
ABSTRACT
Therapeutic antibodies must have “drug-like” properties. These include high affinity and specificity for the intended target, biological activity, and additional characteristics now known as “developability properties”: long-term stability and resistance to aggregation when in solution, thermodynamic stability to prevent unfolding, high expression yields to facilitate manufacturing, low self-interaction, among others. Sequence-based liabilities may affect one or more of these characteristics. Improving the stability and developability of a lead antibody is typically achieved by modifying its sequence, a time-consuming process that often results in reduced affinity. Here we present a new antibody library format that yields high-affinity binders with drug-like developability properties directly from initial selections, reducing the need for further engineering or affinity maturation. The innovative semi-synthetic design involves grafting natural complementarity-determining regions (CDRs) from human antibodies into scaffolds based on well-behaved clinical antibodies. HCDR3s were amplified directly from B cells, while the remaining CDRs, from which all sequence liabilities had been purged, were replicated from a large next-generation sequencing dataset. By combining two in vitro display techniques, phage and yeast display, we were able to routinely recover a large number of unique, highly developable antibodies against clinically relevant targets with affinities in the subnanomolar to low nanomolar range. We anticipate that the designs and approaches presented here will accelerate the drug development process by reducing the failure rate of leads due to poor antibody affinities and developability.
Abbreviations: AC-SINS: affinity-capture self-interaction nanoparticle spectroscopy; CDR: complementarity-determining region; CQA: critical quality attribute; ELISA: enzyme-linked immunoassay; FACS: fluorescence-activated cell sorting; Fv: fragment variable; GM-CSF: granulocyte-macrophage colony-stimulating factor; HCDR3: heavy chain CDR3; IFN2a: interferon α-2; IL6: interleukin-6; MACS: magnetic-activated cell sorting; NGS: next generation sequencing; PCR: polymerase chain reaction; SEC: size-exclusion chromatography; SPR: surface plasmon resonance; TGFβ-R2: transforming growth factor β-R2; VH: variable heavy; VK: variable kappa; VL: variable light; Vl: variable lambda;
Introduction
Monoclonal antibodies are becoming progressively more important as therapeutics, comprising six of the top 10 best-selling drugs in the United States. As is the case with rules applying to many small-molecule drugs (e.g., Lipinski’s rule of five),Citation1 it has been proposed that therapeutic antibodies should similarly adhere to strict criteria regarding pharmacodynamics, kinetics and formulation.Citation2 Once antibodies are produced against a given target, ensuring they have “drug-like” characteristics appears to greatly improve chances of therapeutic success (Suppl. Figure S1).Citation3 In addition to high affinity, therapeutic success also depends on “developability”, a term coined to describe a favorable set of in vitro biophysical characteristics such as reduced aggregation propensity and polyreactivity, which tend to be associated with improved in vivo properties.
Today, antibody discovery campaigns generally employ display technologies or immunization approaches that include the use of transgenic animals and B cell cloning. While immunization provides a straightforward manner to obtain antibodies directly as IgG, controlling antibody binding properties, particularly the specificity for desired isoforms or epitopes, can be challenging, with additional problems imposed by the potential need for humanization. In contrast, generating antibodies using in vitro display technologies allows the discovery of molecules against non-immunogenic targets or epitopes, particularly those with highly conserved sequences; specificity fine-tuning, enabling the isolation of antibodies recognizing particular conformations or isoforms;Citation4 and the selection of antibodies against highly-toxic antigens.Citation5 Finally, display technologies are more amenable to automation, facilitating high-throughput selection strategies.Citation4 However, it has been reported that the developability characteristics of antibodies isolated using phage display are inferior to those produced by immunizing mice,Citation3,Citation6 with the assumption that the stringent quality control antibodies undergo during the natural process of B-cell maturation ensures the retention of superior biophysical properties. We wondered whether this problem may be the direct result of the designs of prior in vitro display libraries, which inevitably lead to libraries having substantial levels of “contamination” with poorly developable antibodies. In the case of natural libraries, the random nature of variable heavy and variable light (VH/VL) chain pairing may create poorly developable combinations, while synthetic diversity may create artificial complementarity-determining region (CDR) sequences that fold poorly.
We hypothesized that a library comprising a defined collection of natural CDR sequences from which most known sequence-based liabilities were eliminated, embedded within paired frameworks derived exclusively from well-behaved therapeutic molecules, would facilitate the discovery of highly developable antibodies directly from the library. Reasoning that the stringent quality control applied to antibodies undergoing natural B-cell maturation would also apply to their individual CDRs, a library comprising replicated natural CDRs informatically purged of sequence liabilities could provide superior biophysical properties when used as a diversity source. While it remains difficult to predict the relevant properties of therapeutic antibodies on the basis of primary sequence alone, many short sequence liabilities have been identified, particularly those related to chemical instability or polyreactivity, including, for example, N-glycosylation motifs, asparagine deamidation motifs, aspartate isomerization motifs, unpaired cysteines, surface hydrophobic/aromatic patches and others ().
Table 1. Liabilities removed from CDR sequences
Building on pioneer antibody library designs (generation 1)Citation17–22 and other designs that further improved the capabilities of in vitro antibody discovery (generation 2),Citation23–40 here we present a “Generation 3” library architecture that leverages developability data on therapeutic antibodies, next-generation sequencing (NGS) of human repertoires, and high-throughput synthetic oligonucleotide synthesis to create a platform able to yield large numbers of high affinity, developable antibodies against any target.
Results
Library design
The Generation 3 library design involved embedding only defined CDRs from natural antibodies into a genetically diverse panel of developable clinical antibody scaffolds. As HCDR3 diversity far exceeds the capabilities of oligonucleotide array-based synthesis, HCDR3s were generated by PCR directly from CD19+ B-cells purified from donor LeukoPaks. The remaining CDRs were based on replicated natural diversity identified by informatic analysis of the NGS of a previously published library.Citation22 This was informatically purged of sequence-based liabilities () and produced using oligonucleotide array-based synthesis. These replicated natural CDRs were then filtered by yeast display as single CDR libraries to eliminate sequences negatively impacting expression, folding and display, prior to combinatorial assembly of all CDRs into the single-chain variable fragment (scFv) format ().
Figure 1. Schematic representation of library design and assembly. LCDR1-3 and HCDR1-2 are from sequences replicated from the naïve repertoire with the liabilities removed. They also undergo a filtering step using yeast display. HCDR3 is recovered from 10 healthy donors. The pieces are assembled to form the VH and VL and subsequently assembled as a scFv. The CDRs are all embedded in a scaffold derived from a developable therapeutic antibody
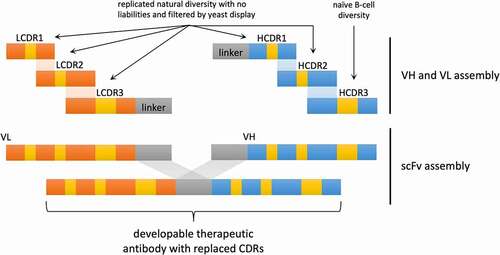
The use of developable antibody scaffolds provides optimal VH/VL pairing within the context of well-behaved clinical antibodies. Replicated natural CDRs allow the elimination of sequence liabilities in all but HCDR3, and avoid problems with traditional intra-CDR combinatorial diversity, such as covariance violations, while maintaining enormous theoretical diversity from inter-CDR combinatorial diversity.
CDR diversity analysis and liabilities in different V families
We analyzed the human antibody repertoire in a phage display library built from 40 healthy human donorsCitation22 using NGS. The CDRs in this dataset were derived from total lymphocytes, and so comprised both naïve and memory B cells containing mutations within their CDRs. We searched this dataset for CDRs devoid of liabilities () and reported them within the context of the germline genes in which they were found (). The HCDR3s were excluded from this analysis. Interestingly, the majority of antibodies (74% of the light chain and 93% of the heavy chains) contained at least one CDR sequence liability.
Figure 2. (a and b) Proportion of chains showing a liability in at least one of the CDRs in the human naïve repertoire (CDR1-2 for heavy chain and CDR1-3 for light chain), as assessed from a phage display library created from 40 healthy human donors. (c, d, and e) Segmentation of liabilities by V family and CDR
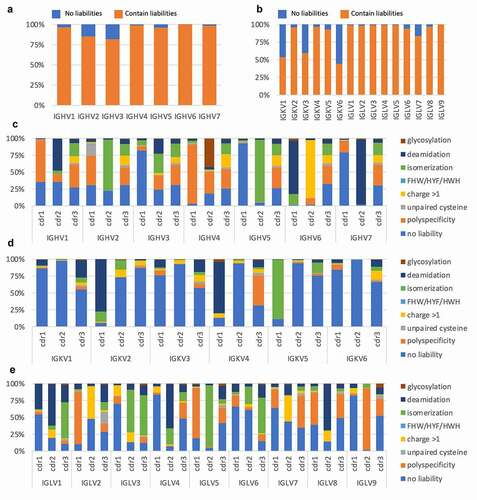
The different heavy and light chain genes showed varying levels of liabilities. For the heavy chain ()), IGHV3 showed the least number of liabilities (82%). In contrast, almost all IGHV6 sequences (99.9%) contained at least one sequence liability. For the light chains ()), IGKV6 had the least number of liabilities, 44%, followed by IGKV1 and IGKV3 with 53% and 60%, respectively. In contrast with IGKV6, IGKV1 and IGKV3 are among the most diverse and highly abundant families of light chain in humans, providing good source material for the development of antibodies.
