
ABSTRACT
Cancer therapeutics approved for clinical application include oncolytic viruses and antibodies, which evolved by nature, but were improved by molecular engineering. Both facilitate outstanding tumor selectivity and pleiotropic activities, but also face challenges, such as tumor heterogeneity and limited tumor penetration. An innovative strategy to address these challenges combines both agents in a single, multitasking therapeutic, i.e., an oncolytic virus engineered to express therapeutic antibodies. Such viro-antibody therapies genetically deliver antibodies to tumors from amplified virus genomes, thereby complementing viral oncolysis with antibody-defined therapeutic action. Here, we review the strategies of viro-antibody therapy that have been pursued exploiting diverse virus platforms, antibody formats, and antibody-mediated modes of action. We provide a comprehensive overview of reported antibody-encoding oncolytic viruses and highlight the achievements of 13 years of viro-antibody research. It has been shown that functional therapeutic antibodies of different formats can be expressed in and released from cancer cells infected with different oncolytic viruses. Virus-encoded antibodies have implemented direct tumor cell killing, anti-angiogenesis, or activation of adaptive immune responses to kill tumor cells, tumor stroma cells or inhibitory immune cells. Importantly, numerous reports have shown therapeutic activity complementary to viral oncolysis for these modalities. Also, challenges for future research have been revealed. Established engineering technologies for both oncolytic viruses and antibodies will enable researchers to address these challenges, facilitating the development of effective viro-antibody therapeutics.
Introduction
Antibodies of various formats are widely approved as cancer therapeutics, including monoclonal antibodies (mAbs) for targeted therapy or immune checkpoint inhibition, antibody-drug conjugates, and recombinant bispecific antibodies that target endogenous T cells to tumors.Citation1 Virotherapy with oncolytic viruses (OVs) is an emerging modality for treatment of cancer, with marketing approval in 2015 of the first OV in the Western hemisphere.Citation2–4 Preclinical and clinical data have revealed that the therapeutic activity of OVs depends on viral oncolysis-triggered antitumoral immune activation, thus encouraging combination immunotherapy with OVs.Citation5–9 Both therapeutic modalities can be combined in a single, multi-effector agent by insertion of antibody-encoding transgenes into OV genomes, thereby facilitating genetic delivery and expression of antibodies in tumors, i.e., viro-antibody therapy. Thus, this approach facilitates higher local antibody concentrations while reducing systemic side effects, and complements viral oncolysis with selected therapeutic modes of action dependent on the choice of antibody.
Both OVs and antibodies evolved by nature, but therapeutic versions have been improved by sophisticated molecular engineering technology. Both facilitate outstanding tumor selectivity and pleiotropic activities, and both can be used to deliver therapeutic payloads into tumors, for example, by insertion of therapeutic genes into the virus or by linkage of drugs to antibodies. Treatment with OVs, i.e., virotherapy, features a unique amplification mechanism by intratumoral virus replication, cell lysis and spread, and thereby triggers in situ tumor vaccination by release of tumor antigens in the context of virus-induced inflammation. Antibodies implement an unparalleled binding specificity and technologies are in place to rapidly obtain antibodies for virtually any target structure.Citation10,Citation11 In cancer therapy, antibodies targeting cell surface markers of cancer cells or cancer-supportive stroma cells (targeted therapy), of immune cells (checkpoint inhibition), or both (bispecific antibodies) are well established. However, the success of both kinds of biotherapeutics is limited by several barriers, including tumor heterogeneity, structural and immunological barriers to OV spread, and poor tumor accumulation and penetration of antibodies. However, as mentioned above, both modalities are amenable to sophisticated engineering approaches that can be exploited toward overcoming these barriers. One innovative engineering approach is to merge both therapeutics into a viro-antibody therapy, i.e., the genetic delivery of recombinant antibodies with accordingly “armed” OVs that are still able to replicate and lyse tumor cells. From the OV perspective, this strategy allows the killing of bystander cells that are not reached or infected by the OVs themselves. To this end, antibodies might better penetrate the extracellular matrix (ECM) than OVs, implement an additional, but independent level of tumor targeting and allow for the selection and tailoring of effector mechanisms. The latter may include the targeting of cells of the tumor microenvironment (TME), i.e., stroma and immune cells, in addition to tumor cells. From the antibody perspective, genetic delivery allows local and extended expression, thus enabling the accumulation of smaller antibody formats that better penetrate the tumor at higher concentrations than is feasible by systemic application. Moreover, viro-antibody therapy combines different modes of direct and indirect cancer cell killing for enhanced therapeutic potency and reduced chances for development of resistance.
In light of the therapeutic potential and opportunities of viro-antibody therapy, we first provide an overview of OVs, recombinant antibody therapeutics and genetic delivery approaches for therapeutic antibodies. Then, we highlight applied antibody formats, strategies for their expression by OVs, implemented therapeutic modes of action by delivered antibodies, and examples for resulting therapeutic activities. A comprehensive overview of reported antibody-encoding OVs with key results of each report is provided in . Finally, we discuss the major conclusions and challenges resulting from viro-antibody therapy to date and look ahead on future opportunities that further underline the potential of this combination biotherapy.
Table 1. Overview of developed therapeutic antibody-encoding OVs: OVs expressing recombinant IgGs/monoclonal antibodies (Viro-MAb therapy) sorted according to targets and publication date
Table 2. Overview of developed therapeutic antibody-encoding OVs: OVs expressing BiTEs (viro-BiTE therapy), sorted according to target and publication date
Table 3. Overview of developed therapeutic antibody-encoding OVs: OVs expressing immune checkpoint inhibitors (viro-CHECKin therapy), sorted according to target and publication date
Table 4. Overview of developed therapeutic antibody-encoding OVs: OVs expressing anti-angiogenic antibodies (viro-ANGin therapy), sorted according to target and publication date
Table 5. Overview of the developed therapeutic antibody-encoding OVs: OVs expressing cytotoxic immunofusions (viro-iTOX therapy)
Oncolytic viruses, virus engineering, and insertion of therapeutic genes
Virotherapy is an emerging modality of cancer treatment with OVs that selectively infect and lyse cancer cells, thereby replicating and spreading in the tumor.Citation2 Tumor selectivity of OVs is based on intrinsic features of cancer cells, such as unrestricted proliferative signaling, increased nucleotide synthesis, defects of apoptosis pathways and deficient antiviral defense mechanisms.Citation12 OV development has exploited a wide repertoire of viruses with or without envelope, with DNA or RNA genomes, and of a wide spectrum of genome and particle sizes.Citation13 This includes wild-type viruses – usually of animal hosts, such as vesicular stomatitis virus (VSV), Newcastle disease virus (NDV) or parvovirus (PV) – or vaccine strains, such as measles viruses (MeV), that are highly sensitive to cellular antiviral defenses in normal human cells, but replicate and kill cancer cells deficient in antiviral defenses.Citation12 Designer OVs, in contrast, are generated by genetic engineering of viral genomes for tumor-specific cell entry or post-entry replication.Citation13–16 Tumor-specific cell entry of OVs has been achieved particularly for enveloped viruses by fusion of ligands to the cell-binding glycoproteins that are further mutated to avoid binding to natural viral receptors. Especially tumor-specific single-chain antibody fragments (scFvs) have been successfully exploited as ligands for entry targeting of MeV, herpes simplex viruses (HSV), and VSV.Citation13,Citation15 Enhanced entry of non-enveloped OVs, especially adenoviruses (Ads), into cancer cells has been achieved by genetic modification of the viral protein capsid, for example, by insertion of an integrin-binding RGD peptide or switching of cell-binding domains between related viruses binding to different receptors.Citation17,Citation18 Post-entry targeting has been achieved by deletion of viral genes or domains responsible for functions redundant in cancer cells (e.g., induction of S phase or apoptosis inhibition) or by tumor-specific expression of viral genes using cellular promoters (DNA viruses only) or target sites for micro-RNAs overexpressed in healthy tissues.Citation14,Citation16
It is now well established that the therapeutic activity of OVs, at least in part, depends on oncolysis-triggered, tumor-directed innate and adaptive immune responses. This oncolytic vaccination effect results from the release of tumor antigens, as well as pathogen- and damage-associated molecular patterns (PAMPs and DAMPs), during OV-induced immunogenic cell lysis.Citation5–9,Citation19 Importantly, it has been shown that local oncolysis, e.g., after intratumoral OV application, can trigger systemic antitumor immunity in animals and patients, thereby mediating the destruction of metastatic lesions not reached by the OV (“abscopal effect”). This oncolytic vaccination effect also suggests virotherapy as a promising modality for combination immunotherapies. As such, preclinical studies have shown that viral oncolysis can sensitize tumors to immune checkpoint inhibitors (ICIs), which are therapeutic antibodies that block inhibitory immune signaling.Citation20 In the clinic, ICIs have shown unprecedented durable responses in subsets of patients suffering from selected inflamed tumor types; however, most patients do not respond.Citation21 This is especially the case for tumors that are immunologically “cold”, i.e., lacking signatures of immune activity, including infiltrating immune cells. Virotherapy has the potential to switch such tumors to “hot”, i.e., stimulating, via an inflammatory response, the infiltration of immune cells, including tumor antigen-specific T cells, that can be unleashed by ICIs.Citation5–9,Citation19 Preliminary results of ongoing clinical studies exploring the combination of OVs with ICIs are promising.Citation22,Citation23 One interesting approach to viro-antibody therapy described in more detail below is the genetic delivery by OVs of antibodies with ICI activity.
With marketing approval of the first OV in the United States and European Union in 2015, the engineered HSV-1 T-Vec encoding granulocyte-macrophage colony-stimulating factor, virotherapy has reached routine clinical oncology.Citation3 A wide panel of other OVs with various genetic modifications are being investigated in numerous clinical Phase 1–3 studies.Citation4,Citation8 While safety of OVs and therapeutic activity in individual patients has been demonstrated, it is clearly necessary to develop OVs with improved potency in order to realize the full potential of virotherapy in clinical oncology. To this end, established technologies to engineer virus genomes are a clear advantage of the OV drug platform, as they facilitate efforts to overcome barriers, e.g., by improving tumor cell infectivity, or to implement additional modes of action. A prominent example for the latter is the insertion of transgenes into OV genomes in order to express proteins or small regulatory RNAs that improve viral oncolysis or secrete biotherapeutics for complementing oncolysis with the destruction of tumor cells not reached by the OVs themselves.Citation8,Citation13,Citation24 Compared with replication-deficient vectors, such “armed” OVs facilitate dramatically increased expression of biotherapeutics restricted to tumors, where the OV genomes are amplified and OV infections spread.Citation25 Recombinant therapeutic antibodies are powerful candidates for such “armed” OVs for at least four reasons: (1) they implement a paracrine mode of action to target noninfected cancer cells, (2) they possess an additional level of tumor selectivity, (3) structurally nearly identical molecules targeting basically any desired cancer surface marker are available or can be rapidly generated, and (4) they allow customization by adapting the antibody format to make it fit for purpose, for example, by modifying valencies or by fusing effector proteins, all of which have been pursued and reported in viro-antibody therapy, as discussed below.
Therapeutic antibodies
With more than 40 molecules approved for cancer therapy (www.antibodysociety.org/antibody-therapeutics-product-data), antibodies are established treatment options for more than 30 different tumor entities, including numerous hematologic malignancies and solid tumors.Citation1,Citation26 Many more are in different stages of preclinical and clinical development.Citation27 Most of these antibodies recognize tumor-associated antigens on tumor cells. Prominent examples are IgG molecules targeting CD20 to treat B-cell malignancies and antibodies targeting epidermal growth factor receptor (EGFR) or human epidermal growth factor receptor 2 (HER2) approved for the treatment of different types of solid tumors, e.g., colorectal cancer, gastric cancer, lung cancer, and breast cancer. These antibodies utilize the natural functions of IgG molecules to destroy tumor cells by blocking the activity of target structures, by inducing apoptosis, and/or via Fc-mediated effector functions such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement fixation.Citation28 These activities, however, are not always sufficient to efficiently induce a long-lasting tumor regression. Therefore, in a second generation of developments, the mode of action was extended by using the antibody as a targeting module to deliver cytotoxic compounds, such as radionuclides, chemotherapeutics and toxins, to tumor cells.Citation29
A growing number of molecules are used as antibody drug-conjugates (ADCs) to delivery cytotoxic compounds to and into tumor cells.Citation30 More recently, a first recombinant immunotoxin composed of a disulfide-stabilized Fv fragment directed against CD22 fused to a Pseudomonas exotoxin A fragment was approved for the treatment of hairy cell leukemia.Citation31 Conceptually, this delivery approach offers a multitude of possibilities by combining the antigen-binding site of an antibody as a targeting unit with an effector moiety. For example, various so-called immunocytokines are being developed to locally induce and enhance an antitumor immune response by binding to tumor cells or targets expressed by cells of the tumor microenvironment.Citation32 Besides immuno-regulatory fusion partners, other proteins such as death ligands (e.g., TRAIL, TNF) and enzymes (RNases) have been used to generate antibody fusion proteins.Citation33,Citation34
The application of antibodies in tumor therapy was also extended by targeting and inhibiting ligands and receptors involved in tumor angiogenesis.Citation35,Citation36 For example, antibodies targeting vascular endothelial growth factor (VEGF) or VEGFR2 are approved for the treatment of different types of solid tumors.
