operated at 280 nm wavelength followed by the DAWN HELEOS-II/µDAWN multiangle light scattering detector and an Optilab T-rEX differential refractometer (Wyatt Technology). Molar mass distribution of the proteins was calculated in the Astra v7.3 software using the Zimm model. The SEC-MALS plots and analyses are provided as Supplemental Material.
X-ray crystallography and structural analyses
Mouse CD96 was expressed in Expi293 cells treated with kifunensine and purified protein was treated with EndoH. For complex formation, mouse CD96 was added to either mCD96-A Fab or mCD96-B Fab in molar excess and incubated at room temperature for 1 hr. The mCD96-A or mCD96-B complexes were then purified from unbound mouse CD96 by gel filtration using 50 mM NaCl, 10 mM Tris pH 8.0.
mCD96-A:CD96 crystals were grown by sitting drop vapor diffusion at 20°C by mixing 0.2 µL of concentrated protein sample (15 mg/mL) with 0.2 µL of mother liquor (1 M ammonium sulfate, 0.1 M bis-tris pH 5.5, 1% PEG 3350). Crystals were cryo-protected with 15% glycerol and flash frozen in liquid nitrogen. X-ray diffraction data were collected at beamline 17ID (wavelength 1.0 Å) at the Advanced Photon Source (APS) under cryo conditions using a DECTRIS Eiger2 X 9 M detector. Diffraction data were processed with the autoPROCCitation24 toolbox that made use of external programs XDS/XSCALE,Citation25 POINTLESS,Citation26 CCP4,Citation27 and STARANISO (Global Phasing Limited) for ellipsoidal truncation and anisotropic scaling. The mCD96-A complex diffracted to a nominal resolution of 3.12 Å resolution, with anisotropic diffraction limits of 3.35 Å, 2.58 Å, 2.78 Å. The mCD96-A complex was determined by molecular replacement with Phaser using the variable heavy and light domains from PDB IDs 3MJ8 and 4AMK, respectively, as well as the constant domains from PDB ID 4NM4. A mouse CD96 model based on the human CD96 structure (PDB ID 6ARQ) was also used as a search model. Four mCD96-A:CD96 complexes were found in the asymmetric unit. The model was iteratively built using CootCitation28 and refined in Phenix.Citation29 In the final mCD96-A complex structure, 94.5% of the residues are in favored regions of the Ramachandran plot with 0.7% outliers, as calculated by MolProbity.Citation30 X-ray diffraction data collection and refinement statistics are reported in Table S3.
mCD96-B:CD96 crystals were similarly grown in a mother liquor consisting of 0.2 M sodium citrate tribasic dihydrate, 20% w/v polyethylene glycol 3,350. Crystals were cryo-protected in 15% ethylene glycol and flash frozen in liquid nitrogen. X-ray diffraction data were similarly collected and processed as for the mCD96-A complex. The mCD96-B complex diffracted to a nominal isotropic resolution of 2.45 Å resolution, with anisotropic diffraction limits of 2.32 Å, 2.12 Å, 2.70 Å. The mCD96-B complex was determined by molecular replacement with Phaser using the Fab variable and constant domains PDB IDs 4AIZ and 1NC2, respectively. A mouse CD96 model based off of the human CD96 structure (PDB ID 6ARQ) was also used as a search model. Two mCD96-B Fabs bound to a mouse CD96 dimer was found in the asymmetric unit. The model was iteratively built using CootCitation28 and refined in Phenix.Citation29 In the final mCD96-B complex structure, 95.2% of the residues are in favored regions of the Ramachandran plot with 0.3% outliers, as calculated by MolProbity.Citation30 X-ray diffraction data collection and refinement statistics are reported in Table S3.
The molecular epitope and paratope were calculated using Areaimol of the CCP4 suite.Citation27 PyMOL was used to render structure figures. Kabat numbering was applied to the variable domains of the Fabs. Coordinates and structure factors have been deposited in the RCSB Protein Data Bank under accession codes 7S11 (mCD96-A: mouse CD96 complex) and 7S13 (mCD96-B: mouse CD96 complex).