While the number of VH and VL genes lacking sequence liabilities was extremely low, the number of CDRs lacking sequence liabilities was significantly higher, with means/medians of 31%/23%, 68%/79% and 37%/39% for VH (CDR1-2 only), Vk and Vλ CDRs, respectively. Breaking down the analysis by each CDR (), different patterns emerge. For example, while 47% of IGHV1 CDR2s contain asparagine deamidation motifs, only 5% have aspartate isomerization motifs, while in the CDR2 of IGHV2, the opposite is true, with 2% and 76% for deamidation and isomerization motifs, respectively. It is important to note that the liabilities were analyzed in the listed order, so if a CDR contained more than one liability, only the first one encountered in the sequence was counted. This analysis therefore underestimates the total number of sequence liabilities and likely skews their representation according to the order in which they were analyzed ().
Clinical scaffold selection
To create a library of “drug-like” antibodies, we reasoned that using scaffolds from antibodies that were already drugs – either approved for human therapy or in advanced stage clinical trials – would be better than using scaffolds, including germlines, with poor or unknown therapeutic outcomes. Using a previously published dataset as a starting point,Citation3 we assessed the abundance of each germline V gene in the natural human repertoire, in clinical antibodies overall, and those identified as developable. To characterize developability in this data set, we used the “red flag” descriptor, defined as a biophysical characteristic falling in the bottom 10th percentile of the clinical antibodies (, and Jain et al.Citation3), and directly analogous to Lipinsky’s rule of five.Citation1 The presence of red flags correlates with a reduced likelihood of an antibody being approved for clinical use (Suppl. Figure S1 and Jain et al.Citation3).
Figure 3. Analysis of the clinical antibody heavy (a) and light (b) V germline genes in comparison with the human naïve repertoire and the frequency of developable clinical antibodies (defined as having no more than one red flag). Chosen V domains are highlighted in red. (c) V germline genes of the clinical antibodies selected to be used as scaffolds in the new phage display library
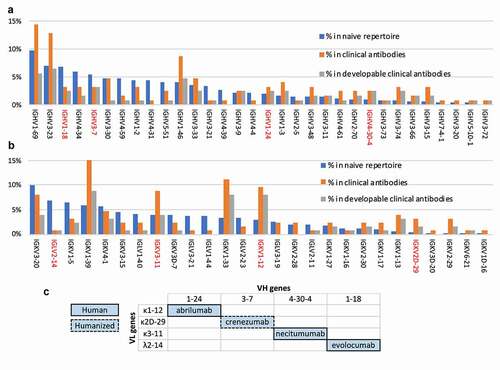
The prevalence of IGHV1 genes in the clinical antibody repertoire, particularly IGHV1-69 and IGHV1-46, can be explained by the fact that many of these antibodies were generated from immunized mice and later humanized, and it is known that IGHV1 family genes are often used in the mouse repertoire.Citation41 However, antibodies containing these commonly used VH1 genes were often identified in the Jain et al. datasetCitation3 as containing biophysical liabilities. The role of murine antibodies in human therapeutics also explains why most therapeutic antibodies use a kappa light chain rather than lambda.Citation42 Due to the biases imposed by mouse-derived antibodies, and the subsequent preferences motivated by this historical precedent, the dataset presents limited information on other germlines such as IGHV6 and IGHV7 of the heavy chain and lambda light chain genes.
Finally, we selected four different therapeutic scaffolds from a variety of germline families to maximize structural and sequence diversity in the library and, therefore, improve the ability to select against different antigens ()). The four antibodies selected to serve as scaffolds in the library were: abrilumab (IGHV1-24, IGKV1-12), crenezumab (IGHV3-7, IGKV2D-29), necitumumab (IGHV4-30-4, IGKV3-11), and evolocumab (IGHV1-18, IGLV2-14) ()). These were representative of three heavy chain germline families (IGHV1, 3, and 4), three kappa light chain families (IGκV1-3) and one lambda light chain family (IGκV1-3). The germlines chosen to represent a particular family were picked for having (where possible) a high proportion of developable antibodies in relation to the number of the clinical antibodies using that germline gene (e.g., the chosen IGHV1-18 is used by four therapeutic mAbs of which three have good biophysical properties, while IGHV1-2 is also found in four therapeutic mAbs, only one of which has good biophysical properties) (, gray and orange bars, respectively).
Creation of highly functional single-CDR scFv libraries
The proportion of sequences in an antibody library that makes functional molecules is an important aspect that remains poorly understood. Functionality, in this case, goes beyond assessing the percentage of antibodies with open reading frames, and includes key biophysical properties that enable an antibody to be used as a therapeutic molecule.Citation3,Citation7,Citation15,Citation43 Sequences encoding molecules with developability issues, or that are improperly folded or displayed, reduce a library’s functional diversity, however high the genetic open reading frame diversity may be. This affects selection outcomes and may result in antibodies that cannot be used for downstream development without further engineering. In contrast to using degenerate oligonucleotides, for this library we produced defined sequences by oligonucleotide array synthesis for HCDR1-2 and LCDR1-3 corresponding to individual CDRs found in our NGS dataset. This provides substantial advantages: 1) the inclusion of only naturally occurring CDR sequences ensures CDRs are derived from B-cell receptors able to provide tonic survival signals,Citation44 and are hence inherently well folded; 2) the avoidance of inadvertent use of aberrant sequences, encoding co-variance violations, that may occur in degenerate oligos; 3) the exclusion of sequence liabilities; and 4) a more uniform CDR diversity distribution in the library, as opposed to that found in natural diversity in which germline sequences heavily dominate the CDR1 and CDR2 repertoires. As synthesis of defined oligonucleotides in arrays is limited to less than 1 million, we sourced HCDR3s from human donor B-cells, reasoning that liabilities occurring only in HCDR3s (and not the remaining CDRs) would be less detrimental to overall antibody developability, and could be more easily eliminated if necessary.
To further optimize the oligo pool for sequences encoding CDRs able to express well within the context of our chosen scaffolds, we included a yeast display enrichment step. For each of the four therapeutic scaffolds we generated five libraries containing diversity in only one CDR at a time (single-CDR libraries). Yeast display vectors for each antibody scaffold, reformatted as scFv, were created (). Additionally, five versions of each scaffold were created, where a Type II restriction site was inserted at one of the CDR positions, to enable scarless cloning of the CDR pools by in vivo yeast homologous recombination. Single-CDR libraries gave us the ability to probe the entire diversity at each CDR position, within the context of a known developable clinical scaffold, enabling efficient sampling and filtering of all the CDRs at each position. While single-CDR libraries have relatively low diversities at any single site, the combinatorial diversity of all the V-gene specific CDRs (excluding HCDR3) was as high as 1018 ().
Table 2. Number of CDR sequences without liabilities synthetized as oligonucleotides. Theoretical diversity given is the product of the diversity of LCDR1-3 and HCDR1-2
Figure 4. Schematic representation of how each single-CDR library was built and sorted. (a) Design of the six yeast display vectors created for each of four scaffolds: one vector has the original clinical antibody reformatted as a single chain and the other five have one CDR replaced by Type II restriction enzyme sites for scarless insertion of CDR libraries and filtering, represented by a white gap. (b) Workflow for creation and filtering of each single-CDR library: liability-free, replicated natural CDRs are inserted into the open yeast display vector by homologous recombination and filtered for high expression using FACS or MACS. (c) Flow cytometry analysis of the four chosen therapeutic antibodies displayed as scFv on the yeast surface. (d) Flow cytometry analysis of the five single-CDR libraries corresponding to abrilumab (Lib1), comparing the parent therapeutic scaffold with the non-enriched libraries and the FACS/MACS libraries enriched for higher levels of display
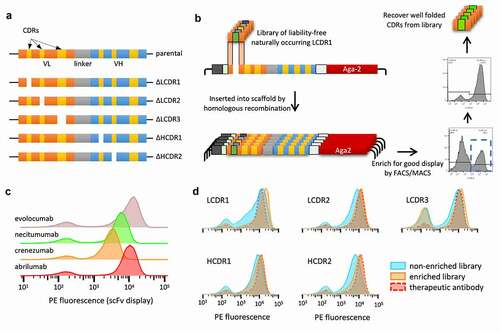
We generated 20 yeast display libraries with 106–107 transformants to cover the maximum diversity of each library at least 10 times. After 48 h of selective pressure using synthetic dropout media, scFv expression was induced with galactose. Fused to the C-terminal of each scFv, we included the SV5 tag for scFv surface detection, followed by the Aga2 protein, responsible for anchoring the scFv to the yeast surface.Citation45 Cells stained with an anti-SV5 antibody conjugated to phycoerythrin (PE) that showed high display signals were sorted using fluorescence-activated cell sorting (FACS) ()), while the LCDR3 libraries were enriched using magnetic-activated cell sorting (MACS) due to their larger diversity (approaching 105 variants). Comparison of the libraries by flow cytometry before and after sorting shows that this step was effective in depleting the population of nonfunctional molecules ()). Further, this process improved the overall display of the libraries, favoring clones with a higher propensity for display () and Suppl. Figure S2), providing profiles similar to the original therapeutic antibody scaffold.