A rapidly expanding area for use of therapeutic antibodies is in the field of immuno-oncology. Here, antibodies are developed to initiate or foster an antitumor immune response.Citation37 This can be achieved, for example, with bispecific antibodies capable of redirecting immune effector cells, especially T-cells, to tumor cells by binding with one arm to a surface antigen on tumor cells and with the second arm to a trigger molecule on the immune effector cell, e.g., CD3 on T-cells. Here, new tools to genetically engineer molecules with the desired composition have emerged in recent years.Citation38 The first genetically engineered bispecific antibody, blinatumomab, is a tandem scFv molecule (bispecific T cell engager, BiTE) directed against CD19 and CD3 for the treatment of acute lymphoblastic leukemia approved in 2014. Many more are in clinical development, most of them differing from this small BiTE format by comprising an Fc region (quite often with silenced effector functions) as a half-life extension module.
As a further immune-oncology approach, already introduced above, several antibodies are approved as ICIs to block inhibitory signals, for example, through binding of CTLA-4 or PD-1 on T-cells, or PD-L1 on tumor cells, to restore a potent antitumor T-cell response through recognition of MHC-displayed peptides on tumor cells.Citation20 Other antibodies targeting next-generation immune checkpoint molecules, such as LAG-3, TIGIT, TIM-3, NKG2A, and CD47 are in clinical development.Citation39,Citation40
In summary, the field of therapeutic antibodies has rapidly expanded during the past two decades into different directions with the aim to enhance and employ novel modes of action for the development of novel antibodies and antibody derivatives following a “fit for purpose” strategy. However, antibodies still face various hurdles to be efficacious and safe therapeutics.Citation41 One of the main limitations of antibodies, especially for the treatment of solid tumors, lies in their often rather poor and heterogeneous tumor penetration after systemic application,Citation42 which is a result of physical barriers and a high interstitial fluid pressure limiting extravasation and interstitial diffusion of large antibody molecules. This can be improved by using smaller antibody fragments, such as single-domain antibodies, scFv and Fabs,Citation43,Citation44 but removal of the Fc region eliminates Fc-mediated effector functions and FcRn-mediated recycling response for the long half-life of IgGs.Citation45 Additionally, biochemical properties intrinsic to the antibody, such as surface charge, can influence the pharmacokinetic properties and their tissue distribution.Citation46,Citation47 Furthermore, affinity for the target antigen has been shown to limit penetration of antibodies into the tumor tissue.Citation48,Citation49 Thus, antibodies with high affinity accumulate preferably around the blood vessels, known as binding-site barrier effect.Citation50,Citation51 A further limitation results from “on target, off tumor” activities, due to the lack of tumor-specific targets, which can lead to adverse effects as shown for bispecific T-cell engagers and CAR-T therapeutics, which can induce T-cell reactivities against normal tissues.Citation52,Citation53 Several strategies have been developed to increase tumor selectivity. This includes the introduction of locks into the antigen-binding site, which can be conditionally removed through proteolytic cleavage within the tumor or as a response to hypoxia or low pH.Citation54 Alternatively, intratumoral injections of antibody drugs are used to increase local accumulation.Citation55 A promising approach toward addressing some of the mentioned limitations of antibody therapy of cancer is genetic antibody delivery, especially when targeted to solid tumors.
Genetic antibody delivery – lessons from gene therapy
Antibody gene transfer was initially pursued using replication-deficient viral vectors as passive immunization against infectious diseases or for delivery of therapeutic tumor-targeted antibodies,Citation56–65 and more recently (transient) delivery of antibodies by mRNA has also been pursued.Citation66 Gene therapy studies showed that the kinetics of gene expression were determined by the choice of vector. Specifically, Ad vectors mediate fast, strong, but, due to immune-mediated virus elimination, transient expression in animal models after intravenous (i.v.) injection. In contrast, adeno-associated virus (AAV) vectors enabled delayed, but persistent expression,Citation59,Citation61,Citation65 but clinical studies revealed that AAV vectors are immunogenic in humans.Citation66 Furthermore, gene expression could be strongly increased by choosing appropriate promoters, posttranscriptional stabilization and codon optimization.Citation67 As discussed below, control of antibody gene expression is also a critical parameter for viro-antibody therapy, for which gene expression strength and kinetics are primarily determined by virus replication. Importantly, antibody gene therapy addressed a problem also of relevance for viro-antibody therapy: the genetic delivery of full-length antibodies requires the co-expression of immunoglobulin (Ig) heavy and light chains, ideally at similar concentrations to ensure optimal efficiency and minimize adverse effects. Initial approaches explored the expression of both Ig chains as separate transcription units using two promoters and polyadenylation sequences (see, e.g., refs57, 58, 60). Such expression cassettes depend on extensive genomic space, which may compromise virus titers due to packaging limits, as observed for AAV vectors.Citation60 Alternative approaches link heavy- and light-chain genes in one bicistronic mRNA via an internal ribosome entry site (IRES)Citation56 or in a single open reading frame (ORF) using viral 2A peptides mediating a “ribosomal skip”, i.e., a specific peptide bond is not formed during translation.Citation63 The former approach has the disadvantage that IRESs are also quite long sequences and IRES-mediated translation of the downstream Ig chain is less efficient than cap-dependent translation of the upstream chain.Citation68 The 2A approach ensures an expression ratio of close to 1:1 but is hampered by immunogenicity and/or structural constraints resulting from retained amino acids of the 2A sequence. To overcome this limitation, the insertion of a cleavage site for endogenous proteases has been explored.Citation63 Of note, the expression of recombinant single-chain or homo-dimeric antibody derivatives, including derived fusion proteins, circumvents problems related to co-expression.
Strongly attenuated derivatives of vaccinia viruses (VVs), which are also in development as OVs (see below), have been explored as gene transfer vectors for antibody delivery. One report explored the expression of a membrane-bound IgG (with two separate transcription units encoding heavy and light chains) or an scFv in macrophages or T cells, resulting in antibody-mediated binding and subsequent killing of cancer cells.Citation57,Citation58 As an alternative approach, polyclonal stimulation of T cells by VV gene transfer vectors encoding membrane-bound, T-cell receptor (TCR)-specific IgGs was investigated, demonstrating strong expression in tumor cells mediating recruitment and activation of T cells and tumor rejection.Citation69
The combination of antibody gene transfer and virotherapy has been explored by applying two separate Ad-derived viruses, namely a replication-deficient antibody-encoding Ad vector and an oncolytic Ad.Citation70–73 Thus, in co-infected cancer cells, the antibody-encoding Ad vector can replicate via complementation by the oncolytic Ad. This co-infection approach allows for flexibility in the combination of OVs with antibody-delivering vectors, e.g., for testing different combinations of OVs and (vector-encoded) antibodies. Also, oncolytic Ads were combined with Ad vectors encoding two different antibodies or antibodies in combination with cytokines in order to implement different antibody- and cytokine-mediated modes of action.Citation71,Citation72 In contrast to the single agent viro-immunotherapy approaches discussed below, this combination approach, especially its clinical translation, is hampered by the necessity to produce and apply two separate virus products and the expected rarity of co-infections in vivo.
Therapeutic antibody-encoding OVs
Viro-antibody therapy has been pursued with increasing intensity in the past decade, with studies investigating different OVs, antibody formats and antibody-mediated modes of action (). The following sections highlight the principles and key achievements of viro-antibody therapy research according to the major parameters, i.e., the explored viruses, antibody expression strategies, antibody formats, therapeutic targets and effector mechanisms. The accompanying provide a comprehensive overview of the developed therapeutic antibody-encoding OVs with key results.
Figure 1. Viro-antibody therapy – an overview. Transgenes encoding recombinant therapeutic antibodies are inserted into the genome of oncolytic viruses (OVs) by exploitation of diverse strategies for transgene expression (see ). OV-encoded antibodies are produced locally in the tumor by infected cancer cells and expression lasts as long as active OV infection and spread is ongoing. The produced antibodies, dependent on their format, valency and size (see ), perfuse the tumor, bind their target on (noninfected) cancer cells or cancer-associated cells, and trigger their direct or indirect killing via diverse modes of action (see )
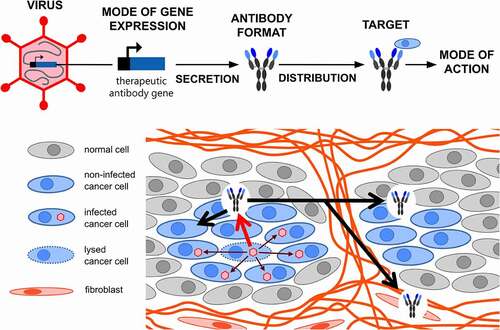
Figure 2. Viruses and modes of antibody expression exploited for viro-antibody therapy. A panel of viruses with or without envelope and with DNA (double-strand) or RNA genomes (negative strand, continuous for MeV, NDV, VSV or segmented for influenza virus) have been exploited for viro-antibody therapy (left panel). Dependent on the virus chosen, different strategies have been pursued for the insertion of antibody genes into the viral genomes toward efficient and/or replication-dependent gene expression and minimizing the required genomic space (right panel). ab, antibody; Env, envelope; HSV, herpes simplex virus; MeV, measles virus; NDV, Newcastle disease virus; ORF, open reading frame; VSV, vesicular stomatitis virus
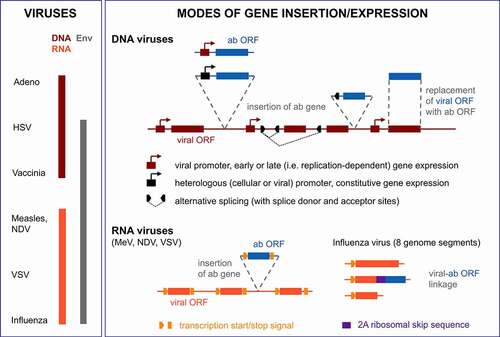
Figure 3. Antibody formats utilized in viro-antibody therapy. BiTE, bispecific T cell engager; CH/CL, constant domain of heavy/light chain; Fab, antigen-binding fragment; Fc, constant fragment of antibody; IgG, immunoglobulin G; RNase, ribonuclease; scFv, single chain variable fragment; sdAb, single domain antibody (variable fragment of camelid antibody heavy chain); VH/VL, variable domain of heavy/light chain. For the scFv fusion proteins, proteins might be fused at the C-terminus of the scFv (cytokine, trimerization peptide and FasL) or at the N-terminus (RNase); scFvs might be in VH-VL or VL-VH configuration
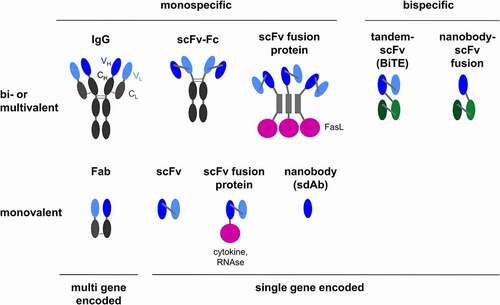
Figure 4. Target cells and modes of action of OV-encoded antibodies. The depicted cancer targets and direct or immune- or stroma-mediated modes of action have been reported. For some of the depicted modes of action only examples of the explored antibody formats are depicted (see for a comprehensive list). CAF, cancer-associated fibroblast; TAM, tumor-associated macrophage; TECs, tumor endothelial cells
When exploring viro-antibody therapy in immunocompetent syngeneic models, it is important to consider that these models face limitations with respect to OV transduction, replication and oncolysis. This results from the overall species-specificity of virus replication, meaning that human-infecting OVs are usually strongly attenuated in murine cells, including murine cancer cells. Thus, studies of viro-antibody therapy in syngeneic mouse models underestimate the oncolysis effect. In this light, xenograft models with additionally applied human immune cells are being explored (see viro-BiTE therapy, ). Of note, patient-derived ex vivo models, for example, pleural effusion and peritoneal ascites, containing cancer cells, stroma cells and immune cells, are increasingly valued for analysis of OVs including immune effects, as reported for studies exploring oncolytic Ad-encoded BiTEs.Citation74–76
We note that we do not discuss the insertion of recombinant antibody genes into OV genomes pursued to target virus cell entry to tumor cells, either by genetically fusing tumor-specific scFvs to viral glycoproteinsCitation77–79 or by virus-encoded, antibody-based bispecific adapter proteins.Citation80,Citation81 Also not covered are fusion proteins containing IgG Fc domains, but not antigen-binding antibody domains. Such approaches aim at Fc-mediated protein multimerization and stabilization, modulation of protein size and thus biodistribution, and/or Fc-mediated effector functions.Citation82 Applications include the expression of decoy receptors consisting of extracellular receptor domains fused to Fcs, e.g., TGF-β inhibitorsCitation83–86 and recombinant checkpoint inhibitors, such as the Fc-fused extracellular domain PD-1.Citation87
Therapeutic antibody-encoding OVs: viruses and expression strategies
Of the established repertoire of different viruses used for virotherapy, most allow the insertion of therapeutic antibody genes, as they fulfill the requirements of available genomic space, knowledge of viral genome organization and replication, and established technology for genome engineering (including reverse genetics for RNA viruses). Thus, it is not surprising that a panel of OVs have been explored for application in viro-antibody therapy: Ads, VV, HSV, MeV, VSV, NDV, and influenza virus (). The properties of the chosen virus (or of the specific strain or mutant), i.e., infection efficiency, mode of tumor-targeting, the kinetics of genome amplification and of viral spread, determine the kinetics, strength and tumor-selectivity of antibody expression. However, direct comparisons of different viruses, as for Ad and AAV vectors in gene therapy, remain to be performed for viro-antibody therapy. The choice of OV moreover determines both the feasible gene expression strategy and the genomic space available for transgene insertion, the latter being restricted by the packaging capacity of the virus particle (). Powerful viral gene expression mechanisms can be exploited for efficient and/or replication-dependent, thus tumor-specific antibody expression, as discussed below.