Cell line generation
CHO-s cells (ThermoFisher) were cultured in CD CHO supplemented with 1x Hypoxythymine and 8 mM Glutamax in shaker flasks with 8% CO2. The cells were transfected with a plasmid containing the full-length mouse CD96 and a hygromycin resistance marker using a Lonza Nucleofector II. At 48 hours post transfection, cells were placed under selection with 600 µg/ml hygromycin. Following selection, the cells were checked by fluorescence-activated single-cell sorting for expression using an anti-mouse CD96-PE antibody (Ebioscience #12-0960-80).
In vitro cell binding and blocking assays
The mCD96-A variants and mCD96-B as IgGs or monovalent antibodies were tested for binding to CHO cells expressing mouse CD96. 2 × 105 CHO cells were incubated in 96-well plates with the antibodies, which were titrated at a 1:4 dilution across eight points starting at 20 µg/mL in 1x PBS + 5% fetal bovine serum (FBS) for 30 minutes at 4°C in the dark and were washed two times. Binding of the antibodies was detected using an Alexa Fluor 647 AffiniPure Fab fragment Donkey anti-mouse IgG (H + L) (Jackson ImmunoResearch, code 715–607-003) and were read on a Cytoflex-S instrument (Beckman Coulter).
Mouse CD155 was enzymatically biotinylated with BirA (Avidity) and tetramers were formed with an Alexa Fluor 647 streptavidin (ThermoFisher). For the blocking assays, 2 × 105 CHO cells were incubated with the antibodies for 10 minutes at room temperature, which were titrated at a 1:4 dilution across eight points starting at 100 µg/mL in 1x PBS + 5% FBS. 1 µg/mL CD155-tetramer was then added and cells were incubated for 30 minutes at 4°C in the dark and washed two times. Blocking of mouse CD155 was read on Cytoflex-S.
Abbreviations: CDR, complementarity-determining region; CHO, Chinese hamster ovary; Fab, antigen-binding fragment; Ig, immunoglobulin; KD, dissociation constant; MFI, mean fluorescence intensity; SEC-MALS, size exclusion chromatography with multi-angle light scattering; SPR, surface plasmon resonance
Supplemental Material
Download Zip (6.5 MB)Acknowledgments
The authors thank SJ Diong, Amanda Rhea, Elly Seo, Christina Milburn, Sean West, Andy Deng, Gamze Camdere, Andrew Drake, and Diksha Gupta for reagents, technical advice, and scientific discussions. The authors thank Steven Sheriff, Jodi Muckelbauer, and the staff at the IMCA-CAT at the Argonne National Laboratory for assistance in x-ray data collection and beamline support.
Disclosure statement
All authors are current or former employees of Bristol Myers Squibb.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–8. doi:10.1016/S0065-2776(06)90008-X.
- Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004. doi:10.1016/j.immuni.2016.05.001.
- Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2018;29:71–83. doi:10.1093/annonc/mdx686.
- Manieri NA, Chiang EY, Grogan JLTIGIT, Key A. Inhibitor of the cancer immunity cycle. Trends Immunol. 2017;38:20–28. doi:10.1016/j.it.2016.10.002.
- Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276:112–20. doi:10.1111/imr.12518.
- Jin HS, Park Y. Hitting the complexity of the TIGIT-CD96-CD112R-CD226 axis for next-generation cancer immunotherapy. BMB Rep. 2021;54:2–11. doi:10.5483/BMBRep.2021.54.1.229.
- Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15:431–38. doi:10.1038/ni.2850.
- Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–37. doi:10.1016/j.ccell.2014.10.018.
- Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, Yao S, Bevers S, Edil BH. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med. 2016;213:167–76. doi:10.1084/jem.20150785.