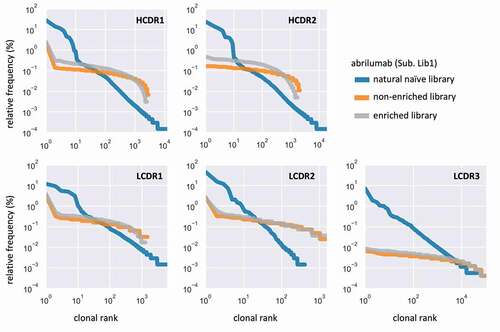
The light and heavy chain CDRs for each library were sequenced (MiSeq) to assess CDR diversity and clonal distribution. The newly produced single-CDR libraries (, orange and gray lines) show a substantially flatter distribution relative to the natural library (, blue line). The population after enrichment (, orange) deviates slightly from its original form. Similar results are observed in the other libraries (Suppl. Figures S3–5).
Figure 5. Comparison of CDR distribution and dominance between the natural naïve library, the non-enriched library and enriched single-CDR libraries. Data are shown for the libraries built using the abrilumab scaffold (Lib1, IGHV1-24, IGKV1-12)
Recovery of HCDR3 diversity from human donors
As a source of HCDR3 diversity we used peripheral blood leukapheresis (LeukoPak) samples from 10 healthy adult human donors (Suppl. Table S1), with the number of viable nucleated cells ranging from 1 × 109 to 5 × 109 per donor. To ensure a more uniform HCDR3 diversity distribution, and hence better diversity sampling in the library, we purified cells using paramagnetic beads recognizing the CD19 marker.Citation46 This protein is expressed in all B-cell developmental stages except plasmacytes, which are actively producing and secreting large quantities of antibodies. Including plasmacytes (CD19−) would skew the library toward these antibody sequences as a result of the larger amount of antibody mRNA in their cytoplasm when compared to other developmental B cell stages (CD19+) ()). From a starting population of 3.3 × 1010 viable white blood cells, a total of 1.9 × 109 B-cells were recovered after purification (5.64% yield), in line with known B cell abundance.
Figure 6. (a) Schematic representation of HCDR3 diversity rescue from 10 human donors. First, peripheral blood is submitted to leukapheresis; then recovered cells are purified by magnetic activated cell sorting (MACS) recognizing the CD19 marker for B-cells. The RNA is extracted, reverse transcribed with an IgM CH1 specific primer and the HCDR3 is amplified by PCR with primers specific to different germline families used in the library. (b) Saturation analysis of the HCDR3 deep sequencing results. (c) HCDR3 amino acid length distribution in library
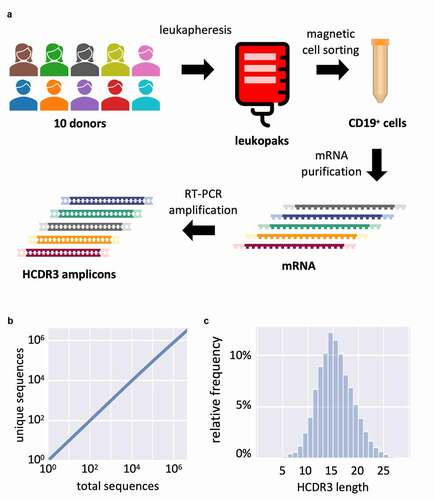
Total RNA was purified from the cells and enriched for polyA+ RNA. We reverse-transcribed the mRNA using an IgM CH1 specific primer. HCDR3s were PCR amplified using forward primers specific to the framework 3 of each germline used (IGHV1, 3, and 4) and a single reverse primer specific for the 3ʹ end of the IGHJ segment. The amplicons were sequenced using MiSeq for quality control. From a total of 4,489,674 analyzed HCDR3s, we found 3,255,058 unique sequences. However, accumulation analysis revealed that the accrual of sequenced clones was far from saturation, indicating vast undersampling of the population, and a true diversity far higher than that measured by NGS ()). Our previous experience using the same methods with other libraries shows that a diversity >108 unique HCDR3s is anticipated when measured by NovaSeq.Citation40 Analysis of the HCDR3 sequence length revealed a normal distribution with a mode at 14 amino acids, consistent with the human repertoire ()).
Phage display library creation and validation
To create the final phage display library, we PCR amplified the CDRs plus their flanking regions from each single-CDR enriched library. Three fragments containing LCDR1, LCDR2 and LCDR3 were first combined by overlap PCR to create the light chain plus the linker downstream. Next, two fragments containing HCDR1 and HCDR2 were combined with the natural HCDR3 amplicons to create the heavy chain with a linker upstream. Finally, light and heavy chains were assembled using the linker region as anchor to create the four individual scFv libraries (). After digestion with restriction enzymes BssHII and NheI, inserts were ligated into the phage display vector, pDAN5,Citation22 and transformed into E. coli TG1, yielding a combined total of 9 × 109 transformants. Subsequently, each library was superinfected with the helper phage M13K07 for phage particle production.
To test the library performance, we employed a selection strategy using two rounds of phage display followed by two or more rounds of yeast display ()).Citation47 As targets, we selected four human proteins of therapeutic interest: interferon α-2 (IFN2a);Citation48 granulocyte-macrophage colony-stimulating factor (GM-CSF);Citation49,Citation50 interleukin-6 (IL6);Citation51,Citation52 and transforming growth factor β-R2 (TGFβ-R2).Citation53 Briefly, the recombinant proteins were biotinylated in vitro using sulfo-NHS chemistry and captured using streptavidin-conjugated magnetic beads. The combined phage from the four libraries was incubated with the coated beads; non-bound clones were washed away, and the remaining phage were eluted with HCl and rescued with E. coli. After two rounds, the scFv inserts were amplified and transferred to a yeast display system by in vivo homologous recombination. Yeast cells expressing the scFv were incubated with biotinylated antigen and labeled to detect the scFv display (anti-SV5-tag PE) and antigen binding (streptavidin-Alexa 633). Cells binding to the antigen were sorted by flow cytometry. Antigen concentration was decreased in each yeast selection round (100 nM: all antigens, 10 nM: all antigens and 1 nM: GM-CSF and IFN-2α).
Figure 7. (a) Schematic representation of the selection process combining two rounds of phage display and yeast display. (b) Flow cytometry analysis of the final selected populations against each antigen at varying concentrations. Display is detected with anti-SV5 antibody labeled with PE and binding is detected with streptavidin labeled with alexa-633. (c) Levenshtein distance of merged CDRs between clones selected against GM-CSF. (d) Levenshtein distance of HCDR3 between clones selected against GM-CSF. (e) Surface plasmon resonance affinity plot for test clones from GM-CSF, IFN-2⍺, IL6 and TGFβ-R2. The diagonal lines (isoaffinity) represent the affinity (KD) of the antibodies, x-axis show dissociation constant (kd) and y-axis shows association constant (ka)
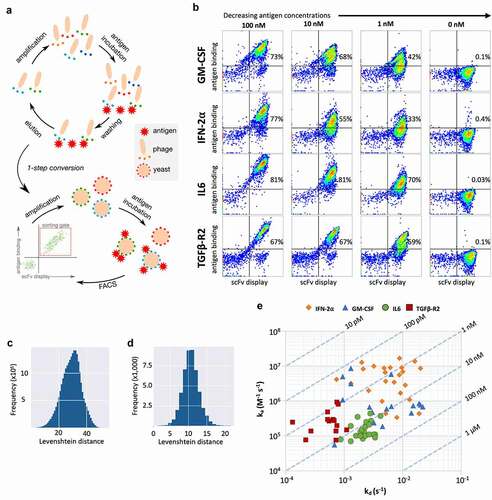
We analyzed the binding profile of the final resulting populations by flow cytometry when incubated with different antigen concentrations ()), including a no-antigen control (0 nM) to check for nonspecific binding to secondary reagents. As expected, we observed decreased binding with decreasing antigen concentration. Nonetheless, at 1 nM we observed a significant population bound to the antigen (GM-CSF: 42%; IFN-2α: 33%, IL6: 70%; TGFβ-R2: 59%) whereas the binding signal was negligible when no antigen is present (GM-CSF: 0.1%; IFN-2α: 0.4%, IL6: 0.03%; TGFβ-R2: 0.1%), indicating the selection of high-affinity specific antibodies.