Most viro-antibody therapy studies explored OVs with DNA genomes: Ads, VVs and HSVs (). They allow for the insertion of large heterologous DNA sequences and the utilization of diverse transgene expression strategies. To this end, the most straightforward strategy is to insert additional transcription units with promoter/enhancer, antibody ORF and polyadenylation sequences. For strong but constitutive expression, heterologous viral promoters of cytomegalovirus (CMV), respiratory syncytial virus (RSV) or Moloney murine leukemia virus have been used in OVs derived from Ad or HSV.Citation74–76,Citation88–93 In VV-derived OVs, natural or synthetic VV promoters were used, including late, i.e., replication-dependent, promoters.Citation94–105 The expression of two scFvs by a single OV has been reported for a VV using two viral promoters and insertion sites.Citation102 For Ads, besides separate transcription units with CMV promoter, for which the orientation might be critical,Citation74–76 the insertion of antibody genes into endogenous viral transcription units have been established. To this end, antibodies are expressed either from ORFs replacing viral ORFsCitation106–108 or by alternative splicing using splice acceptor sites upstream of the antibody ORF.Citation74,Citation75,Citation109–114 These approaches need less genomic space. Moreover, insertion of transgenes into late transcription units enables replication-dependent gene expression,Citation25 which is tumor-restricted dependent on the level of tumor-selectivity of the OV.Citation115
Replication-dependent antibody expression from late viral promoters, directly, as established for oncolytic VV, or via alternative splicing, as established for oncolytic Ads, might affect therapeutic activity and/or side effects of viro-antibody therapy (or of therapeutic gene-expressing OVs in general). Constitutive promoters can mediate a faster onset of effector functions, as results for CMV promoter-driven versus replication-coupled expression by oncolytic Ads indicate: the former approach resulted in a more rapid T cell activation and target cell killing by virus-encoded BiTEs in co-cultures.Citation74 On the other hand, replication-dependent antibody expression might be an advantage when expression in normal cells needs to be avoided, i.e., especially after systemic OV application. This is because constitutive promoters mediate transgene expression also in healthy cells transduced by OVs (if they are not entry-targeted), even if they do not replicate. For example, the above study with oncolytic Ads reported GFP expression in macrophages of malignant exudates with CMV promoter-driven GFP expression, but not with replication-coupled GFP expression. A study investigating the same oncolytic Ad format for CMV-driven or replication-dependent expression of a fibroblast-targeted BiTE, directed against the fibroblast-activating protein (FAP) and CD3, observed toxicity for normal fibroblasts in co-cultures with T cells (no tumor cells) only for the CMV promoter virus.Citation75 In this setting, the oncolytic Ad can enter normal fibroblasts, but cannot replicate and lyse these cells. Thus, fibroblast killing results from BiTE expression, which is active only for the CMV virus, and BiTE-mediated T cell cytotoxicity. This result is in so far relevant, as systemically applied conventional FAP-targeted therapeutics have been reported to induce toxicity in FAP+ cells of the bone marrow.Citation116 In conclusion, the opportunity to implement replication-dependent versus immediate antibody expression for some OVs has functional implications, but more studies are needed to exploit and optimize this mechanism for improving the therapeutic window of viro-antibody therapy.
Late expression during the OV replication cycle might also be necessary for therapeutic antibodies that otherwise interfere with viral replication, thereby impairing virus manufacturing as well as oncolysis in situ. As such, high-quality virus preparations of oncolytic Ads encoding an immunoRNase for targeted cancer cell killing required tight replication-dependent expression, as strong expression, both by replication-deficient vectors or OVs, produced viruses of low infectivity.Citation109 Still, the immunoRNase was expressed by the optimized Ad at levels sufficient to mediate enhanced therapeutic efficacy in vitro and in vivo.
Antibody expression by OVs is not restricted to DNA viruses but has been demonstrated also for RNA viruses MeV, VSV, NDVs, and influenza virus (). For viruses with non-segmented genomes (MeV, VSV, NDV), the expression strategy is to insert additional transcription cassettes including viral gene start and stop elements into the RNA genome. Gene expression strength is determined by the insertion site/position within the virus genome considering expression gradients from the 3ʹ to the 5ʹ genome terminus. Influenza viruses possess a segmented genome of eight segments. ORFs encoding heavy and light chains of an IgG were fused via a “ribosomal skip” 2A sequence to viral ORFs of different segments.Citation117
Considering that antibodies are naturally produced by specific and highly differentiated plasma cells, it is worth mentioning that reported viro-antibody therapy studies clearly show that functional therapeutic antibodies can also be produced in cancer cells of diverse tissue origin in vitro and in tumors of animal models in vivo. One study suggested that antibody production by tumor cells requires active cell lysis, as antibodies were not released from cancer cells after transduction with a replication-deficient, and thus not lytic Ad vector.Citation108 However, another study detected antibodies in supernatants after transduction of a different tumor cell line with a replication-deficient AdCitation109; thus, viral cell lysis might not be a requirement for antibody release from cancer cells. It remains to be investigated in more detail how cell lysis affects biosynthesis and release of antibody from infected tumor cells and whether modulation of cell lysis kinetics by OV engineering, as reported for transgene-encoding Ads,Citation118 allows for further increasing antibody production.
When inserting transgenes into OV genomes, adverse effects to virus replication kinetics or integrity must be considered. These might result from the disruption of viral gene regulation circuits, from surpassing the genome packaging limit of virus particles, or from the activity of the encoded therapeutic protein. Note that most of the reported viro-antibody studies demonstrate the expression and function of recombinant antibodies in vitro and show that OV replication and tumor cell lysis are not affected by insertion of antibody genes and their expression. Only exemptions to the latter aspect will be discussed in the following sections or are mentioned in . Interestingly, a study exploring an oncolytic Ad encoding a FAP-specific BiTE was actually reported to increase infectious virus particle production in vivo.Citation104 This is likely a consequence of the BiTE-mediated killing of cancer-associated fibroblasts and resulting destruction of the extracellular matrix, thereby eliminating a barrier to intratumoral spread of viral infection. This hypothesis is supported by reports of enhanced intratumoral infection of oncolytic Ads encoding matrix-degrading enzymes, such as relaxin or hyaluronidase.Citation119,Citation120
Therapeutic antibody-encoding OVs: antibody formats, targets and therapeutic strategies
Various viro-antibody therapy approaches for expression of therapeutic antibodies and derivatives have been reported. These have explored OVs encoding full-length IgGs, Fabs, scFvs, bispecific tandem scFvs (BiTEs), nanobodies, or scFv fusion proteins () that directly target tumor cells (tumor surface antigens), tumor supporting fibroblasts (α-FAP), tumor angiogenesis (α-VEGF), or immune cells (ICIs, BiTEs) (). In the following, we highlight key properties and results of reported viro-antibody therapy studies beginning with OVs encoding full-length IgGs/mAbs (see for an overview of all reported studies) and continuing with different virus formats according to effector strategies, i.e., T cell engagement, immune checkpoint inhibition, anti-angiogenesis and direct tumor cell killing.
Expression of recombinant monoclonal antibodies (viro-MAb therapy)
As highlighted above, a panel of mAbs has been approved and is widely used in clinical oncology in order to directly target and kill cancer cells, de-block immune cells for antitumor immunity (checkpoint inhibition), or deprive growth factors, such as VEGF for inhibiting tumor angiogenesis. Several reported viro-antibody studies aimed at establishing the genetic delivery of full-length IgGs by OVs exploring these different modes of action (viro-MAb therapy). These studies included a model antibody to demonstrate proof-of-principle,Citation117 an experimental therapeutic antibody targeting the tumor vascular antigen fibronectin extradomain B,Citation121 IgGs with specificity for tumor targets (CD147),Citation122 or targets of approved mAbs (CTLA-4, PD-1, VEGF),Citation92,Citation103,Citation106,Citation111 and the approved antibody trastuzumab.Citation108 Overall, the reports established that the genetic delivery of full-length IgGs is feasible for both RNA viruses, as reported for NDVCitation121,Citation122 and influenza A,Citation117 and for DNA viruses, reported for Ad,Citation106–108,Citation111 VV,Citation103,Citation105 and HSV.Citation92 Strategies for the required co-expression of IgG heavy and light chains were the insertion of two separate transcription units for NDV and VV, fusion to two viral ORFs of different gene segments using viral 2A sequences for influenza A virus or co-expression via an IRES or a 2A sequence for Ads. Co-expression in VV via separate transcription units was improved with optimized promoter choice, which avoided misassembled by-products that were considered to be homodimers of (over-) expressed Ig light chains.Citation103
Viro-MAb therapy studies demonstrated that genetic delivery by OVs enables selective, strong and prolonged expression of active IgGs in tumors. Tumor-selective antibody expression was shown for oncolytic NDV and Ad after in vitro infection of tumor versus normal cells, with a more than 2000-fold window of tumor-specificity observed for the oncolytic Ad.Citation111,Citation121 In particular, strong antibody expression mediated by the replicative nature of OVs was confirmed in a xenograft model allowing for efficient Ad replication that showed an 81-fold higher antibody expression after i.t. injection of an oncolytic Ad compared with a matching replication-deficient Ad vector.Citation106 IgG concentrations were 43-fold higher in the tumor than in plasma. A follow-up study with the same oncolytic Ad and expression strategy to deliver a recombinant derivative of trastuzumab allowed a comparison of a recombinant OV-encoded IgG, purified using a standard laboratory procedure, with the matching commercial mAb,Citation108 revealing that the activity of the oncolytic Ad-encoded antibody was similar regarding direct growth inhibition or ~30% regarding ADCC. Equivalent or even superior activity of OV-encoded compared with a matching hybridoma-produced or commercial mAb were also reported in studies using VV or influenza virus.Citation103,Citation117 The oncolytic Ad-encoded trastuzumab, after i.t. injection in a mouse xenograft model, produced higher antibody concentrations in tumors and dramatically higher tumor-to-blood antibody concentration ratios compared with intraperitoneally (i.p.) injected commercial mAb.Citation108 Durability of antibody expression by viro-MAb therapy in an immunocompetent organism was reported using an IgG-encoding oncolytic VV in a syngeneic mouse model showing, after i.t. virus injection, IgG expression for 11 and 5 d in tumors or serum, respectively, with a peak at day 5. These kinetics mirrored virus replication.Citation103 IgG concentrations in tumors were higher and longer lasting compared with mice that were injected i.t. with 10 μg of a matching commercial mAb, whereas serum antibody concentrations were 10-fold lower at day 1, but higher at later times.
Antibody-mediated therapeutic activity via different modes of action has been demonstrated for IgG-encoding OVs. The expression of trastuzumab by the oncolytic Ad resulted in enhanced tumor growth inhibition in a xenograft model when compared with monotherapies.Citation108 The activation of patient-derived T cells by cell culture supernatants of infected tumor cells in vitro has been reported for the oncolytic Ad encoding checkpoint inhibitor anti-CTLA-4.Citation106 Furthermore, OVs encoding an anti-murine PD-1, CTLA-4 or TIGIT antibody enabled tumor growth inhibition and survival superior to parental virus and for the PD-1 virus similar to combined parental virus with repeated high dose systemic mAb.Citation103,Citation105,Citation117 Similar results were obtained for an oncolytic HSV-2 encoding an anti-human PD-1 antibody in a mouse model with humanized PD-1.Citation92
Expression of BiTEs (viro-BiTE therapy)
BiTEs are another type of genetically engineered antibodies besides mAbs that has been approved for cancer treatment. They are defined not only by the antibody format, two scFvs linked via a flexible peptide linker (tandem scFv, scFv2) (), but also by their specificities and mode of action: they bind T cells via CD3 and tumor targets via specific cell surface molecules. Thus, BiTEs activate T cells in the presence of target cells, irrespective of the TCR-specificity of T cells and irrespective of MHC expression by target cells. The latter makes BiTEs an especially interesting therapeutic approach for immunotherapy of cancers that lack MHC expression. Blinatumomab, a BiTE with specificity for the B cell marker CD19, is approved for treatment of relapsed or refractory B-ALL. However, BiTEs face the problems of (1) short half-lives in serum due to their small size, thus requiring continuous infusion; (2) immune-hostile TMEs in solid tumors lacking T cells required for retargeting by BiTEs or containing exhausted T cells; and/or (3) severe off-tumor toxicities. Viro-antibody therapy is a promising strategy to overcome some of the drawbacks of conventional BiTE therapy by intratumoral and extended BiTE expression (viro-BiTE therapy).Citation123,Citation124 This mode of delivery facilitates enhanced accumulation and penetration of BiTEs in the tumor and minimizes systemic BiTE distribution to reduce off-tumor activities and adverse effects. Moreover, OVs induce inflammation, thereby triggering infiltration of T cells including potentially powerful antiviral T cells that can be redirected to tumor targets by the BiTEs. In light of these advantages, several viro-BiTE therapy studies have been reported investigating BiTE-encoding oncolytic VV, Ad, MeV, and HSV-1 ().