- Chiang EY, de Almeida PE, de Almeida Nagata DE, Bowles KH, Du X, Chitre AS, Banta KL, Kwon Y, McKenzie B, Mittman S, et al. CD96 functions as a co-stimulatory receptor to enhance CD8(+) T cell activation and effector responses. Eur J Immunol. 2020;50:891–902. doi:10.1002/eji.201948405.
- Johnston RJ J, Lee PSs L, Strop P, Smyth MJ S. Cancer immunotherapy and the Nectin family. Annual Rev Cancer Biol. 2021;5:203–19. doi:10.1146/annurev-cancerbio-060920-084910
- Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. doi:10.1038/ni.1674.
- Holmes VM, Maluquer de Motes C, Pt R, Roldan J, Ak B, JS O, Krummenacher C. Interaction between nectin-1 and the human natural killer cell receptor CD96. PLoS One. 2019;14:e0212443. doi:10.1371/journal.pone.0212443.
- Liu J, Qian X, Chen Z, Xu X, Gao F, Zhang S, Zhang R, Qi J, Gao GF, Yan J. Crystal structure of cell adhesion molecule nectin-2/CD112 and its binding to immune receptor DNAM-1/CD226. J Immunol. 2012;188:5511–20. doi:10.4049/jimmunol.1200324.
- Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS, Ahlsen G, Troyanovsky RB, Troyanovsky SM, Honig B, et al. Nectin ectodomain structures reveal a canonical adhesive interface. Nat Struct Mol Biol. 2012;19:906–15. doi:10.1038/nsmb.2366.
- Samanta D, Ramagopal UA, Rubinstein R, Vigdorovich V, Nathenson SG, Almo SC. Structure of Nectin-2 reveals determinants of homophilic and heterophilic interactions that control cell-cell adhesion. Proc Natl Acad Sci U S A. 2012;109:14836–40. doi:10.1073/pnas.1212912109.
- Deuss FA, Watson GM, Fu Z, Rossjohn J, Berry R. Structural basis for CD96 immune receptor recognition of Nectin-like protein-5, CD155. Structure. 2019;27:219–28 e3. doi:10.1016/j.str.2018.10.023.
- Deuss FA, Watson GM, Goodall KJ, Leece I, Chatterjee S, Fu Z, Thaysen-Andersen M, Andrews DM, Rossjohn J, Berry R. Structural basis for the recognition of nectin-like protein-5 by the human-activating immune receptor, DNAM-1. J Biol Chem. 2019;294:12534–46. doi:10.1074/jbc.RA119.009261.
- Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, Wiesmann C, Bazan JF, Eaton DL, Grogan JL. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U S A. 2012;109:5399–404. doi:10.1073/pnas.1120606109.
- Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of nectin-2 by the natural killer cell receptor T cell immunoglobulin and ITIM domain (TIGIT). J Biol Chem. 2017;292:11413–22. doi:10.1074/jbc.M117.786483.
- Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–74. doi:10.1006/viro.2001.1320.
- Blattner C, Lee JH, Sliepen K, Derking R, Falkowska E, De La Pena AT, Cupo A, Julien JP, van Gils M, Lee PS, et al. Structural delineation of a quaternary, cleavage-dependent epitope at the gp41-gp120 interface on intact HIV-1 Env trimers. Immunity. 2014;40:669–80. doi:10.1016/j.immuni.2014.04.008.
- Wang F, Tsai JC, Davis JH, Chau B, Dong J, West SM, Hogan JM, Wheeler ML, Bee C, Morishige W, et al. Design and characterization of mouse IgG1 and IgG2a bispecific antibodies for use in syngeneic models. MAbs. 2020;12:1685350. doi:10.1080/19420862.2019.1685350.
- Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. doi:10.1107/S0907444911007773.
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337.
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi:10.1107/S0907444905036693.
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi:10.1107/S0907444910045749.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi:10.1107/S0907444910007493.
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi:10.1107/S0907444909052925.
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi:10.1107/S0907444909042073.