To further analyze the enriched populations, we performed PacBio NGS (IL6, GM-CSF, IFN-2α). When we used the pairwise string edit distance of the six concatenated CDRs to compare all identified antibodies from selections (); GMCSF output shown), the majority differed by 20 to 40 amino acids. When comparing HCDR3 only ()), we observe a difference of 8 to 13. These data indicate that the selected clones are not derivatives or small variations of a common antibody, but rather distinct sequences. Interestingly, we have also observed that for each one of the antigens one of the sublibraries was preferentially selected (GM-CSF: sublib.-4; IFN-2α: sublib.-1, IL6: sublib.-2) (Suppl. Figure S6).
To probe for antibody equilibrium dissociation constants (KD), we used high-throughput surface plasmon resonance (SPR).Citation54 Unique clones identified from a 96-clone pool from the final sorted population were converted to scFv-Fc fusions and expressed using S. cerevisiae YVH10.Citation55,Citation56 To capture the antibodies to the SPR chip we first coupled a polyclonal anti-human Fc to the gold nanolayer. Unpurified yeast expression supernatants containing the scFv-Fc molecules were flowed onto the chip for capture, followed by antigens (analyte) at increasing concentrations. Association and dissociation constants were determined using a first-order kinetic model (“one-to-one”) as implemented in the analysis software provided by the equipment manufacturer ()).
We tested a total of 81 different antibodies (GM-CSF: 19; IFN-2α: 24, IL6: 24; TGFβ-R2: 14). Of these, 16 (20%) showed sub-nanomolar affinities and 48 showed sub-10 nM affinities (59%), with average/median affinities of 12.3 nM/9.8 nM for GM-CSF, 5 nM/1.3 nM for IFN-2α, 17.5 nM/15.3 nM for IL6 and 3 nM/2.1 nM for TGFβ-R2 ()). The additional selective pressure applied to the IL6 population at 1 nM caused the enrichment of truncated clones that dominated the sorted population, hence the 1 nM output was omitted from the SPR analysis. Nonetheless, with a straightforward selection protocol and without further affinity maturation/engineering, the platform demonstrated a capacity to deliver high-affinity binders against different therapeutic targets.
Developability
Additionally, we determined whether the elimination of sequence liabilities from CDRs resulted in the generation of antibodies that were developable as well as having high affinity. Clones from each library were produced as human IgG1 in HEK293 cells and purified by affinity chromatography. To gather relevant developability information about the selected antibodies, we chose the following non-overlapping and scalable assays: differential scanning fluorimetry for assessment of thermostability/melting temperature (Tm),Citation57 enzyme-linked immunoassay (ELISA) polyspecificity assay, affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS)Citation58,Citation59 and AC-SINS in the presence of 300 mM of ammonium sulfate for antibody self-interaction/aggregation-prone behavior,Citation60 size-exclusion chromatography (SEC) after freeze-thaw cycles and after accelerated stability assay (exposure to high temperature [37°C] for 4 weeks).Citation3
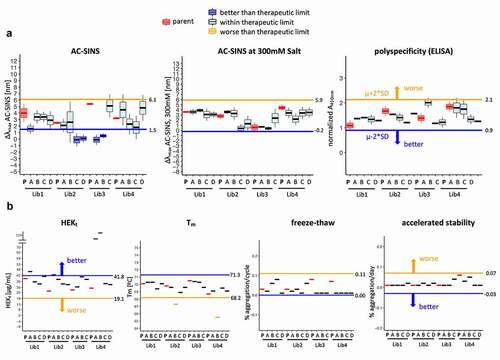
To have a baseline for comparison, we also tested the clinical antibodies used as scaffolds (parentals – )). We classified the measurements into three categories: 1) better than parental, when the test antibody showed a result that differed by more than two times the standard deviations of the corresponding therapeutic scaffold in the direction of better developability; 2) within clinical range, when the measurement was within two times of the standard deviation of the therapeutic antibodies used as scaffolds for the generation 3 library; and 3) outside clinical range, when the measurement was outside the range of two times the standard deviation of the therapeutic antibody scaffold in the direction of poor developability.
Of the 105 measurements collected for the antibodies selected from the libraries, 97% scored as well as, or better than, the measurements of the corresponding highly developable clinical parental antibody, with only two of the antibodies showing Tm (67.3°C and 65.5°C) slightly below the therapeutic range (≥68.5°C) ( and Suppl. Figure S7). Remarkably, many antibodies showed signals that were better than the clinical parent in the assays: 4 of 15 antibodies on AC-SINS and Hek titer ( and Suppl. Figure S7). At the antibody level, 13 of the 15 tested antibodies (87%) had no measured biophysical liability whatsoever, while the remaining 2 antibodies had a single biophysical liability. In conclusion, the antibodies from the library behaved very favorably when tested for developability and compared to the clinical molecules.
Figure 8. Developability profile of selected clones from the library. Measurement(s) of the selected clones (named A to D) are compared to the parental clinical scaffold (named P) and the therapeutic limit. The therapeutic limit is defined as two times the standard deviation of all measurements of the parental mAbs (Lib1-P, Lib2-P, Lib3-P, Lib4-P), represented by the horizontal line extending across each plot in the direction of better (blue) or worse (orange) developability. (a) AC-SINS, AC-SINS at 300 mM salt and polyspecificity results are derived from independent experiments (N = 3 for AC-SINS at 300 mM salt and AC-SINS; N = 2 for polyspecificity). The middle line, box limits and whiskers of the boxplot represent the mean, one standard deviation and two standard deviations of the repeat measurements, respectively. (b) HEKt, Tm, Freeze-Thaw and AS represent the final measured or calculated values from single experiments and depicted by thick horizontal line. Colors indicate whether the test mAb measurement(s) is better (dark blue box; dark blue line), worse (light orange box; light orange line) or within two standard deviations (light blue box; black line) of the therapeutic limit. The parental mAbs (red box; red line) are distinctly colored to provide reference for the test mAbs
Lastly, we also tested if scFv selected from the generation 3 library could be converted to IgG and still show binding to the intended antigen. scFv sequences identified after panning against four additional therapeutically relevant antigens were converted to IgG. We observed a conversion rate ranging from 74% to 92% (), depending on the antigen. However, it should be noted that the tested antibodies were not previously validated in the scFv format, a negative binding result as IgG could reflect either a loss of binding activity during conversion, or that these antibodies were not true binders (i.e., background). Nonetheless, the results show that a high percentage of antibodies can be effectively converted from scFv to IgG and retain functionality.
Table 3. Binding of clones to intended target in the IgG format measured by ELISA assay. Molecules were tested solely in the IgG format, not undergoing previous validation as scFvs
Discussion
Since the introduction of in vitro antibody discovery, many antibody library architectures have been described.Citation19,Citation22,Citation24–26,Citation28,Citation32,Citation34,Citation39,Citation61–64 Previous sources of library diversity have comprised: 1) natural full length VH/VL repertoires amplified from donors (natural libraries); 2) CDR-only repertoires amplified from lymphocytes and inserted into a synthetic scaffold (natural combinatorial CDR libraries); 3) degenerate oligonucleotides, designed with various levels of sophistication, inserted into synthetic scaffolds (synthetic libraries); or, 4) semi-synthetic, in which CDR3 diversity is natural, and CDR1/2 diversity is derived from degenerate oligonucleotides. In none of these was it possible to definitively eliminate sequence liabilities. This is the first report of a semi-synthetic library architecture where all CDR diversity corresponds to natural CDRs and sequence-based liabilities are removed from 5 of the 6 CDRs.
High affinity, improved developability and broad diversity have been described as together defining the “holy grail” for next-generation antibody libraries.Citation65 Many of the published libraries described above are of comparable size (number of transformants) and the best among them have delivered some antibodies with comparable affinities to those described in this work. However, we show that by removing sequence liabilities from CDRs and selecting scaffolds derived from therapeutic antibodies, it is possible to routinely obtain both high affinity and highly developable antibodies directly from selections, results expected to close the gap between antibodies generated in vitro and in vivo.Citation59,Citation66
Achieving clinical success with an antibody depends on many different factors beyond the intrinsic properties of the molecule being tested.Citation67 In most cases it is not possible to determine whether an antibody failed a clinical trial due to poor developability given the data currently available. Poorly developable antibodies should show abnormal behavior in earlier development stages, such as pre-clinical and formulation (e.g., a polyspecific antibody may show faster than normal clearance in pre-clinical studies),Citation66 and it is clear that the proportion of poorly developable antibodies decreases with clinical stage and approval (Suppl. Figure S1 and Jain et al.Citation3 figure S1). Examples such as sirukumab and bococizumab show that poorly developable antibodies may eventually make their way to clinical trials, generating huge human and financial costs that could have been avoided by early developability assessments.