Efficacy of viro-BiTE therapy has been demonstrated for four different kinds of tumor targets: 1) direct targeting of cancer cells has been explored with OVs carrying a transgene encoding a BiTE directed against EphA2,Citation98 EGFR,Citation110,Citation112,Citation114 EpCAM,Citation74 CEA,Citation125 or CD20;Citation125 2) targeting of cancer-associated fibroblasts with a FAP-specific BiTE;Citation75,Citation104,Citation113 3) targeting of tumor-supportive M2 macrophages with a BiTE specific for the M2 marker folate receptor (FR)-β;Citation76 and recently 4) simultaneous targeting of cancer cells and immunosuppressive cells via a BiTE directed to PD-L1, that can be regarded as a pan-cancer marker.Citation93 The latter study explored a bispecific nanobody-scFv format as T cell engager, with the nanobody specific for PD-L1, in addition to the standard tandem scFv BiTE (see ), demonstrating comparable activity in co-culture models (see below).
The activation of T cells and resulting killing of target cells has been reported for OVs encoding BiTEs with specificity for the targets EphA2,Citation98 EGFR,Citation110,Citation112 EpCAM,Citation74 or CEACitation125 in co-cultures of tumor cells or recombinant cells expressing the corresponding BiTE-target with peripheral blood mononuclear cells (PBMCs) or PBMC-derived T cells, for OVs encoding BiTEs with specificity for FAP in co-cultures that in addition contain fibroblasts,Citation75,Citation113 and for OVs encoding BiTEs with specificity for PD-L1 in co-cultures containing M2-like macrophages instead of or in addition to cancer cells.Citation93 Investigations of viro-BiTE therapy with oncolytic Ads in clinically relevant co-cultures derived from pleural effusion- and/or peritoneal ascites demonstrated T cell activation and depletion of either cancer cells using an EpCAM-BiTE, fibroblasts using an FAP-BiTE, M2 macrophages using an FR-β-BiTE, or cancer cells and M2 macrophages using an PD-L1-BiTE or PD-L1 nanobody T cell engager.Citation74–76,Citation93 Here, the viro-BiTE treatments showed results similar to recombinant BiTE. Of note, these studies demonstrate that the viro-BiTE approach facilitates activation of patient-derived autologous T cells and that it is effective even in presence of the immunosuppressive tumor exudates. Notably, for the approach using the PD-L1-specific T cell engagers, superior T cell activation was reported in the presence of immunosuppressive ascites, likely resulting from increased expression of the BiTE target PD-L1. Thus, these BiTE- or nanobody T cell engager-encoding OVs converted an immunosuppressive tumor microenvironment into enhanced immunotherapeutic activity.Citation93 Furthermore, for the FAP- and FR-β-targeted viro-BiTE approach, TME repolarization toward a pro-inflammatory state was reported via analysis of T cell transcriptomes and/or repolarization of the remaining macrophages to the M1 phenotype, respectively.Citation75,Citation76 The FAP study has prompted a clinical trial with oncolytic Ad, EnAd, encoding a FAP-specific BiTE and further immunostimulatory proteins (PsiOxus, virus NG-641, NCT 04053283).
In vivo therapeutic activity of viro-BiTE therapy was shown in tumor xenograft models with PBMCs or PBMC-derived T cells for oncolytic BiTE-encoding VV (injected i.v.), Ad (injected i.t. or i.p. in carrier cells), and MeV (injected i.t.).Citation98,Citation110,Citation113,Citation114,Citation125 These studies also revealed infiltration and activation of T cells and, for a BiTE specific for human and mouse FAP, reduced mouse FAP expression likely resulting from depletion of tumor-associated fibroblasts that are of mouse origin. In an effort to address tumor heterogeneity, one study investigated the combination of cell therapy using FR-α-directed CAR-T cells, which triggered tumor escape, with viro-BiTE therapy using an oncolytic Ad encoding an EGFR-specific BiTE.Citation112 For this combination therapy, redirection to EGFR-positive tumor cells and activation of CAR-T cells, notably including the CAR-negative fraction of CAR-T cell preparations, was demonstrated in vitro and decreased tumor growth with increased survival compared with monotherapies in a xenograft tumor model. CAR-T cell infiltration was increased for tumors not strongly expressing FR-α.
Viro-BiTE therapy was also explored in immunocompetent syngeneic animal models, demonstrating the induction of T cell infiltration, T cell activation and therapeutic responses, as observed after i.t. injection of a FAP-BiTE-encoding VV or a CD20-BiTE-encoding MeV.Citation104,Citation125 FAP cells were depleted by the VV/FAP-BiTE treatment and the MeV/CD20-BiTE induced protective immunity resulting in the rejection of tumor cell rechallenge. Of note, the rechallenge was performed with matching tumor cells not expressing the BiTE target. Moreover, the MeV/CD20-BiTE treatment was superior to direct BiTE injection in the immunocompetent model. These observations point at an OV-triggered tumor vaccination effect responsible for the observed protective immunity and are supportive of the complementarity of oncolysis and BiTE treatment in the immunocompetent context. Another interesting observation was that this approach was active in a tumor model with low baseline T cell infiltration, but not in another model with high tumor-infiltrating lymphocytes content. Thus, analysis of the TME might inform about the responsiveness of tumors to viro-antibody therapy or rather suggest alternative treatments, such as OVs encoding different therapeutic proteins.
Expression of immune checkpoint inhibitors (viro-CHECKin therapy)
ICIs represent a major breakthrough in cancer treatment, as introduced above. With the success of ICIs and identification of their limitations, OVs have received considerable attention of cancer immunotherapy research toward sensitizing patients to ICIs that are unresponsive to ICI monotherapy. Specifically, several OVs have been reported in preclinical models to switch immune-“cold”, thus ICI unresponsive tumors to “hot” by triggering intratumoral inflammation and activation and recruitment of tumor antigen-specific T cells, thereby facilitating enhanced therapeutic efficacy of combination treatment with OV and ICI.Citation5,Citation6,Citation19 Results from early clinical trials indicate improved therapeutic responses of OV and ICI combination treatment in patients.Citation22,Citation23 In fact, the analysis of combination treatment with ICIs has emerged as a standard experiment widely used by labs analyzing the therapeutic activity of OVs. These studies, together with reports of effective intratumoral injection or expression of low dose ICIs,Citation126–129 established a clear rationale for viro-antibody therapy with ICI antibodies (viro-CHECKin therapy) implementing a single-agent OV-ICI combination therapy featuring local, genetic antibody delivery. This approach, in addition to enhancing therapeutic outcome compared with monotherapies, aims at reducing the severe systemic side effects of standard ICI therapy. Viro-CHECKin therapy has been explored with many OVs, i.e., Ad, VV, MeV, HSV, NDV, VSV, and influenza virus (). Furthermore, different antibody formats (IgGs [see also viro-MAb therapy above], scFvs, scFv-Fc fusion proteins, and scFv-cytokine fusion proteins []) and targets (CTLA-4, PD-1, PD-L1, and TIGIT), have been investigated. It should be mentioned that alternative ICI formats independent of antibody-derived variable domains have been explored for genetic delivery by OVs, using a soluble checkpoint molecule, exemplified by soluble PD-1 or soluble PD-1 fused to an Fc domain.Citation87,Citation130
Viro-CHECKin therapy studies demonstrated activation of T cells, including T cells from cancer patients, in vitro,Citation106,Citation131 and therapeutic activity superior to parental viruses in syngeneic models in vivo,Citation91,Citation92,Citation103,Citation105,Citation117,Citation132–134 indicating that effective levels of antibodies were expressed in the tumor. Some studies reported therapeutic outcomes similar to parental virus in combination with systemic or local ICI treatment.Citation91,Citation92,Citation103,Citation134 Moreover, delayed growth of untreated contralateral tumorsCitation91,Citation117 and rejection of tumor cell rechallengeCitation89,Citation91,Citation92,Citation105,Citation134 revealed the establishment of systemic and long-term immunity. Mechanistic insights were provided by a study using an oncolytic HSV encoding an anti-PD-1 scFv that showed increased cross-presentation of a model tumor antigen by dendritic cells and enhanced T cell activation in a syngeneic tumor model.Citation91 Furthermore, an increased overall T cell infiltration, but reduced Treg infiltration, by viro-CHECKin therapy was reported using an oncolytic VV encoding an anti-PD-1 IgG and an oncolytic MeV encoding anti-CTLA-4 scFv-Fc or anti-PD-L1 scFv-Fc fusion proteins. Notably, viro-CHECKin therapy enhanced T cell infiltration compared with parental OV alone in the MeV study, but not in the VV study, as oncolytic VV alone, but not MeV alone, triggered massive T cell infiltration already.Citation103,Citation133 In consequence, the choice of therapeutic genes for effective viro-antibody therapy should consider the OV vector and its specific oncolytic and immunological properties.
Interestingly, one viro-CHECKin therapy study compared different antibody formats of the same specificity, i.e., IgG and scFv. Using an oncolytic VV, this study showed stronger expression of the scFv, but similar tumor growth inhibition for both formats, which was superior to parental virus. A survival advantage compared with parental virus was shown to be significant only for the IgG format, but not for either the scFv format or the combination of parental virus with systemic anti-PD-1 treatment.Citation103
The choice of the OV-encoded ICI determines the outcome of viro-CHECKin therapy, as revealed by a study that compared in a syngeneic mouse model oncolytic MeVs encoding anti-CTLA-4 versus anti-PD-L1, both in the scFv-Fc format.Citation133 Compared with control virus, the anti-CTLA-4 virus delayed tumor progression but did not significantly prolong survival, whereas the anti-PD-L1 virus prolonged survival without reducing early tumor growth. Tumor-specific splenocytes were activated early after treatment with the anti-CTLA-4 virus and late after anti-PD-L1 virus treatment. In contrast to other viro-CHECKin studies (see above), in the MeV study the combination of MeV with systemic mAb therapy was superior to genetic antibody delivery, with differences being significant for the anti-CTLA-4 virus, but not the anti-PD-L1 virus. The authors speculated that their results are due to different modes of action: the CTLA-4 checkpoint is active earlier in the immune response, especially during T cell activation in secondary lymphoid organs, while the PD-1 checkpoint is active during execution of T cell activity in the tumor. Therefore, anti-PD-L1 effects might appear later during treatment and the anti-CTLA-4 approach might benefit more from systemic antibody therapy, pointing at potential limitations of viro-antibody therapy, whenever systemic rather than intratumoral activity of antibodies is required.
Viro-CHECKin therapy can be combined with further therapeutics or treatment modalities to improve therapeutic outcome. One approach was inspired by the observation of increased tumor infiltration by CD155+ myeloid-derived suppressor cells after viro-CHECKin therapy with an oncolytic HSV encoding anti-PD-1 scFv in a syngeneic mouse model, in spite of improved therapeutic outcome (see ref.91). Correspondingly, anti-PD-1-encoding HSV was combined with an anti-TIGIT antibody that blocks the CD155-binding checkpoint molecule expressed on T cells. The combination resulted in an infiltration of tumor-specific T cells and therapeutic activity superior to both monotherapies and also to the combination of the parental virus with anti-TIGIT.Citation91 A different combination strategy was to combine viro-CHECKin therapy with radiation therapy. This was inspired by the observation that triple treatment of oncolytic NDV, irradiation and anti-CTLA-4 showed increased complete response rates compared with double treatments in a syngeneic mouse model. The combination of an anti-CTLA-4 scFv-encoding NDV combined with irradiation in the syngeneic model resulted in a similar survival compared with triple treatment with parental virus, irradiation and anti-CTLA-4, which was significantly improved compared with anti-CTLA-4 treatment alone.Citation135
A distinct viro-CHECKin combination treatment was based on genetic fusion of an ICI scFv with a cytokine (immunocytokine), representing yet another recombinant antibody format and mode of action (). Specifically, oncolytic NDVs encoding anti-PD1 or anti-PD-L1 scFvs fused to single-chain murine IL-12, scmIL-12, were explored.Citation132 In addition, this study investigated the combination of viro-CHECKin therapy targeting PD1/PD-L1 with systemic anti-CTLA-4 treatment. In a syngeneic mouse model, treatment with NDV-anti-PD1, NDV-anti-PD1-scmIL-12 and NDV-anti-PD-L1-scmIL-12 resulted in superior survival compared with parental virus, but only when combined with systemic anti-CTLA4. Moreover, NDV-anti-PD-L1-scmIL-12 was superior to NDV-anti-PD-L1 in the presence, not absence, of anti-CTLA-4 and triggered an especially strong increase in CD8 and TNF expression as determined by gene expression analysis. This study also reported an oncolytic NDV encoding an anti-CD28 scFv as T cell costimulatory superagonist, with or without fused IL-12, representing yet another mode of action of viro-antibody therapy, “viro-CoStim therapy” (see ). For the NDV-anti-CD28-scFv-scmIL-12 virus, superior survival outcome in comparison to parental virus was observed, but again only in the presence of anti-CTLA-4. The clearly different outcomes of viro-antibody therapies in the presence versus absence of anti-CTLA-4 in this study underscore the more global concept that the efficacy of OV-encoded checkpoint inhibitors or co-stimulatory molecules is determined by the overall immunological status, systemically or in the TME.
