
ABSTRACT
Bioconjugates are an important class of therapeutic molecules. To date, O-glycan-based metabolic glycoengineering has had limited use in this field, due to the complexities of the endogenous O-glycosylation pathway and the lack of an O-glycosylation consensus sequence. Here, we describe the development of a versatile on-demand O-glycosylation system that uses a novel, widely applicable 5 amino acid O-glycosylation tag, and a metabolically engineered UDP-galactose-4-eperimase (GALE) knock-out cell line. Optimization of the primary sequence of the tag enables the production of Fc-based proteins with either single or multiple O-glycans with complexity fully controlled by media supplementation. We demonstrate how the uniformly labeled proteins containing exclusively N-azido-acetylgalactosamine are used for CLICK chemistry-based bioconjugation to generate site-specifically fluorochrome-labeled antibodies, dual-payload molecules, and bioactive Fc-peptides for applications in basic research and drug discovery. To our knowledge, this is the first description of generating a site-specific O-glycosylation system by combining an O-glycosylation tag and a metabolically engineered cell line.
INTRODUCTION
Bioconjugates are increasingly used in research, imaging, diagnostics, and drug development to improve potency, impart novel unique functionalities, and to simplify workflows.Citation1 Examples include antibody-drug conjugates (ADCs),Citation2 GalNAcylated-nucleic acids,Citation3 and peptide-Fc conjugates,Citation4 which are already used as therapeutic agents. A typical bioconjugate is composed of two bio-orthogonal components. The first is a scaffold that contains a reactive group that is either naturally present (e.g., native lysines and cysteines) or that has been engineered into the molecule (e.g., non-natural amino acids and transaminase tags). The second molecule must then be able to specifically react with the scaffold reactive group in such a way that it generates a bond and forms a bioconjugate molecule. The four main challenges in the discovery and development of this class of molecules are as follows: 1) site-specificity of conjugation, 2) product homogeneity, 3) efficiency of conjugation reaction, and 4) production yields.Citation5
Numerous strategies have been developed that co-opt the endogenous biosynthetic machinery to generate biomolecules containing bio-orthogonal functional groups that can be used for site-specific bioconjugation.Citation5 Such approaches exploit the endogenous pathways at three different levels: 1) transcriptionally by the expansion of the genetic code and codon reassignment;Citation6 2) translationally by the evolution of bio-orthogonal aminoacyl-tRNA synthetase/tRNA pairs,Citation7 which facilitate the incorporation of unnatural amino acids; and 3) post-translationally, by modifying glycosylation,Citation8 phosphorylation,Citation9 and lipidationCitation10 patterns, among others. Metabolic glyco-engineering (MGE) approaches (reviewed by Agatemor et al.Citation8) that manipulate cellular metabolism to generate functionalized glycoproteins have become an attractive method for the generation of bioconjugates because they bypass technical challenges of other techniques, such as the requirement for the transfection of multicomponent codon reassignment machinery for unnatural amino acid incorporation or complex post-production steps required by in vitro enzymatic post-translational approaches.
Of the many MGE approaches, the engineering of endogenous O-glycosylation pathways has been the least utilized, primarily due to the complexity and enzymatic redundancy of the intracellular pathwaysCitation8 that hamper the production of site-specifically modified proteins with homogeneous O-glycan profiles. To address this challenge, many groups have attempted to generate simplified expression systems through genome engineering,Citation11,Citation12 or by using lower eukaryoticCitation13 and bacterial cellsCitation14 that allow the user to better define the resulting glycoforms. However, such approaches require extensive engineering of the production host or only provide limited control over the type of glycans added.
O-glycosylation in eukaryotes is initiated by the covalent attachment of monosaccharides to serine or threonine residues in fully folded proteins, subsequent to N-glycosylation in the Golgi compartment.Citation15 To date, no well-defined O-glycosylation consensus sequence has been identified at the amino acid level, although several groups have demonstrated that high proline, serine and threonine content enhances O-glycosylation,Citation16 typified by sequences present in mucins termed variable number of tandem repeats (VNTR).Citation17 In so-called mucin-type O-glycosylation, the most common O-glycosylation in eukaryotic cells, O-glycan structures are formed by stepwise sequential monosaccharide condensations to an initiating N-acetylgalactosamine (GalNAc) sugar.Citation18,Citation19 The initiating GalNAc moiety (also referred to as the Tn antigen) in this pathway is typically further extended by the addition of either sialic acid (to form sialyl-Tn antigen) or sugars that can be further extended, such as galactose, GalNAc, GlcNAc, and fucose, to form more complex oligosaccharides.