The developability assays used here were selected based on their widespread acceptance and non-redundant ability to assess relevant properties in a large study of the biophysical characteristics of clinical-stage antibodies.Citation3 Some act as surrogates for properties desired in an antibody but not directly quantifiable as a meaningful characteristic. Melting temperature (Tm), for example, is often considered an important parameter when generating new libraries for therapeutic purposes.Citation32,Citation33,Citation61 However, the analysis shows no correlation between the Tm of therapeutic antibodies and other relevant properties such as aggregation and specificity, and only a weak correlation with antibody expression titer (Spearman coefficient = 0.35). For example, rilotumumab (anti-hepatocyte growth factor, Amgen Inc), while being highly thermostable (Tm 79°C), performed poorly in an accelerated stability study in solution (the worst of all the tested antibodies), and among the scaffolds we chose, evolocumab has the lowest reported Tm (65°C), while having the highest reported expression titer (260.7 mg/ml).Citation3
Of additional interest, non-approved antibodies often show poor results in AC-SINS and SGAC-SINS when compared to approved antibodies, suggesting a predictive power to these measurements (Suppl. Figure S8a). In addition, phage-derived antibodies seem to perform worse in these tests when compared to antibodies developed in vivo (Suppl. Figure S8b). One can speculate that this is simply because the datasetCitation3 has many more approved antibodies derived from animals than from phage display, skewing the data. However, if we look only at approved antibodies, those that are phage-derived still perform markedly worse (Suppl. Figure S8c). The antibodies described here show good results in these tests, despite being developed using phage (and yeast) display, indicating that the present library format in combination with sequential phage and yeast display are able to deliver antibodies as good or better than current in vivo technology. Further testing on 86 different antibodies discovered from other selection campaigns carried out using this library showed that 14% of antibodies had AC-SINS results worse than the therapeutic range (<6.6 nm),Citation3 compared to 20% for a natural library (Suppl. Figure S9), with the median AC-SINS values for all antibodies derived from the Generation 3 platform (2.1 nm) lower than those (3.0 nm) derived from a natural phage library also selected using only phage display. This suggests the problem is not inherent to display technology, but more a function of antibody library design. , perhaps facilitated by the quality control exerted by the yeast endoplasmic reticulum and secretory pathway.Citation68,Citation69 Nonetheless, different antigens may present unique challenges (e.g., charged or hydrophobic patches) and may require specific strategies to overcome biases and obtain developable leads.
Developability problems can be, and often are, addressed downstream of the selection process by discarding antibodies containing sequence liabilities after in vitro selection or in vivo development, an approach that effectively reduces functional library size. Instead of discarding binders with developability issues, further engineering to improve the biophysical properties of promising leads can also be undertaken – of note, the existence of a sequence liability (e.g., NxS glycosylation site) does not necessarily mean that the antibody will be poorly developable (e.g., glycosylation leading to immunogenicity). However, assessing the nature of the liabilities and engineering if necessary is a process that may consume months and incur substantial additional expense. Furthermore, mutations that improve developability may negatively impact affinity, and vice-versa, making this approach particularly difficult and not always successful.Citation12,Citation70–72
Libraries in which the majority of sequence liabilities have been eliminated have the advantage that functional diversity will be greater than libraries in which such sequence liabilities need to be engineered away or followed closely. With regulatory agencies demanding greater product quality and safety profiles (ICH guidelines Q8, Q9, Q11), the advantage of upfront, improved developability is of paramount importance. Sequence liabilities that have the potential to result in loss in efficacy or increased immunogenicity risk are considered critical quality attributes (CQAs), requiring identification, monitoring, mechanistic understanding and analysis, process control strategies and risk management throughout the development lifecycle. As such, the removal or absence of CQAs has a huge financial benefit by reducing development timelines and resource burden, as well as de-risking the likelihood of failure in the clinic.
In this work, some of the motifs flagged as sequence liabilities, particularly those identified as CQAs, may be considered self-evident, while others are not. The detrimental effect of exposed unpaired Cys residues in a CDR requires little explanation,Citation73 whereas motifs such as Gly-GlyCitation11 pose a more delicate argument. Given the technical impossibility of achieving the total theoretical diversity (~1022 to ~1026, including HCDR3) of the library, we reasoned that the exclusion of any CDR sequence containing any motif supported by the literature, would have no detrimental effect on the functionality of the final library, an assumption supported by the quality of the antibodies selected. The vast excess of untapped theoretical diversity means that additional motifs can be easily eliminated from future libraries, as they become identified. Noteworthy exceptions are methionine and tryptophan oxidation,Citation74 since removal of every CDR containing one of these amino acids would dramatically reduce final diversity.
The filtering of the CDRs using yeast display may also have had a positive influence on the developability of the antibodies in our Generation 3 library. Secreted proteins have to pass a stringent quality control process in the endoplasmic reticulum in order to be exported. Proteins that fail maturation are targeted for degradation.Citation75 We hypothesize that by producing libraries with diversity in only one CDR and using yeast display as a tool to filter for high-expressing/displaying sequences, the resulting antibodies obtained from combinatorial assembly of these filtered CDRs would be more likely to have favorable characteristics related to folding, secretion and thermostability, since these characteristics may be linked to display levels.Citation69,Citation76,Citation77 We believe the filtering process eliminates natural CDR sequences that do not fold well within the desired scaffold, as well as possible oligonucleotide synthesis or PCR errors that may cause similar problems.
It has been suggested that antibody pharmacokinetics are not altered in mice by Fv glycosylationCitation78 and the addition of glycosyl groups has even been used as a strategy to increase the solubility of antibodies.Citation12 In fact, 15 to 25% of circulating IgGs contain glycans in their variable domains.Citation79 Nonetheless, their presence may be detrimental for therapeutic antibodies. Different glycosylation patterns in the variable regions, particularly in the CDRs, may not only create heterogeneity in manufacture and hence binding and affinity, but also cause unwanted immunogenicity. For instance, cetuximab, an anti-EGFR therapeutic mAb, has been shown to induce IgE-mediated anaphylaxis in up to 22% of treated individualsCitation80 and a deeper analysis revealed that the glycans present in the variable domain were responsible for this hypersensitivity.Citation81
We have not assessed immunogenicity in this work. However, the fact that all scaffolds are from well-behaved human therapeutic antibodies and the CDRs are based on human sequences from peripheral B cells, together resulting in antibodies with improved developability characteristics, may reduce the need to implement extensive efforts to mitigate immunogenicity. Furthermore, the removal of sequences mediating post-translational modifications such as glycosylation, asparagine deamidation and aspartate isomerization in CDR L1-L3 and H1-H2 are expected to reduce the potential for immunogenicity.Citation82,Citation83 However, general conclusions regarding antibody immunogenicity have to be made with some caution, since even fully human antibodies may elicit immune responses.Citation65,Citation84–88
By implementing the desired developability properties into the antibody library platform used for antibody lead generation, we have shown that one can retrieve a wide panel of “drug-like” antibodies directly from in vitro selection campaigns. We anticipate that the use of the designs and approaches presented here will enable scientists to considerably reduce the failure rate of leads and shorten development times by, for example, eliminating time-consuming processes such as iterative rounds of affinity maturation followed by cycles of developability engineering. In fact, the inherent liability-free diversity in these libraries can also be used to affinity-mature antibodies without introducing sequence liabilities. Furthermore, use of this platform may improve the performance of bispecifics by ensuring the pairing of domains that are well-behaved biophysically.
Materials and methods
Natural HCDR3 diversity recovery
Fresh leukapheresis products from 10 different healthy human donors (StemExpress, #LE001F) were used to generate the HCDR3 diversity for the library. The samples were pooled into two groups (A and B) and were kept separate until the final scFv assembly. CD19+ cells from each pool were purified using paramagnetic beads coated with CD19-specific antibodies (CD19 MicroBeads human, Miltenyi, #130-050-301) and magnetic-activated cell sorting. Purification was carried out according to the manufacturer’s protocol and as described by Ferrara et al.Citation46 The number of viable cells were counted using a hemocytometer and a 0.4% trypan blue solution (Gibco, 15250061). Total RNA from cells was purified using high-capacity spin columns (RNeasy Maxi Kit, Qiagen, #75162) and subsequently enriched for mRNA using a resin specific for polyA+ (Oligotex mRNA Midi Kit, Qiagen, #70042). The variable region of the heavy chain was reverse transcribed using a reverse primer specific to the IgM CH1 region (5ʹGGAAAAGGGTTGGGGCGGAT3ʹ) and reverse transcriptase (SuperScript™ IV First-Strand Synthesis System, Invitrogen, #18091200). Finally, heavy chain CDR3 sequences were amplified by PCR employing high-fidelity DNA polymerase (Q5 DNA Polymerase, NEB, #M0491L), four different forward primers specific to the end of framework 3 region of the heavy chain and one reverse primer specific to the heavy joining (J) gene segment. All procedures were carried as instructed by the reagents’ manufacturers, unless otherwise noted.