Expression of anti-angiogenic antibodies (viro-ANGin therapy)
Another viro-antibody therapy approach aims at inhibiting tumor angiogenesis (viro-ANGin therapy). To this end, oncolytic VV and Ad encoding recombinant antibodies binding and inhibiting the angiogenic growth factor VEGF have been engineered () based on the clinically established conventional antibody therapy with Avastin® (bevacizumab).Citation136 An oncolytic Ad encoding a VEGF-binding IgG is discussed above. Several other studies with oncolytic VV explored human and mouse VEGF-specific scFvs, and demonstrated superior or more rapid tumor growth inhibition compared with parental virus after systemicCitation94,Citation97,Citation102 or intratumoralCitation99 injection in human xenograft models. In a lung cancer xenograft model, a reduction in malignant effusion was observed.Citation97 Further analyses revealed a reduced blood vessel density in infected, but not in noninfected, areas of tumorsCitation94,Citation99,Citation102 and a noticeable reduced vascular flow in tumors,Citation99 with oncolytic VV alone triggering either an increase or a decrease of vascular density dependent on the tumor model. These results indicate that diffusion of OV-encoded scFv from infected tumor regions was insufficient to block VEGF in noninfected areas of the tumor. Of note, i.v. injection of VVs expressing the anti-VEGF scFv gene from strong promoters resulted in antibody concentrations 7 days post-infection that were 12–15 times higher in infected areas of tumors compared with sera,Citation94 underscoring the potential of viro-antibody therapy for targeted delivery of therapeutic antibodies to tumors.
A single OV can feature two distinct antibody-mediated modes of action (see ) by co-expression of different recombinant antibodies, i.e., as a “viro-double-antibody therapy.” This has been explored for oncolytic VVs expressing an anti-VEGF scFv together with either an anti-EGFR nanobody (a single-domain antibody format, see ) or an anti-FAP scFv.Citation102 Viruses encoding anti-EGFR or anti-FAP alone showed more rapid tumor growth inhibition than the parental virus in xenograft models. Mechanistic studies revealed for the anti-EGFR virus an inhibition of proliferation similar to parental virus in infected tumor areas, but superior to parental virus in noninfected areas, indicating spread of the virus-encoded nanobody. The anti-FAP virus resulted in a reduction of FAP+ and CD31+ cells in both infected and noninfected tumor areas when compared with the parental virus, also indicating antibody spread. The anti-VEGF/anti-EGFR and anti-VEGF/anti-FAP double-antibody viruses showed superior tumor growth inhibition to parental VVs, however, reaching significance only in comparison to the VV not encoding an Ab. Importantly, the anti-VEGF/anti-EGFR virus combined suppression of both proliferation and angiogenesis, demonstrating “double-antibody” activity.
Viro-ANGin therapy has also been combined with radiotherapy, considering that irradiation-induced VEGF expression by tumor cells triggers radioresistance of endothelial cells. An oncolytic VV-encoded anti-VEGF scFv was reported to reverse VEGF-mediated radioresistance of ECs, but not tumor cells, in vitro. Moreover, radio-viro-ANGin therapy with focal fractionated irradiation achieved superior tumor growth inhibition with durable responses when compared with parental virus alone, irradiation alone, and with parental virus and irradiation combined.Citation96 This was correlated with superior reduction in blood vessel density and reduced VEGF levels in tumors. Of note, this study also detected irradiation-enhanced VV infection in vivo, which might contribute to superior outcome of combination therapy.
Expression of cytotoxic antibody fusion proteins (viro-iTOX therapy)
Recombinant antibody fusion molecules can direct effector functions of fused proteins to tumor antigens or combine them with antibody-mediated effector activities. Such immunofusion molecules represent yet another antibody format that has been explored in the context of viro-antibody therapy ( and ). One example is the expression of immunocytokines,Citation32 as discussed above for OVs encoding fusion proteins of scFvs that combine checkpoint inhibition or costimulation with IL-12 activity. Two other studies explored OVs expressing tumor antigen-specific scFvs fused to cytotoxic proteins for direct bystander killing of cancer cells (viro-iTOX therapy): One study engineered an oncolytic Ad encoding an immunoRNase consisting of a cetuximab-derived scFv fused to an RNase that triggers cell death after scFv-mediated cellular uptake.Citation109 This approach required an optimized transgene expression strategy by the Ad to avoid interference of the immunoRNase with virus replication and infectious particle production (as discussed above). However, with the optimized expression strategy, this viro-iTOX therapy enabled a potent and specific bystander killing of EGFR-positive target cells in vitro and tumor growth inhibition after intratumoral injection in a xenograft model in vivo. The second study explored an oncolytic HSV encoding a HER2-specific scFv fused to an extracellular fragment of the apoptosis-inducing FasL and a collagen-derived trimerization domain.Citation88 This immunotoxin features a trimeric killer molecule with limited killing activity on its own, but potent killing of HER2+ target cells mediated by multivalent antigen-binding. Expression of this “immunokiller” gene by the oncolytic HSV triggered apoptosis, but also somewhat reduced virus replication in vitro, likely due to its pro-apoptotic activity. This viro-iTOX therapy inhibited tumor growth after intratumoral injection in a xenograft (low dose) and a syngeneic mouse model (high dose), the latter using a virus strain adapted to the mouse tumor cell line by in vivo passaging.
Of note, immunofusion approaches are relevant also for tumors resistant to the parental antibody due to defects in downstream signaling of the target molecule, e.g., Ras mutations mediating resistance to cetuximab even when EGFR is still expressed. This is because of the different mode of action, i.e., cell killing by the scFv-targeted cell killing moiety. This has been demonstrated for viro-iTOX therapy with the immunoRNase-encoding Ad facilitating the destruction of cetuximab-resistant cancer cells.Citation109
Conclusion, challenges and outlook
Viro-antibody cancer therapy uses OVs for intratumoral genetic and amplified delivery of therapeutic antibodies or, in other words, arms OVs with a gene encoding a therapeutic antibody to complement viral oncolysis with a tumor-targeted therapeutic mode of action. The emerging field has reached three major preclinical research milestones since the first publication in 2008.Citation121 First, functional therapeutic antibodies of different formats can be expressed in and released from cancer cells infected with different OVs in vitro and in vivo. To this end, gene expression strategies have been established for the expression of polymeric and single-chain antibodies in a manner compatible with productive OV replication. Interestingly, one study even reported that antibody expression indirectly improves viral oncolysis by antibody-mediated ECM destruction.Citation104 Antibody expression by OVs was shown to be much stronger compared with replication-deficient vectors of the same virus platform.Citation106 Moreover, tumor–blood ratios of antibody concentrations after OV delivery were reported to be dramatically higher compared with conventional application.Citation103,Citation106 Second, viro-antibody therapy can implement several antibody-mediated modes of action, including direct tumor cell killing (some viro-MAb therapies, viro-iTOX therapy), anti-angiogenesis (viro-ANGin therapy) or indirect and systemic killing of tumor cells, tumor stroma cells or inhibitory immune cells by activation of adaptive immune responses (viro-BiTE therapy, viro-CHECKin therapy). For the latter, several studies used sophisticated patient-derived ex vivo models derived from tumor ascites or pleural effusions containing tumor, stroma and immune cells, or mouse models with human tumor and immune cell xenografts (see ). Third, after genetic delivery to tumors by OVs, therapeutic antibodies improved therapeutic outcome, i.e., showed therapeutic activity complementary to viral oncolysis, as detected by increased tumor destruction or growth inhibition, improved survival advantage, and/or higher cure rates. For viro-BiTE therapy or viro-CHECKin therapy, long-term antitumor immunity was demonstrated.Citation89,Citation91,Citation125,Citation134 Therapeutic activity has been reported to be similar or superior to combined application of parental OV with standard antibody treatment.Citation91,Citation92,Citation102,Citation134,Citation135 Still, for specific settings, when systemic antibody activity is required, as it is believed to be the case for anti-CTLA4-mediated immune checkpoint inhibition, conventional antibody application in addition to OV injection might be superior to OV-encoded delivery.Citation133 However, this approach requires the development of two biotherapeutics. In this light, it is not surprising that the therapeutic outcome of viro-antibody therapy was further improved by conventional application of anti-CTLA-4, or of another immune checkpoint inhibitor, anti-TIGIT, as reported for OVs encoding PD-1, PD-L1-, or CD28-specific scFvs or scFv-cytokine fusions.Citation91,Citation132 Other studies demonstrated effective combination regimen of viro-CHECKin therapy with radiotherapy and of viro-BiTE therapy with CAR-T cell application.Citation112,Citation135
Research discussed here has also revealed several challenges that remain for viro-antibody therapy development: The above-mentioned achievements have been reported for selected combinations of OV platforms, antibody formats and modes of action. Moreover, direct comparisons of different virus platforms, antibody formats or modes of action in a given viro-antibody therapy approach or tumor model have, with one exception,Citation103 not been done. With respect to virus platforms, gene therapy provides a lesson, having shown clear differences between AAV and Ad vectors with respect to the kinetics and durability of antibody expression.Citation59,Citation61,Citation65,Citation138 Certainly, differences between OVs relevant for specific viro-antibody therapy approaches exist and need further exploration, such as differences in OV-triggered immune cell infiltration determining the outcome of antibody-mediated immune cell activation or defining the required complementary mode of action, i.e., recruitment versus activation of T cells. Notably, analyses of tumor perfusion and biodistribution of OV-encoded antibodies and of their possible improvement by switching antibody formats have been reported rarely or not at all, respectively. There are some indirect indications from reported data: OV-mediated expression of anti-VEGF scFvs was reported in several studies to result in reduced blood vessel density in infected, but not noninfected, tumor areas (see ), indicating limited perfusion of the scFv in the tumor. In contrast, one study showed that a FAP-specific scFv or an EGFR-binding nanobody encoded by the same OV triggered depletion of FAP+ cells and reduced tumor cell proliferation also in noninfected tumor regions.Citation102 These results point at the antibody target and/or the antibody format being important parameters for intratumoral spread of antibody activity. Overall, these reports provide a clear rationale for further mechanistic studies as starting point for improving tumor perfusion by OV-encoded antibodies. Furthermore, studies to explore how antibody engineering can be exploited to reduce or increase release into the bloodstream are warranted for therapeutic approaches for which systemic toxicities need to be avoided or systemic activity is of therapeutic benefit, respectively. Finally, activity of viro-antibody therapy in patients remains to be shown. Initial clinical studies by PsiOxus exploring oAds encoding an anti-CD40 antibody or an anti-FAP BiTE and further immunostimulatory factors, are presently recruiting (NCT 03852511, NCT 04053283), but further strengthening of translational activities is warranted.
Future research and development of viro-antibody therapy will certainly benefit from established technologies for engineering of both OVs and antibodies. Such engineering technologies represent tools to (1) address the above-mentioned challenges and overcome emerging roadblocks and (2) seize opportunities for design of novel viro-antibody therapies toward the development and translation of antibody-encoding OVs as one-agent multifunctional biotherapeutics featuring fine-tuned antibody expression, distribution, potency and tumor-specificity.
On the OV side of future viro-antibody therapy research, besides comparative studies to identify the best fit OV for individual therapeutic approaches and tumor entities, virus engineering provides opportunities to further improve antibody expression. While antibody delivery in viro-antibody therapy is inherently coupled to virus infection, replication and spread, modification of gene expression control may improve strength and/or modulate the kinetics of antibody expression. The latter is of critical interest to avoid premature destruction of OV-producing cancer cells by direct or indirect antibody-mediated cell killing. For example, it has been shown that the timing of immune checkpoint inhibition by antibodies relative to oncolysis is critical for therapeutic outcome, as it determines the strength of anti-viral versus antitumor immune activation.Citation139 Also, as OV replication efficiency in individual patients is difficult to predict, safety switches blocking either OV replication or antibody expression in case of toxicity will be a relevant research topic, especially for viro-CHECKin and viro-BITE therapy.Citation123 OV engineering might also provide an opportunity to increase antibody release from OV infected cells by modulation of how and with what kinetics infected tumor cells are lysed (see, e.g., ref118). Lastly, for therapeutic approaches that benefit from extended or repeated antibody delivery, strategies facilitating repeated application of antibody-encoding OVs are of interest, e.g., by switching of OV (sub-) type or platform.