Deep sequencing and design of HCDR1-2 and LCDR1-3
A 40-donor phage display scFv libraryCitation22 was used to profile the diversity of LCDR1-3 and HCDR1-2. Light and heavy chains were amplified separately by PCR and sequenced using MiSeq (2x250bp) and NovaSeq (2x150bp) (Illumina). For MiSeq, the paired-ends were assembled using PandaSeq.Citation89 Sequences were quality filtered using the FASTX-Toolkit with a minimum quality of 25 and minimum percent of bases that must retain this quality set to 90%. All DNA reads were annotated with IgBlastCitation90 using IMGT human antibody germline database and CDR definitions,Citation91,Citation92 except for LCDR2 where the Kabat definition was used. We excluded all CDR amino acid sequences having liabilities, anomalous length, or less than four reads (). Oligonucleotides corresponding to those identified for HCDR1-2 and LCDR1-3 after the elimination steps as described in the above examples were synthesized (Twist, Inc., San Francisco, CA), resulting in a total of 337,697 oligonucleotides coding for the selected CDRs. The CDR coding sequence in these oligonucleotides was flanked by 5ʹ and 3ʹ sequences homologous to the framework vectors, into which the CDR coding sequences were cloned. The homologous sequences were used for both amplification and insertion of the oligonucleotides into the yeast display vectors.
Generation of yeast libraries with replicated natural diversity and filtering
Each of the 20 CDR collections were amplified with specific primers for the flanking regions using Q5 polymerase (NEB #M0491L). For each of the four libraries created using the four scaffolds, six cassettes encoding scFv corresponding to each of the scaffolds were synthesized. One of the six encodes the non-modified scFv, while the other five were each modified by replacing the original CDRs (excluding HCDR3) with a combination of restriction sites including two inverted BsaI sites, an additional SfiI site to ensure cleavage of the vector and serve as a spacer between the BsaI sites, a frameshift and an ocher stop codon to prevent expression of background sequence. Each of these modified polynucleotides encoding the scaffolds was cloned into a yeast display vector, and the presence of the stop codon in this sequence prevented the expression of the scaffold on the yeast surface until the modified CDR is replaced with a functional CDR. In all constructs, an SV5 tag sequence was present downstream of the scFv. The 20 plasmids were digested with BsaI-HF-v2 (NEB, #R3733L) and SfiI (NEB, # R0123L) and co-transformed in Saccharomyces cerevisiae strain EBY100 with the corresponding CDR amplicon collection. All 20 libraries were subject to selection and growth in selective dropout media for 48 hours, then induced for scFv expression with galactose. Cells were stained with anti-SV5 antibody conjugated with R-PE. For the CDR 1–2 (heavy and light) libraries, the top 2% of the scFv-expressing population were sorted using FACS. For the LCDR3 libraries, scFv expressing cells were captured with paramagnetic-beads coated with anti-PE antibody (Miltenyi, #130-105-639) and purified using MACS.
Phage display library construction
CDRs from each of the filtered libraries were amplified along with the flanking framework regions (or scFv linker). VL and VH were assembled separately in a 3-fragment PCR reaction (CDR1, CDR2 and CDR3 fragments) using the framework regions as priming sequences. Subsequently, VL and VH were assembled in a 2-fragment PCR reaction using the scFv linker as priming sequence. All PCR reactions used Q5 polymerase. The scFv amplicons were digested using BssHII and NheI restriction enzymes (NEB, #R0199L and #R0131L) and ligated to the pDAN5 phagemid vectorCitation22 previously digested with the same enzymes. Ligation products were transformed into E. coli TG1 strain (Lucigen, #60502-2) by electroporation, plated onto 2xYT agar containing 3% glucose, 1.5% sucrose and 100 mg/ml of carbenicillin and grown overnight at 37°C. Next day, plates were scraped, and bacteria for each library was frozen individually in 2xYT 16% glycerol at −80°C.
Antibody selections and deep sequencing analysis
Phage particles were produced by inoculating bacteria from each of the 4 libraries into 2xYT media + 3% dextrose + 100 ug/ml carbenicillin and growing at 37°C until OD600nm = 0.5. The bacteria were then infected with the helper phage M13K07 (MOI = 10) and grown overnight in 2xYT media + 100 ug/ml carbenicillin at 25°C for phage production. Next day, phage were precipitated from the supernatant using the PEG/NaCl method. Selections were carried out by performing two rounds of phage display and then cloning the output into our yeast display platform where the libraries were sorted at decreasing antigen concentrations, as previously described.Citation47 After the final round of yeast display sorting, the plasmid was purified from the yeast using glass beads and alkaline lysis and the scFvs were amplified by PCR and sequenced using PacBio Sequel, with a 1 M SMRT Cell.
Affinity measurements by surface plasmon resonance
Individual scFv clones were subcloned into a yeast expression vector containing a human IgG1 Fc region. The vectors were transformed into Saccharomyces cerevisiae strain YVH10. scFv-Fc fusions were expressed for 72 h at 20°C in the presence of galactose. We used a Carterra LSA surface resonance machine for the measurements. Briefly, anti-Human IgG Fc (Southern Biotech, #2048-01) was chemically coupled to a HC200M chip following manufactures protocols. Crude yeast supernatants containing the scFv-Fc fusions were arrayed on the chip. Antigen was injected at seven different concentrations (300, 100, 33, 11.1, 3.7, 1.2, 0.4 nM) to determine association and dissociation rates. All analyses were performed using Carterra software.
Developability assays
All assay were performed as described before,Citation3 with modifications or additional experimental procedures detailed here.
Freeze-thaw
Freeze/thaw SEC experiments were carried similar to those described previously.Citation93 Briefly, aggregation/degradation profiles of the antibodies were tested over a 10-day period in two separate storage conditions maintained at refrigeration (4°C) or frozen at −20°C at 1 mg/mL. Samples were frozen and thawed at different days 0, 1, 2, 5, and 10 days. Day 10 samples were analyzed by size-exclusion high-performance liquid chromatography (HPLC) and detected using a diode array detector (SE-HPLC-DAD) at 214 and 280 nm. All SE-HPLC-DAD methods were conducted with samples diluted to 0.1 mg/mL with 150 mM NaCl, 50 mM Phosphate, pH 7.0, carried out with on Agilent 1100 HPLC. All % monomers, aggregates and degradants were determined using the Area Under the Curve (AUC) with the ChemStation platform.
AC-SINS at 300 mM salt concentration
– In contrast to Jain et al., we opted to conduct the AC-SINS experiment with a single reported ammonium sulfate concentration at 300 mM. We reported the spectral shift value, similar to the value reported from AC-SINS, as described in greater detail by Jain et al.Citation3
Supplemental Material
Download Zip (4 MB)Acknowledgments
We thank Dr. Yasmina Abdiche and Dr. Daniel Bedinger from Carterra, Inc. for the assistance with surface plasmon resonance affinity measurements.
Disclosure statement
AAT, MFE and ARMB are listed as inventors on patents US10,954,508 and WO2020014143A1
ARMB, SD, MFE, FF, and LN are shareholders of Specifica, Inc.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–16. doi:10.1016/S0169-409X(00)00129-0.
- Wang W, Wang E, Balthasar J. Monoclonal Antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58. doi:10.1038/clpt.2008.170.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci. 2017;114(5):944–49. doi:10.1073/pnas.1616408114.
- Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29(3):245–54. doi:10.1038/nbt.1791.
- Naranjo L, Ferrara F, Blanchard N, Demangel C, D’Angelo S, Erasmus MF, Teixeira AA, Bradbury ARM. Recombinant antibodies against mycolactone. Toxins. 2019;11:346.
- Spencer S, Bethea D, Raju TS, Giles-Komar J, Feng Y. Solubility evaluation of murine hybridoma antibodies. mAbs. 2012;4(3):319–25. doi:10.4161/mabs.19869.
- Kumar S, Singh SK. Developability of biotherapeutics. Computational approaches. CRC Press; Boca Raton, Florida, USA: 2015.
- Gavel Y, Heijne G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng Des Sel. 1990;3(5):433–42. doi:10.1093/protein/3.5.433.
- Lowenthal MS, Davis KS, Formolo T, Kilpatrick LE, Phinney KW. Identification of novel N-glycosylation sites at noncanonical protein consensus motifs. J Proteome Res. 2016;15(7):2087–101. doi:10.1021/acs.jproteome.5b00733.
- Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Mølhøj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. Plos One. 2014;9(6):e100736. doi:10.1371/journal.pone.0100736.
- Kelly RL, Le D, Zhao J, Wittrup KD. Reduction of nonspecificity motifs in synthetic antibody libraries. J Mol Biol. 2018;430(1):119–30. doi:10.1016/j.jmb.2017.11.008.
- Wu S-J, Luo J, O’Neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel PEDS. 2010;23(8):643–51. doi:10.1093/protein/gzq037.
- Bethea D, Wu S-J, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’Neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel. 2012;25(10):531–38. doi:10.1093/protein/gzs047.
- Yadav S, Sreedhara A, Kanai S, Liu J, Lien S, Lowman H, Kalonia DS, Shire SJ. Establishing a link between amino acid sequences and self-associating and viscoelastic behavior of two closely related monoclonal antibodies. Pharm Res. 2011;28(7):1750–64. doi:10.1007/s11095-011-0410-0.
- Alam ME, Geng SB, Bender C, Ludwig SD, Linden L, Hoet R, Tessier PM. Biophysical and sequence-based methods for identifying monovalent and bivalent antibodies with high colloidal stability. Mol Pharm. 2018;15(1):150–63. doi:10.1021/acs.molpharmaceut.7b00779.
- Brych SR, Gokarn YR, Hultgen H, Stevenson RJ, Rajan R, Matsumura M. Characterization of antibody aggregation: role of buried, unpaired cysteines in particle formation. J Pharm Sci. 2010;99(2):764–81. doi:10.1002/jps.21868.
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. J Mol Biol. 1991;222(3):581–97. doi:10.1016/0022-2836(91)90498-U.
- Barbas CF, Bain JD, Hoekstra DM, Lerner RA. Semisynthetic combinatorial antibody libraries: a chemical solution to the diversity problem. Proc Natl Acad Sci. 1992;89(10):4457–61. doi:10.1073/pnas.89.10.4457.
- Little M, Welschof M, Braunagel M, Hermes I, Christ C, Keller A, Rohrbach P, Kürschner T, Schmidt S, Kleist C, et al. Generation of a large complex antibody library from multiple donors. J Immunol Methods. 1999;231(1–2):3–9. doi:10.1016/S0022-1759(99)00164-7.
- Hanes J, Schaffitzel C, Knappik A, Plückthun A. Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat Biotechnol. 2000;18(12):1287–92. doi:10.1038/82407.
- Griffiths’ AD, Williams SC, Hartley’ O, Tomlinson IM, Crosbyl WL, Jones’ PT, Low M, Allison TJ, Hoogenboom HR, Coxl JPL, et al. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994;13:3245–60. doi:10.1002/j.1460-2075.1994.tb06626.x.
- Sblattero D, Bradbury A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 2000;18(1):75–80. doi:10.1038/71958.
- Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JMW, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, et al. Flow-cytometric isolation of human antibodies from a nonimmune saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21(2):163–70. doi:10.1038/nbt785.
- Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338(2):299–310. doi:10.1016/j.jmb.2004.02.050.
- Rothe C, Urlinger S, Löhning C, Prassler J, Stark Y, Jäger U, Hubner B, Bardroff M, Pradel I, Boss M, et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol. 2008;376(4):1182–200. doi:10.1016/j.jmb.2007.12.018.
- Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GMR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci. 2009;106(48):20216–21. doi:10.1073/pnas.0909775106.
- Ge X, Mazor Y, Hunicke-Smith SP, Ellington AD, Georgiou G. Rapid construction and characterization of synthetic antibody libraries without DNA amplification. Biotechnol Bioeng. 2010. doi:10.1002/bit.22712.
- Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, Nörenberg S, Stark Y, Kölln J, Popp A, et al. HuCAL PLATINUM, a synthetic fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol. 2011;413(1):261–78. doi:10.1016/j.jmb.2011.08.012.
- Villa A, Lovato V, Bujak E, Wulhfard S, Pasche N, Neri D. A novel synthetic naïve human antibody library allows the isolation of antibodies against a new epitope of oncofetal fibronectin. mAbs. 2011;3(3):264–72. doi:10.4161/mabs.3.3.15616.
- Mahon CM, Lambert MA, Glanville J, Wade JM, Fennell BJ, Krebs MR, Armellino D, Yang S, Liu X, O’Sullivan CM, et al. Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J Mol Biol. 2013;425(10):1712–30. doi:10.1016/j.jmb.2013.02.015.
- Schwimmer LJ, Huang B, Giang H, Cotter RL, Chemla-Vogel DS, Dy FV, Tam EM, Zhang F, Toy P, Bohmann DJ, et al. Discovery of diverse and functional antibodies from large human repertoire antibody libraries. J Immunol Methods. 2013;391(1–2):60–71. doi:10.1016/j.jim.2013.02.010.
- Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, Berenguer M, Poujol D, Stehle J, Stark Y, et al. A fully synthetic human fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs. 2013;5(3):445–70. doi:10.4161/mabs.24218.
- Weber M, Bujak E, Putelli A, Villa A, Matasci M, Gualandi L, Hemmerle T, Wulhfard S, Neri D, Highly Functional A. Synthetic phage display library containing over 40 billion human antibody clones. Plos One. 2014;9:e100000. doi:10.1371/journal.pone.0100000.
- Bai X, Kim J, Kang S, Kim W, Shim H, Novel A. Human scFv library with non-combinatorial synthetic CDR diversity. Plos One. 2015;10(10):e0141045. doi:10.1371/journal.pone.0141045.
- Li K, Zettlitz KA, Lipianskaya J, Zhou Y, Marks JD, Mallick P, Reiter RE, Wu AM. A fully human scFv phage display library for rapid antibody fragment reformatting. Protein Eng Des Sel. 2015;28(10):307–16. doi:10.1093/protein/gzv024.
- Pasello M, Zamboni S, Mallano A, Flego M, Picci P, Cianfriglia M, Scotlandi K. Design and construction of a new human naïve single-chain fragment variable antibody library, IORISS1. J Biotechnol. 2016;224:1–11. doi:10.1016/j.jbiotec.2016.02.034.
- Maruthachalam BV, El-Sayed A, Liu J, Sutherland AR, Hill W, Alam MK, Pastushok L, Fonge H, Barreto K, Geyer CR. A single-framework synthetic antibody library containing a combination of canonical and variable complementarity-determining regions. ChemBioChem. 2017;18(22):2247–59. doi:10.1002/cbic.201700279.
- Schofield DJ, Pope AR, Clementel V, Buckell J, Chapple SD, Clarke KF, Conquer JS, Crofts AM, Crowther SRE, Dyson MR, et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007;8(11):R254. doi:10.1186/gb-2007-8-11-r254.
- Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, Rem L, Frans N, Daukandt M, Pieters H, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 2005;23(3):344–48. doi:10.1038/nbt1067.
- Erasmus MF, D’Angelo S, Ferrara F, Naranjo L, Teixeira AA, Buonpane R, Stewart SM, Nastri HG, Bradbury ARM. A single donor is sufficient to produce a highly functional in vitro antibody library. Commun Biol. 2021;4(1):1–16. doi:10.1038/s42003-021-01881-0.
- Collins AM, Wang Y, Roskin KM, Marquis CP, Jackson KJL. The mouse antibody heavy chain repertoire is germline-focused and highly variable between inbred strains. Philos Trans R Soc B Biol Sci. [Internet] 2015; 370(1676):20140236. Available from: [cited 2019 Sep 18]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4528413/.
- Ramsden DA, Wu GE. Mouse kappa light-chain recombination signal sequences mediate recombination more frequently than do those of lambda light chain. Proc Natl Acad Sci U S A. 1991;88:10721–25. doi:10.1073/pnas.88.23.10721.
- Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting antibody developability profiles through early stage discovery screening. mAbs. 2020;12(1):1743053. doi:10.1080/19420862.2020.1743053.
- Yasuda S, Zhou Y, Wang Y, Yamamura M, Wang J-Y. A model integrating tonic and antigen-triggered BCR signals to predict the survival of primary B cells. Sci Rep. 2017;7(1):14888. doi:10.1038/s41598-017-13993-x.
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15(6):553–57. doi:10.1038/nbt0697-553.
- Ferrara F, Kolnik M, D’Angelo S, Erasmus FM, Vorholt D, Bradbury ARM. Rapid purification of billions of circulating CD19+ B cells directly from leukophoresis samples. New Biotechnol. 2018;46:14–21.
- Ferrara F, Naranjo LA, Kumar S, Gaiotto T, Mukundan H, Swanson B, Bradbury ARM. Using phage and yeast display to select hundreds of monoclonal antibodies: application to antigen 85, a tuberculosis biomarker. PLOS ONE. 2012;7:e49535. doi:10.1371/journal.pone.0049535.
- Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, Neuwelt CM, Robbie G, White WI, Higgs BW, et al. Sifalimumab, a human anti–interferon-α monoclonal antibody, in systemic lupus erythematosus: a phase i randomized, controlled, dose-escalation study. Arthritis Rheum. 2013;65(4):1011–21. doi:10.1002/art.37824.
- Guo X, Higgs BW, Bay-Jensen A-C, Wu Y, Karsdal MA, Kuziora M, Godwood A, Close D, Ryan PC, Roskos LK, et al. Blockade of GM-CSF pathway induced sustained suppression of myeloid and T cell activities in rheumatoid arthritis. Rheumatol Oxf Engl. 2018;57(1):175–84. doi:10.1093/rheumatology/kex383.
- Hamilton JA. GM-CSF as a target in inflammatory/autoimmune disease: current evidence and future therapeutic potential. Expert Rev Clin Immunol. 2015;11(4):457–65. doi:10.1586/1744666X.2015.1024110.
- Rincon M. Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012;33(11):571–77. doi:10.1016/j.it.2012.07.003.
- Rossi J-F, Lu Z-Y, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21(6):1248–57. doi:10.1158/1078-0432.CCR-14-2291.