Recent progress in the development of antibody therapies provides a plethora of opportunities for future viro-antibody therapy research, not only regarding therapeutic concepts but also approaches to increase efficacy and safety. This includes engineering of antibody molecules to meet pharmaceutical and clinical requirements by improving biochemical and biophysical properties of the antibodies, e.g., to increase stability, and to reduce immunogenicity. Furthermore, functional properties of the antibodies are now routinely adapted to the therapeutic needs, including modifications of the Fc region to tailor antibody effector functions. For example, IgG molecules intended to recruit immune effector cells through binding to Fcγ receptors can benefit from an increased and selective binding to certain Fcγ receptors, e.g., FcγRIII on NK cells to increase ADCC, while strategies where such immune effector cells are detrimental will benefit from silenced Fc regions.Citation140,Citation141 Many of these modifications are accessible through genetic engineering. However, it should also be mentioned that some modifications, such as glyco-engineering or conjugation of therapeutic compounds, are excluded from viro-antibody therapy.
An increasing number of new targets is being evaluated for cancer therapy.Citation142 This pipeline is fueled by novel tools of antibody discovery, e.g., moving from target-led strategies to phenotypic screening approaches,Citation26,Citation143 including targets associated with the various stages of the metastatic process and the tumor microenvironment.Citation144 Thereby, the “high-hanging fruits” regarding target biology, target exposure and antibody modes of action are being pursued. The development of intracellularly produced antibodies, so-called intrabodies, which have recently been shown to work in vivo,Citation145,Citation146 provides opportunities for developing a different kind of viro-antibody therapy. As such, the expression of cytosolic, nuclear or vesicular intrabodies facilitates the targeting of intracellular host restriction factors and of cellular molecules involved in immune activation at the protein level. By this means, specific epitopes, conformations, or post-translational modifications may be targeted and protein–protein interactions might be blocked. A recent study points at possible applications in virotherapy: it demonstrated increased replication of a VSV mutant by knockdown of interferon-α secretion using an ER intrabody with specificity for several interferon-α isoforms.Citation147
Antibodies against novel targets, but also antibodies against established and clinically used targets, can be further improved by a fine-tuning of antibody properties, e.g., considering affinity, valency, effector functions, and epitope specificity.Citation148,Citation149 Here, bi- or multi-specific antibodies for combinatorial targeting of cell surface antigens and/or soluble ligands are currently under investigation, also allowing tumor heterogeneity to be addressed.Citation150 However, bispecific antibodies offer much more. Through their dual-binding mode, bispecific antibodies can exert many new modes of action beyond those of natural antibodies, including, for example, target-mediated uptake and inhibition of intracellular structures, forced internalization and degradation, ligand or co-factor mimicry resulting in cis activation of target molecules, and targeted clustering of surface receptors in trans, just to name a few of them.Citation38 Here, an entire zoo of bispecific antibody formats is available to adapt the molecular configuration to the therapeutic need.Citation38 Many of these bispecific antibody formats are evaluated in clinical studies for T-cell retargeting. Thus, it can be expected that, besides the BiTE format currently used in combination with OVs, alternative formats with favorable functional properties will be implemented in the future, for example, increasing valency and tuning affinity for target antigens to increase potency and tumor cell selectivity. Furthermore, strategies are available to endow bispecific T-cell engagers with mechanisms for conditional activation at the tumor site, e.g., by local liberation of the binding site through proteolytic cleavage or as a response to pH or hypoxia.Citation54 Finally, the exploitation of antibody fusion proteins for targeted delivery of effector moieties offers further opportunities. With a plethora of immune-regulating ligands available, new immunocytokines might be used to foster an antitumor immune response or to inhibit immuno-suppressive activities, e.g., of regulatory T-cells, in OV therapy.Citation32,Citation151
In summary, viro-antibody therapy research has established preclinical proof of principle for a panel of OVs, antibody formats and modes of action, and has identified challenges for further research and translation. Importantly, available comprehensive tools and strategies for engineering of both OVs and antibodies will allow researchers to address these challenges toward the development of improved and/or novel antibody-encoding OVs for effective viro-antibody therapy.
List of abbreviations
AAV, adeno-associated virus; Ad, adenovirus; ADC, antibody drug–conjugate; ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; BiTE, bispecific T cell engager; DAMPs damage-associated molecular patterns; ECM, extracellular matrix; Fab, antigen-binding antibody fragment; FAP, fibroblast-activating protein; HSV, herpes simplex viruses; IAV, influenza A virus; ICIs, Immune checkpoint inhibitors; Ig, immunoglobulin; IRES, internal ribosome entry site; mAbs, monoclonal antibodies; MDSC, myeloid-derived suppressor cells; MenSC, menstrual blood-derived mesenchymal stem cells; MeV, measles virus; NDV, Newcastle disease virus; OVs, oncolytic viruses; PAMPs, pathogen-associated molecular patterns; PBMC, peripheral blood mononuclear cells; scFv, single-chain antibody fragments; TME, tumor microenvironment; VV, vaccinia virus; VSV, vesicular stomatitis virus
Acknowledgments
We thank Johannes P.W. Heidbüchel for critical reading of the manuscript.
Disclosure statement
G.U. is founder and CMO/CSO of CanVirex AG, a company developing oncolytic measles viruses as (cancer) immunotherapeutics. R.E.K. is co-inventor on patent/-applications covering therapeutic antibodies and proteins and reports licensing agreements and research support from Baliopharm, BioNTech, and Oncomatryx and consultancy fees from Oncomatryx, Roche, and Immatics. D.M.N. declares no conflict of interest.
Additional information
Funding
References
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, and Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1.
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–29. doi:10.1038/nbt.2287.
- Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer. 2016;4(1):53. doi:10.1186/s40425-016-0158-5.
- Pol JG, Levesque S, Workenhe ST, Gujar S, Le Boeuf F, Clements DR, Fahrner JE, Fend L, Bell JC, Mossman KL, et al., Trial Watch: oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology. 2018;7(12):e1503032. doi:10.1080/2162402X.2018.1503032.
- Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62. doi:10.1038/nrd4663.
- Breitbach CJ, Lichty BD, Bell JC. Oncolytic viruses: therapeutics with an identity crisis. EBioMedicine. 2016;9:31–36. doi:10.1016/j.ebiom.2016.06.046.
- Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: priming time for cancer immunotherapy. BioDrugs. 2019;33(5):485–501. doi:10.1007/s40259-019-00367-0.
- Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019;18(9):689–706. doi:10.1038/s41573-019-0029-0.
- Fisher K, Hazini A, Seymour LW. Tackling HLA deficiencies head on with oncolytic viruses. Cancers (Basel). 2021;13(4). doi:10.3390/cancers13040719.
- Kennedy PJ, Oliveira C, Granja PL, Sarmento B. Monoclonal antibodies: technologies for early discovery and engineering. Crit Rev Biotechnol. 2018;38(3):394–408. doi:10.1080/07388551.2017.1357002.
- Laustsen AH, Greiff V, Karatt-Vellatt A, Muyldermans S, Jenkins TP. Animal immunization, in vitro display technologies, and machine learning for antibody discovery. Trends Biotechnol. 2021. doi:10.1016/j.tibtech.2021.03.003.
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, and Bell JC Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–25. doi:10.1038/77558.
- Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–40.
- Dorer DE, Nettelbeck DM. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv Drug Deliv Rev. 2009;61(7–8):554–71. doi:10.1016/j.addr.2009.03.013.
- Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol. 2014;12(1):23–34. doi:10.1038/nrmicro3140.
- Ruiz AJ, Russell SJ. MicroRNAs and oncolytic viruses. Curr Opin Virol. 2015;13:40–48. doi:10.1016/j.coviro.2015.03.007.
- Rivera AA, Davydova J, Schierer S, Wang M, Krasnykh V, Yamamoto M, Curiel DT, and Nettelbeck DM. Combining high selectivity of replication with fiber chimerism for effective adenoviral oncolysis of CAR-negative melanoma cells. Gene Ther. 2004;11(23):1694–702. doi:10.1038/sj.gt.3302346.
- Kaufmann JK, Nettelbeck DM. Virus chimeras for gene therapy, vaccination, and oncolysis: adenoviruses and beyond. Trends Mol Med. 2012;18(7):365–76. doi:10.1016/j.molmed.2012.04.008.
- Russell SJ, Barber GN. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell. 2018;33(4):599–605. doi:10.1016/j.ccell.2018.03.011.
- Gravbrot N, Gilbert-Gard K, Mehta P, Ghotmi Y, Banerjee M, Mazis C, et al. Therapeutic monoclonal antibodies targeting immune checkpoints for the treatment of solid tumors. Antibodies (Basel). 2019;8(4):1-54.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–68.
- Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al., Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109–1119.e10. doi:10.1016/j.cell.2017.08.027.
- Chesney J, Puzanov I, Collichio F, Milhem MM, Hauschild A, Chen L, Sharma A, Garbe C, Singh P, and Mehnert JM. Patterns of response with talimogene laherparepvec in combination with ipilimumab or ipilimumab alone in metastatic unresectable melanoma. Br J Cancer. 2019;121(5):417–20. doi:10.1038/s41416-019-0530-6.
- Pearl TM, Markert JM, Cassady KA, Ghonime MG. Oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol Ther Oncolytics. 2019;13:14–21. doi:10.1016/j.omto.2019.03.001.
- Rivera AA, Wang M, Suzuki K, Uil TG, Krasnykh V, Curiel DT, and Nettelbeck DM. Mode of transgene expression after fusion to early or late viral genes of a conditionally replicating adenovirus via an optimized internal ribosome entry site in vitro and in vivo. Virology. 2004;320(1):121–34. doi:10.1016/j.virol.2003.11.028.
- Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9(1):86–120. doi:10.1007/s13238-017-0457-8.
- Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1):1860476. doi:10.1080/19420862.2020.1860476.
- Shah A, Rauth S, Aithal A, Kaur S, Ganguly K, Orzechowski C, Varshney GC, Jain M, and Batra SK. The current landscape of antibody-based therapies in solid malignancies. Theranostics. 2021;11(3):1493–512. doi:10.7150/thno.52614.
- Goldenberg DM, Sharkey RM. Using antibodies to target cancer therapeutics. Expert Opin Biol Ther. 2012;12(9):1173–90. doi:10.1517/14712598.2012.693472.
- Birrer MJ, Moore KN, Betella I, Bates RC. Antibody-drug conjugate-based therapeutics: state of the science. J Natl Cancer Inst. 2019;111(6):538–49. doi:10.1093/jnci/djz035.
- Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Robak T, le Coutre PD, Gjertsen BT, Troussard X, Roboz GJ, Karlin L, et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol. 2021;14(1):35. doi:10.1186/s13045-020-01004-y.
- Kontermann RE. Antibody-cytokine fusion proteins. Arch Biochem Biophys. 2012;526(2):194–205. doi:10.1016/j.abb.2012.03.001.
- de Bruyn M, Bremer E, Helfrich W. Antibody-based fusion proteins to target death receptors in cancer. Cancer Lett. 2013;332(2):175–83. doi:10.1016/j.canlet.2010.11.006.
- Jordaan S, Akinrinmade OA, Nachreiner T, Cremer C, Naran K, Chetty S, and Barth S. Updates in the development of immunornases for the selective killing of tumor cells. Biomedicines. 2018;6(1). doi:10.3390/biomedicines6010028.
- Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. The Oncologist. 2015;20(6):660–73. doi:10.1634/theoncologist.2014-0465.
- Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. 2020;77:1745–70.
- Lee A, Sun S, Sandler A, Hoang T. Recent progress in therapeutic antibodies for cancer immunotherapy. Curr Opin Chem Biol. 2018;44:56–65. doi:10.1016/j.cbpa.2018.05.006.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol. 2020;11:167.
- Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. 2019;19(10):568–86. doi:10.1038/s41568-019-0183-z.
- Dalziel M, Beers SA, Cragg MS, Crispin M. Through the barricades: overcoming the barriers to effective antibody-based cancer therapeutics. Glycobiology. 2018;28:697–712.
- Beckman RA, von Roemeling R, Scott AM. Monoclonal antibody dose determination and biodistribution into solid tumors. Ther Deliv. 2011;2(3):333–44. doi:10.4155/tde.10.91.
- Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52:3402–08.
- Colcher D, Pavlinkova G, Beresford G, Booth BJ, Choudhury A, Batra SK. Pharmacokinetics and biodistribution of genetically-engineered antibodies. Q J Nucl Med. 1998;42:225–41.
- Nessler I, Khera E, Vance S, Kopp A, Qiu Q, Keating TA, Abu-Yousif AO, Sandal T, Legg J, Thompson L, et al., Increased tumor penetration of single-domain antibody-drug conjugates improves in vivo efficacy in prostate cancer models. Cancer Res. 2020;80(6):1268–78. doi:10.1158/0008-5472.CAN-19-2295.
- Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21(12):2153–63. doi:10.1021/bc100261d.
- Kraft TE, Richter WF, Emrich T, Knaupp A, Schuster M, Wolfert A, and Kettenberger H. Heparin chromatography as an in vitro predictor for antibody clearance rate through pinocytosis. MAbs. 2020;12(1):1683432. doi:10.1080/19420862.2019.1683432.
- Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, Marks JD, and Weiner LM. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750–55.
- Rudnick SI, Lou J, Shaller CC, Tang Y, Klein-Szanto AJ, Weiner LM, Marks JD, and Adams GP. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71(6):2250–59. doi:10.1158/0008-5472.CAN-10-2277.
- Saga T, Neumann RD, Heya T, Sato J, Kinuya S, Le N, Paik CH, and Weinstein JN. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. Proc Natl Acad Sci U S A. 1995;92(19):8999–9003. doi:10.1073/pnas.92.19.8999.
- Tang Y, Cao Y. Modeling the dynamics of antibody-target binding in living tumors. Sci Rep. 2020;10(1):16764. doi:10.1038/s41598-020-73711-y.
- Staflin K, Zuch de Zafra CL, Schutt LK, Clark V, Zhong F, Hristopoulos M, Clark R, Li J, Mathieu M, Chen X, et al., Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight. 2020;5(7):1-15.
- Middelburg J, Kemper K, Engelberts P, Labrijn AF, Schuurman J, Van Hall T. Overcoming challenges for CD3-bispecific antibody therapy in solid tumors. Cancers (Basel). 2021;13(2). doi:10.3390/cancers13020287.
- Lin WW, Lu YC, Chuang CH, Cheng TL. Ab locks for improving the selectivity and safety of antibody drugs. J Biomed Sci. 2020;27(1):76. doi:10.1186/s12929-020-00652-z.
- De Lombaerde E, De Wever O, De Geest BG. Delivery routes matter: safety and efficacy of intratumoral immunotherapy. Biochim Biophys Acta Rev Cancer. 2021;1875(2):188526. doi:10.1016/j.bbcan.2021.188526.
- Chen JD, Yang Q, Yang AG, Marasco WA, Chen SY. Intra- and extracellular immunization against HIV-1 infection with lymphocytes transduced with an AAV vector expressing a human anti-gp120 antibody. Hum Gene Ther. 1996;7(13):1515–25. doi:10.1089/hum.1996.7.13-1515.
- Paul S, Bizouarne N, Dott K, Ruet L, Dufour P, Acres RB, and Kieny MP. Redirected cellular cytotoxicity by infection of effector cells with a recombinant vaccinia virus encoding a tumor-specific monoclonal antibody. Cancer Gene Ther. 2000;7(4):615–23. doi:10.1038/sj.cgt.7700161.
- Paul S, Snary D, Hoebeke J, Allen D, Balloul JM, Bizouarne N, Dott K, Geist M, Hilgers J, Kieny MP, et al. Targeted macrophage cytotoxicity using a nonreplicative live vector expressing a tumor-specific single-chain variable region fragment. Hum Gene Ther. 2000;11(10):1417–28. doi:10.1089/10430340050057495.
- Noël D, Pelegrin M, Kramer S, Jacquet C, Skander N, Piechaczyk M. High in vivo production of a model monoclonal antibody on adenoviral gene transfer. Hum Gene Ther. 2002;13(12):1483–93. doi:10.1089/10430340260185111.
- Lewis AD, Chen R, Montefiori DC, Johnson PR, Clark KR. Generation of neutralizing activity against human immunodeficiency virus type 1 in serum by antibody gene transfer. J Virol. 2002;76(17):8769–75. doi:10.1128/JVI.76.17.8769-8775.2002.
- Schnepp BC, Johnson PR. Vector-mediated in vivo antibody expression. Microbiol Spectr. 2014;2(4):Aid-0016-2014. doi:10.1128/microbiolspec.AID-0016-2014.
- Yang L, Wang P. Passive immunization against HIV/AIDS by antibody gene transfer. Viruses. 2014;6(2):428–47. doi:10.3390/v6020428.
- Fang J, Qian JJ, Yi S, Harding TC, Tu GH, VanRoey M, and Jooss K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat Biotechnol. 2005;23(5):584–90. doi:10.1038/nbt1087.
- Jiang M, Shi W, Zhang Q, Wang X, Guo M, Cui Z, Su C, Yang Q, Li Y, Sham J, et al. Gene therapy using adenovirus-mediated full-length anti-HER-2 antibody for HER-2 overexpression cancers. Clin Cancer Res. 2006;12(20):6179–85. doi:10.1158/1078-0432.CCR-06-0746.
- Skaricic D, Traube C, De B, Joh J, Boyer J, Crystal RG, and Worgall S. Genetic delivery of an anti-RSV antibody to protect against pulmonary infection with RSV. Virology. 2008;378(1):79–85. doi:10.1016/j.virol.2008.04.016.
- Schlake T, Thess A, Thran M, Jordan I. mRNA as novel technology for passive immunotherapy. Cell Mol Life Sci. 2019;76:301–28.
- Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2011;481(7379):81–84. doi:10.1038/nature10660.
- Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1(4):376–82. doi:10.1006/mthe.2000.0050.
- Paul S, Regulier E, Rooke R, Stoeckel F, Geist M, Homann H, Balloul JM, Villeval D, Poitevin Y, Kieny MP, et al. Tumor gene therapy by MVA-mediated expression of T-cell-stimulating antibodies. Cancer Gene Ther. 2002;9(5):470–77. doi:10.1038/sj.cgt.7700461.
- Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, and Suzuki M. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77(8):2040–51. doi:10.1158/0008-5472.CAN-16-1577.
- Rosewell Shaw A, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, Brenner MK, and Suzuki M. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. 2017;25(11):2440–51. doi:10.1016/j.ymthe.2017.09.010.
- Porter CE, Rosewell Shaw A, Jung Y, Yip T, Castro PD, Sandulache VC, Sikora A, Gottschalk S, Ittman MM, Brenner MK, et al. Oncolytic adenovirus armed with bite, cytokine, and checkpoint inhibitor enables CAR T cells to control the growth of heterogeneous tumors. Mol Ther. 2020;28(5):1251–62. doi:10.1016/j.ymthe.2020.02.016.
- Arnone CM, Polito VA, Mastronuzzi A, Carai A, Diomedi FC, Antonucci L, Petrelli LL, Vinci M, Ferrari F, Salviato E, et al. Oncolytic adenovirus and gene therapy with EphA2-BiTE for the treatment of pediatric high-grade gliomas. J Immunother Cancer. 2021;9(5). doi:10.1136/jitc-2020-001930.
- Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, Miller P, Kanellakis N, Ashfield R, Fisher KD, et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med. 2017;9(8):1067–87. doi:10.15252/emmm.201707567.
- Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, Campo L, Bryant RJ, Verrill C, Lambert A, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78(24):6852–65. doi:10.1158/0008-5472.CAN-18-1750.
- Scott EM, Jacobus EJ, Lyons B, Frost S, Freedman JD, Dyer A, Khalique H, Taverner WK, Carr A, Champion BR, et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J Immunother Cancer. 2019;7(1):320. doi:10.1186/s40425-019-0807-6.
- Bucheit AD, Kumar S, Grote DM, Lin Y, von Messling V, Cattaneo RB, and Fielding AK. An oncolytic measles virus engineered to enter cells through the CD20 antigen. Mol Ther. 2003;7(1):62–72. doi:10.1016/S1525-0016(02)00033-3.
- Campadelli-Fiume G, Petrovic B, Leoni V, Gianni T, Avitabile E, Casiraghi C, and Gatta V. Retargeting strategies for oncolytic herpes simplex viruses. Viruses. 2016;8(3):63. doi:10.3390/v8030063.
- Hanauer JDS, Rengstl B, Kleinlützum D, Reul J, Pfeiffer A, Friedel T, Schneider IC, Newrzela S, Hansmann ML, Buchholz CJ, et al. CD30-targeted oncolytic viruses as novel therapeutic approach against classical Hodgkin lymphoma. Oncotarget. 2018;9(16):12971–81. doi:10.18632/oncotarget.24191.
- Hemminki A, Dmitriev I, Liu B, Desmond RA, Alemany R, Curiel DT. Targeting oncolytic adenoviral agents to the epidermal growth factor pathway with a secretory fusion molecule. Cancer Res. 2001;61:6377–81.
- van Beusechem VW, Mastenbroek DC, van den Doel PB, Lamfers ML, Grill J, Würdinger T, Haisma HJ, Pinedo HM, and Gerritsen WR. Conditionally replicative adenovirus expressing a targeting adapter molecule exhibits enhanced oncolytic potency on CAR-deficient tumors. Gene Ther. 2003;10(23):1982–91. doi:10.1038/sj.gt.3302103.
- Gil M, Bieniasz M, Wierzbicki A, Bambach BJ, Rokita H, Kozbor D. Targeting a mimotope vaccine to activating Fc gamma receptors empowers dendritic cells to prime specific CD8+ T cell responses in tumor-bearing mice. J Immunol. 2009;183(10):6808–18. doi:10.4049/jimmunol.0900364.
- Seth P, Wang ZG, Pister A, Zafar MB, Kim S, Guise T, and Wakefield L. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-beta receptor II and human immunoglobulin Fc for breast cancer therapy. Hum Gene Ther. 2006;17(11):1152–60. doi:10.1089/hum.2006.17.1152.
- Hu Z, Zhang Z, Guise T, Seth P. Systemic delivery of an oncolytic adenovirus expressing soluble transforming growth factor-β receptor II-Fc fusion protein can inhibit breast cancer bone metastasis in a mouse model. Hum Gene Ther. 2010;21(11):1623–29. doi:10.1089/hum.2010.018.
- Hu Z, Gerseny H, Zhang Z, Chen YJ, Berg A, Zhang Z, Stock S, and Seth P. Oncolytic adenovirus expressing soluble TGFβ receptor II-Fc-mediated inhibition of established bone metastases: a safe and effective systemic therapeutic approach for breast cancer. Mol Ther. 2011;19(9):1609–18. doi:10.1038/mt.2011.114.
- Yang Y, Xu W, Peng D, Wang H, Zhang X, Wang H, Xiao F, Zhu Y, Ji Y, Gulukota K, et al. An oncolytic adenovirus targeting transforming growth factor β inhibits protumorigenic signals and produces immune activation: a novel approach to enhance anti-PD-1 and anti-CTLA-4 therapy. Hum Gene Ther. 2019;30(9):1117–32. doi:10.1089/hum.2019.059.
- Wang G, Kang X, Chen KS, Jehng T, Jones L, Chen J, Huang XF, and Chen SY. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun. 2020;11(1):1395. doi:10.1038/s41467-020-15229-5.
- Loya SM, Zhang X. Enhancing the bystander killing effect of an oncolytic HSV by arming it with a secretable apoptosis activator. Gene Ther. 2015;22(3):237–46. doi:10.1038/gt.2014.113.
- Passaro C, Alayo Q, De Laura I, McNulty J, Grauwet K, Ito H, Bhaskaran V, Mineo M, Lawler SE, Shah K, et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res. 2019;25(1):290–99. doi:10.1158/1078-0432.CCR-18-2311.
- Thomas S, Kuncheria L, Roulstone V, Kyula JN, Mansfield D, Bommareddy PK, et al.Thomas S, Kuncheria L, Roulstone V, Kyula JN, Mansfield D, Bommareddy PK, Smith H, Kaufman HL, Harrington KJ, and Coffin RS. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer. 2019;7(1):214. doi:10.1186/s40425-019-0682-1.
- Lin C, Ren W, Luo Y, Li S, Chang Y, Li L, Xiong D, Huang X, Xu Z, Yu Z, et al. Intratumoral delivery of a PD-1-blocking scFv encoded in oncolytic HSV-1 promotes antitumor immunity and synergizes with TIGIT blockade. Cancer Immunol Res. 2020;8(5):632–47. doi:10.1158/2326-6066.CIR-19-0628.
- Zhu Y, Hu X, Feng L, Yang Z, Zhou L, Duan X, Cheng S, Zhang W, Liu B, and Zhang K. Enhanced Therapeutic Efficacy Of A Novel Oncolytic Herpes Simplex Virus Type 2 encoding an antibody against programmed cell death 1. Mol Ther Oncolytics. 2019;15:201–13. doi:10.1016/j.omto.2019.10.003.
- Khalique H, Baugh R, Dyer A, Scott EM, Frost S, Larkin S, Lei-Rossmann J, and Seymour LW. Oncolytic herpesvirus expressing PD-L1 BiTE for cancer therapy: exploiting tumor immune suppression as an opportunity for targeted immunotherapy. J Immunother Cancer. 2021;9(4). doi:10.1136/jitc-2020-001292.
- Frentzen A, Yu YA, Chen N, Zhang Q, Weibel S, Raab V, and Szalay AA. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc Natl Acad Sci U S A. 2009;106(31):12915–20. doi:10.1073/pnas.0900660106.
- Patil SS, Gentschev I, Adelfinger M, Donat U, Hess M, Weibel S, Nolte I, Frentzen A, and Szalay AA. Virotherapy of canine tumors with oncolytic vaccinia virus GLV-1h109 expressing an anti-VEGF single-chain antibody. PLoS One. 2012;7(10):e47472. doi:10.1371/journal.pone.0047472.