- Hagopian MM, Brekken RA. Stromal TGFβR2 signaling: a gateway to progression for pancreatic cancer. Mol Cell Oncol. 2015;2:e975606. doi:10.4161/23723556.2014.975606.
- Abdiche YN. High-throughput antibody characterization. Genet Eng Biotechnol News. 2017;37(15):28–29. doi:10.1089/gen.37.15.13.
- Robinson A, Hines V, Wittrup K. Protein disulfide isomerase overexpression increases secretion of foreign proteins in saccharomyces cerevisiae. Bio/Technology. 1994;12(4):381–84. doi:10.1038/nbt0494-381.
- Ferrara F, D’Angelo S, Gaiotto T, Naranjo L, Tian H, Gräslund S, Dobrovetsky E, Hraber P, Lund-Johansen F, Saragozza S, et al. Recombinant renewable polyclonal antibodies. mAbs. 2015;7(1):32–41. doi:10.4161/19420862.2015.989047.
- He F, Woods CE, Becker GW, Narhi LO, Razinkov VI. High-throughput assessment of thermal and colloidal stability parameters for monoclonal antibody formulations. J Pharm Sci. 2011;100(12):5126–41. doi:10.1002/jps.22712.
- Sule SV, Sukumar M, Weiss WF, Marcelino-Cruz AM, Sample T, Tessier PM. High-throughput analysis of concentration-dependent antibody self-association. Biophys J. 2011;101(7):1749–57. doi:10.1016/j.bpj.2011.08.036.
- Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. mAbs. 2014;6(2):483–92. doi:10.4161/mabs.27431.
- Estep P, Caffry I, Yu Y, Sun T, Cao Y, Lynaugh H, Jain T, Vásquez M, Tessier PM, Xu Y. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. mAbs. 2015;7(3):553–61. doi:10.1080/19420862.2015.1016694.
- Valadon P, Pérez-Tapia SM, Nelson RS, Guzmán-Bringas OU, Arrieta-Oliva HI, Gómez-Castellano KM, Pohl MA, Almagro JC. ALTHEA Gold LibrariesTM: antibody libraries for therapeutic antibody discovery. mAbs. 2019;11:516–31. doi:10.1080/19420862.2019.1571879.
- Frenzel A, Schirrmann T, Hust M. Phage display-derived human antibodies in clinical development and therapy. mAbs. 2016;8(7):1177–94. doi:10.1080/19420862.2016.1212149.
- Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindqvist E, Schier R, Hemingsen G, Wong C, Gerhart JC, Marks JD. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci. 1998;95(11):6157–62. doi:10.1073/pnas.95.11.6157.
- Söderlind E, Strandberg L, Jirholt P, Kobayashi N, Alexeiva V, Aberg AM, Nilsson A, Jansson B, Ohlin M, Wingren C, et al. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat Biotechnol. 2000;18:852–56. doi:10.1038/78458.
- Ponsel D, Neugebauer J, Ladetzki-Baehs K, Tissot K. High affinity, developability and functional size: the holy grail of combinatorial antibody library generation. Molecules. 2011;16(5):3675–700. doi:10.3390/molecules16053675.
- Hötzel I, Theil F-P, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs. 2012;4(6):753–60. doi:10.4161/mabs.22189.
- Sun A, Benet LZ. Late-stage failures of monoclonal antibody drugs: a retrospective case study analysis. Pharmacology. 2020;105(3–4):145–63. doi:10.1159/000505379.
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4(3):181–91. doi:10.1038/nrm1052.
- Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol. 1999;292(5):949–56. doi:10.1006/jmbi.1999.3130.
- Julian MC, Li L, Garde S, Wilen R, Tessier PM. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci Rep. 2017;7(1):1–13. doi:10.1038/srep45259.
- Tiller KE, Li L, Kumar S, Julian MC, Garde S, Tessier PM. Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J Biol Chem. 2017;292(40):16638–52. doi:10.1074/jbc.M117.783837.
- Pepinsky RB, Silvian L, Berkowitz SA, Farrington G, Lugovskoy A, Walus L, Eldredge J, Capili A, Mi S, Graff C, et al. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci Publ Protein Soc. 2010;19(5):954–66. doi:10.1002/pro.372.
- Schmiedl A, Breitling F, Winter CH, Queitsch I, Dübel S. Effects of unpaired cysteines on yield, solubility and activity of different recombinant antibody constructs expressed in E. coli. J Immunol Methods. 2000;242(1–2):101–14. doi:10.1016/S0022-1759(00)00243-X.
- Alam ME, Slaney TR, Wu L, Das TK, Kar S, Barnett GV, Leone A, Tessier PM. Unique impacts of methionine oxidation, tryptophan oxidation, and asparagine deamidation on antibody stability and aggregation. J Pharm Sci. 2020;109(1):656–69. doi:10.1016/j.xphs.2019.10.051.
- Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286(5446):1882–88. doi:10.1126/science.286.5446.1882.
- Kowalski JM, Parekh RN, Mao J, Wittrup KD. Protein folding stability can determine the efficiency of escape from endoplasmic reticulum quality control. J Biol Chem. 1998;273(31):19453–58. doi:10.1074/jbc.273.31.19453.
- Kowalski JM, Parekh RN, Wittrup KD. Secretion Efficiency in Saccharomyces cerevisiae of bovine pancreatic trypsin inhibitor mutants lacking disulfide bonds is correlated with thermodynamic stability †. Biochemistry. 1998;37(5):1264–73. doi:10.1021/bi9722397.
- Millward TA, Heitzmann M, Bill K, Längle U, Schumacher P, Forrer K. Effect of constant and variable domain glycosylation on pharmacokinetics of therapeutic antibodies in mice. Biologicals. 2008;36(1):41–47. doi:10.1016/j.biologicals.2007.05.003.
- Bovenkamp FS, Hafkenscheid L, Rispens T, Rombouts Y. The emerging importance of IgG fab glycosylation in immunity. J Immunol. 2016;196(4):1435–41. doi:10.4049/jimmunol.1502136.
- O’Neil BH, Allen R, Spigel DR, Stinchcombe TE, Moore DT, Berlin JD, Goldberg RM. High incidence of cetuximab-related infusion reactions in Tennessee and North Carolina and the association with atopic history. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(24):3644–48. doi:10.1200/JCO.2007.11.7812.
- Chung CH, Mirakhur B, Chan E, Le Q-T, Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-α-1,3-galactose. N Engl J Med. 2008;358(11):1109–17. doi:10.1056/NEJMoa074943.
- Jefferis R. Posttranslational modifications and the immunogenicity of biotherapeutics. J Immunol Res. 2016;2016:1–15. doi:10.1155/2016/5358272.
- Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;2016:1–18. doi:10.1155/2016/1298473.
- Kay J, Matteson EL, Dasgupta B, Nash P, Durez P, Hall S, Hsia EC, Han J, Wagner C, Xu Z, et al. Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: a randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2008;58(4):964–75. doi:10.1002/art.23383.
- Bender NK, Heilig CE, Dröll B, Wohlgemuth J, Armbruster F-P, Heilig B. Immunogenicity, efficacy and adverse events of adalimumab in RA patients. Rheumatol Int. 2006;27(3):269–74. doi:10.1007/s00296-006-0183-7.
- Furie R, Stohl W, Ginzler EM, Becker M, Mishra N, Chatham WW, Merrill JT, Weinstein A, McCune WJ, Zhong J, et al. Biologic activity and safety of belimumab, a neutralizing anti-B-lymphocyte stimulator (BLyS) monoclonal antibody: a phase I trial in patients with systemic lupus erythematosus. Arthritis Res Ther. 2008;10(5):R109. doi:10.1186/ar2506.
- Getts DR, Getts MT, McCarthy DP, Chastain EM, Miller SD. Have we overestimated the benefit of human(ized) antibodies? mAbs. 2010;2(6):682–94. doi:10.4161/mabs.2.6.13601.
- CAT-152 0102 Trabeculectomy Study Group. A phase III study of subconjunctival human anti–transforming growth factor β2 monoclonal antibody (CAT-152) to prevent scarring after first-time trabeculectomy. Ophthalmology. 2007;114(10):1822–1830.e2. doi:10.1016/j.ophtha.2007.03.050.
- Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13(1):31. doi:10.1186/1471-2105-13-31.
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41(W1):W34–40. doi:10.1093/nar/gkt382.
- Lefranc MP. Unique database numbering system for immunogenetic analysis. Immunol Today. 1997;18(11):509. doi:10.1016/S0167-5699(97)01163-8.
- Lefranc M-P, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, Lefranc G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27(1):55–77. doi:10.1016/S0145-305X(02)00039-3.
- Hernandez-Jimenez JA, Martinez-Ortega A, Salmeron-Garcia J, Cabeza JC, Prados, Ortiz R, Navas N. Study of aggregation in therapeutic monoclonal antibodies subjected to stress and long-term stability tests by analyzing size exclusion liquid chromatographic profiles. Int J Biol Macromol. 2018;118(Pt A):511–524. doi:10.1016/j.ijbiomac.2018.06.105. Epub 2018 Jun 23.