- Buckel L, Advani SJ, Frentzen A, Zhang Q, Yu YA, Chen NG, Ehrig K, Stritzker J, Mundt AJ, and Szalay AA. Combination of fractionated irradiation with anti-VEGF expressing vaccinia virus therapy enhances tumor control by simultaneous radiosensitization of tumor associated endothelium. Int J Cancer. 2013;133(12):2989–99.
- Weibel S, Hofmann E, Basse-Luesebrink TC, Donat U, Seubert C, Adelfinger M, Gnamlin P, Kober C, Frentzen A, Gentschev I, et al., Treatment of malignant effusion by oncolytic virotherapy in an experimental subcutaneous xenograft model of lung cancer. J Transl Med. 2013;11(1):106. doi:10.1186/1479-5876-11-106.
- Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014;22(1):102–11. doi:10.1038/mt.2013.240.
- Gholami S, Marano A, Chen NG, Aguilar RJ, Frentzen A, Chen CH, Lou E, Fujisawa S, Eveno C, Belin L, et al. A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res Treat. 2014;148(3):489–99. doi:10.1007/s10549-014-3180-7.
- Adelfinger M, Gentschev I, Grimm de Guibert J, Weibel S, Langbein-Laugwitz J, Härtl B, Murua Escobar H, Nolte I, Chen NG, Aguilar RJ, et al., Evaluation of a new recombinant oncolytic vaccinia virus strain GLV-5b451 for feline mammary carcinoma therapy. PLoS One. 2014;9(8):e104337. doi:10.1371/journal.pone.0104337.
- Adelfinger M, Bessler S, Frentzen A, Cecil A, Langbein-Laugwitz J, Gentschev I, and Szalay AA. Preclinical testing oncolytic vaccinia virus strain GLV-5b451 expressing an anti-VEGF single-chain antibody for canine cancer therapy. Viruses. 2015;7(7):4075–92. doi:10.3390/v7072811.
- Huang T, Wang H, Chen NG, Frentzen A, Minev B, Szalay AA. Expression of anti-VEGF antibody together with anti-EGFR or anti-FAP enhances tumor regression as a result of vaccinia virotherapy. Mol Ther Oncolytics. 2015;2:15003. doi:10.1038/mto.2015.3.
- Kleinpeter P, Fend L, Thioudellet C, Geist M, Sfrontato N, Koerper V, Fahrner C, Schmitt D, Gantzer M, Remy-Ziller C, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death −1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 2016;5(10):e1220467. doi:10.1080/2162402X.2016.1220467.
- Yu F, Hong B, Song X-T. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Transl Med. 2017;3(4):122–32.
- Zuo S, Wei M, He B, Chen A, Wang S, Kong L, Zhang Y, Meng G, Xu T, Wu J, et al. Enhanced antitumor efficacy of a novel oncolytic vaccinia virus encoding a fully monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT). EBioMedicine. 2021;64:103240. doi:10.1016/j.ebiom.2021.103240.
- Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, Escutenaire S, Kanerva A, Pesonen S, Löskog A, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19(10):988–98. doi:10.1038/gt.2011.176.
- Du T, Shi G, Li YM, Zhang JF, Tian HW, Wei YQ, Deng H, and Yu DC. Tumor-specific oncolytic adenoviruses expressing granulocyte macrophage colony-stimulating factor or anti-CTLA4 antibody for the treatment of cancers. Cancer Gene Ther. 2014;21(8):340–48. doi:10.1038/cgt.2014.34.
- Liikanen I, Tähtinen S, Guse K, Gutmann T, Savola P, Oksanen M, Kanerva A, and Hemminki A. Oncolytic adenovirus expressing monoclonal antibody trastuzumab for treatment of HER2-positive cancer. Mol Cancer Ther. 2016;15(9):2259–69. doi:10.1158/1535-7163.MCT-15-0819.
- Fernández-Ulibarri I, Hammer K, Arndt MA, Kaufmann JK, Dorer D, Engelhardt S, Kontermann RE, Hess J, Allgayer H, Krauss J, et al. Genetic delivery of an immunoRNase by an oncolytic adenovirus enhances anticancer activity. Int J Cancer. 2015;136(9):2228–40. doi:10.1002/ijc.29258.
- Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, June CH, and Alemany R. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017;77(8):2052–63. doi:10.1158/0008-5472.CAN-16-1708.
- Marino N, Illingworth S, Kodialbail P, Patel A, Calderon H, Lear R, Fisher KD, Champion BR, and Brown ACN. Development of a versatile oncolytic virus platform for local intra-tumoural expression of therapeutic transgenes. PLoS One. 2017;12(5):e0177810. doi:10.1371/journal.pone.0177810.
- Wing A, Fajardo CA, Posey AD, Jr., Shaw C, Da T, Young RM, Alemany R, June CH, and Guedan S. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. 2018;6(5):605–16. doi:10.1158/2326-6066.CIR-17-0314.
- De Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019;7(1):19. doi:10.1186/s40425-019-0505-4.
- Barlabé P, Sostoa J, Fajardo CA, Alemany R, Moreno R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020;27(5):383–88. doi:10.1038/s41417-019-0110-1.
- Quirin C, Rohmer S, Fernandez-Ulibarri I, Behr M, Hesse A, Engelhardt S, Erbs P, Enk AH, and Nettelbeck DM. Selectivity and efficiency of late transgene expression by transcriptionally targeted oncolytic adenoviruses are dependent on the transgene insertion strategy. Hum Gene Ther. 2011;22(4):389–404. doi:10.1089/hum.2010.100.
- Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, and Rosenberg SA. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210(6):1125–35. doi:10.1084/jem.20130110.
- Hamilton JR, Vijayakumar G, Palese P. A recombinant antibody-expressing influenza virus delays tumor growth in a mouse model. Cell Rep. 2018;22(1):1–7. doi:10.1016/j.celrep.2017.12.025.
- Rohmer S, Quirin C, Hesse A, Sandmann S, Bayer W, Herold-Mende C, Haviv YS, Wildner O, Enk AH, and Nettelbeck DM. Transgene expression by oncolytic adenoviruses is modulated by E1B19K deletion in a cell type-dependent manner. Virology. 2009;395(2):243–54. doi:10.1016/j.virol.2009.09.030.
- Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst. 2006;98(20):1482–93. doi:10.1093/jnci/djj397.
- Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. 2010;18(7):1275–83. doi:10.1038/mt.2010.79.
- Pühler F, Willuda J, Puhlmann J, Mumberg D, Römer-Oberdörfer A, Beier R. Generation of a recombinant oncolytic Newcastle disease virus and expression of a full IgG antibody from two transgenes. Gene Ther. 2008;15(5):371–83. doi:10.1038/sj.gt.3303095.
- Wei D, Li Q, Wang XL, Wang Y, Xu J, Feng F, Nan G, Wang B, Li C, Guo T, et al. Oncolytic Newcastle disease virus expressing chimeric antibody enhanced anti-tumor efficacy in orthotopic hepatoma-bearing mice. J Exp Clin Cancer Res. 2015;34(1):153. doi:10.1186/s13046-015-0271-1.
- Scott EM, Duffy MR, Freedman JD, Fisher KD, Seymour LW. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol Biosci. 2018;18(1). doi:10.1002/mabi.201700187.
- Heidbuechel JPW, Engeland CE. Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies. J Hematol Oncol. 2021;14(1):63. doi:10.1186/s13045-021-01075-5.
- Speck T, Heidbuechel JPW, Veinalde R, Jaeger D, von Kalle C, Ball CR, Ungerechts G, and Engeland CE. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin Cancer Res. 2018;24(9):2128–37. doi:10.1158/1078-0432.CCR-17-2651.
- Tuve S, Chen BM, Liu Y, Cheng TL, Touré P, Sow PS, Feng Q, Kiviat N, Strauss R, Ni S, et al. Combination of tumor site-located CTL-associated antigen-4 blockade and systemic regulatory T-cell depletion induces tumor-destructive immune responses. Cancer Res. 2007;67(12):5929–39. doi:10.1158/0008-5472.CAN-06-4296.
- Simmons AD, Moskalenko M, Creson J, Fang J, Yi S, VanRoey MJ, Allison JP, and Jooss K. Local secretion of anti-CTLA-4 enhances the therapeutic efficacy of a cancer immunotherapy with reduced evidence of systemic autoimmunity. Cancer Immunol Immunother. 2008;57(8):1263–70. doi:10.1007/s00262-008-0451-3.
- Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res. 2013;19(19):5381–89. doi:10.1158/1078-0432.CCR-12-0781.
- Sandin LC, Eriksson F, Ellmark P, Loskog AS, Tötterman TH, Mangsbo SM. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology. 2014;3(1):e27614. doi:10.4161/onci.27614.
- Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble pd1 enhances oncolytic virotherapy. Cancer Res. 2017;77(11):2952–63. doi:10.1158/0008-5472.CAN-16-1638.
- Woo Y, Zhang Z, Yang A, Chaurasiya S, Park AK, Lu J, Kim SI, Warner SG, Von Hoff D, and Fong Y. Novel chimeric immuno-oncolytic virus CF33-hNIS-antiPDL1 for the treatment of pancreatic cancer. J Am Coll Surg. 2020;230(4):709–17. doi:10.1016/j.jamcollsurg.2019.12.027.
- Vijayakumar G, McCroskery S, Palese P. Engineering Newcastle disease virus as an oncolytic vector for intratumoral delivery of immune checkpoint inhibitors and immunocytokines. J Virol. 2020;94(3). doi:10.1128/JVI.01677-19.
- Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, Shevchenko I, Umansky V, Nettelbeck DM, Weichert W, et al., CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014;22(11):1949–59. doi:10.1038/mt.2014.160.
- Wu C, Wu M, Liang M, Xiong S, Dong C. A novel oncolytic virus engineered with PD-L1 scFv effectively inhibits tumor growth in a mouse model. Cell Mol Immunol. 2019;16(9):780–82. doi:10.1038/s41423-019-0264-7.
- Vijayakumar G, Palese P, Goff PH. Oncolytic Newcastle disease virus expressing a checkpoint inhibitor as a radioenhancing agent for murine melanoma. EBioMedicine. 2019;49:96–105. doi:10.1016/j.ebiom.2019.10.032.
- Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, and Chinot OL. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017. doi:10.1016/j.ctrv.2020.102017.
- Zhang Z, Yang A, Chaurasiya S, Park AK, Lu J, Kim SI, et al. CF33-hNIS-antiPDL1 virus primes pancreatic ductal adenocarcinoma for enhanced anti-PD-L1 therapy. Cancer Gene Ther; 2021. doi: 10.1038/s41417-021-00350-4
- Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36. doi:10.1182/blood-2013-01-306647.
- Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res. 2015;21(24):5543–51. doi:10.1158/1078-0432.CCR-14-2009.
- Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi:10.1007/s13238-017-0473-8.
- Liu R, Oldham RJ, Teal E, Beers SA, Cragg MS. Fc-engineering for modulated effector functions-improving antibodies for cancer treatment. Antibodies (Basel). 2020;9(4):1-34.
- Martineau P, Watier H, Pèlegrin A, Turtoi A. Targets for MAbs: innovative approaches for their discovery & validation, LabEx MAbImprove 6(th) antibody industrial symposium, June 25- 26,2018, montpellier. France MAbs. 2019;11(5):812–25. doi:10.1080/19420862.2019.1612691.
- Minter RR, Sandercock AM, Rust SJ. Phenotypic screening-the fast track to novel antibody discovery. Drug Discov Today Technol. 2017;23:83–90. doi:10.1016/j.ddtec.2017.03.004.
- Yang H, Kuo YH, Smith ZI, Spangler J. Targeting cancer metastasis with antibody therapeutics. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2021. e1698. Wiley.
- Marschall AL, Single FN, Schlarmann K, Bosio A, Strebe N, van den Heuvel J, et al.Marschall AL, Single FN, Schlarmann K, Bosio A, Strebe N, van den Heuvel J, Frenzel A, and Dübel S Functional knock down of VCAM1 in mice mediated by endoplasmatic reticulum retained intrabodies. MAbs. 2014;6(6):1394–401. doi:10.4161/mabs.34377.
- Zhang C, Ötjengerdes RM, Roewe J, Mejias R, Marschall ALJ. Applying antibodies inside cells: principles and recent advances in neurobiology, virology and oncology. BioDrugs. 2020;34:435–62.
- Büssow K, Themann P, Luu S, Pentrowski P, Harting C, Majewski M, Vollmer V, Köster M, Grashoff M, Zawatzky R, et al., ER intrabody-mediated inhibition of interferon α secretion by mouse macrophages and dendritic cells. PLoS One. 2019;14(4):e0215062. doi:10.1371/journal.pone.0215062.
- Goulet DR, Atkins WM. Considerations for the design of antibody-based therapeutics. J Pharm Sci. 2020;109:74–103.
- Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 2017;122:2–19. doi:10.1016/j.addr.2016.11.004.
- Runcie K, Budman DR, John V, Seetharamu N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol Med. 2018;24(1):50. doi:10.1186/s10020-018-0051-4.
- Murer P, Neri D. Antibody-cytokine fusion proteins: a novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N Biotechnol. 2019;52:42–53. doi:10.1016/j.nbt.2019.04.002.