The addition of initiating GalNAc to a protein is facilitated by a family of enzymes called the GalNAc transferases (GalNAc-T), which all accept UDP-GalNAc as a substrate and attach it to the hydroxyl group of either serine or threonine to initiate the carbohydrate chain.Citation20 In mammalian cells, UDP-GalNAc building blocks are generated by two pathways. The first is an endogenous hexosamine biosynthesis pathway from glucose, where UDP-GalNAc is generated from UDP-GlcNac by the last enzyme of the Leloir pathway, called UDP-galactose 4-epimerase (GALE).Citation21 The second pathway, often referred to as the salvage pathway, uses externally imported GalNAc, which is then enzymatically processed into UDP-GalNAc by the sequential action of galactokinase-2 (GALK2) and UDP-GalNAc pyrophosphorylase (UAP1). Thus, by knocking out GALE, a cell can be made reliant solely on the salvage pathway for UDP-GalNAc production. This could be exploited for the ‘on demand’ metabolic labeling of natural and engineered O-glycoproteins through restricting externally supplied sugars and is feasible since some engineered GALE knock-out (KO) cell lines have already been shown to function without mucin-type O-glycosylation.Citation22–25
Use of mucin-type O-glycan labeling is particularly attractive for two reasons: 1) the use of GalNAc in other glycosylation pathways is limited (for example, only complex mammalian N-glycans, proteoglycans, and glycolipids contain GalNAc); and 2) the salvage pathway for metabolizing functionalized derivatives of GalNAc, such as N-azido-acetyl-galactosamine (GalNAz), is permissive, enabling the introduction of bioorthogonal handles for streamlined downstream conjugation methodologies, such as CLICK-chemistry.Citation26,Citation27 Although methods for the introduction of natural and modified O-glycans into proteins have been described,Citation16 they do not allow control over the location of O-glycosylation, the number of O-glycosylation sites and complexity of the O-glycans added, or high-efficiency protein production.
Here, we demonstrate that, by using a combination of a metabolically engineered GALE KO expression host and a novel, transferable O-glycosylation tag, we can efficiently produce homogeneous and site-specifically O-glycan-modified, ‘conjugation-ready’ recombinant proteins and give diverse examples of their application. Since our system uses exogenous GalNac and galactose for full O-glycosylation in GALE KO cells and facilitates cross-link conjugation to unnatural sugars, we refer to this technology as GALaXy.
MATERIALS AND METHODS
Metabolic labeling of O-glycans
For metabolic labeling experiments, O-glycan precursors N-acetylgalactosamine (GalNAc) or N-azidoacetylgalactosamine-tetraacylated (Ac4GalNAz) (Sigma-Aldrich or Click Chemistry Tools) were added to cell culture media at a final concentration between 50 and 200 µM. Where appropriate, an additional supplementation of cell culture media with galactose was implemented at a final concentration of 10–100 µM. Supplements were added to cell culture media either at the time of transfection or three to five hours prior to transfection. HEK293F and HEK293F GALE KO cells were cultured in FreeStyle™ 293 expression media (Thermo Fisher Scientific) in the presence of supplements for 6 days. On day 3 of the incubation an equal volume of cell culture media containing 2X concentration of metabolic supplements was added. In the case of cell lines that require serum (CHO K1), cells were serum-starved for 3 hours, thoroughly washed with phosphate-buffered saline (PBS) and placed into serum-free media prior to labeling.
CRISPR GALE KO cell line generation
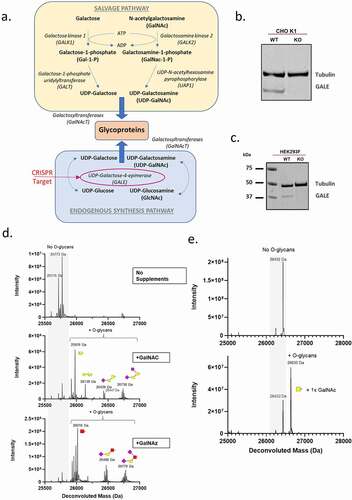
Guide RNAs (sgRNA) targeting the GALE gene were designed using the ATUM CRISPR gRNA Design Tool and cloned into a dual ORF U6-sgRNA/CMV-Cas9-2A-RFP expression vector (pD1301-AP; ATUM). The sequence of the three 20 base pair sgRNAs was as follows: GALE_Cr-1: 5ʹ-CCACACGGTACTGGAGCTGC(NGG)-3ʹ (Exon2), GALE_Cr-2: 5ʹ- GTACTGGAGCTGCTGGAGGC(NGG) – 3ʹ (Exon2) & GALE_Cr-3: 5ʹ-CGGCGGGTCCAGGAACTGAC(NGG) – 3ʹ (Exon3). GALE_CR-1, −2 & 3 target the CHO GALE gene, GALE_Cr-2 & −3 target human GALE. Briefly, gRNAs were co-transfected into CHO K1 cells (GALE_Cr-1, −2 & 3) or HEK293F cells (GALE_Cr-2 & −3) using Lipofectamine2000 (Thermo Fisher Scientific) per manufacturer’s recommendations. RFP-positive cells were sorted into single-cell clones via FACS using a BD FACSAria™ II cell sorter (BD Biosciences). Single-cell clones were then screened for GALE KO by western blot analysis. To simplify KO generation in the HEK293F suspension cell line, the cells were first serum-adapted to adherent culture, transfected as above, screened for knockout, and positive clones were re-adapted to serum-free culture conditions in FreeStyle™ 293 expression media (Thermo Fisher Scientific).
Analysis of de-glycosylated intact mass using LC-MS
The incorporation of O-glycans was assessed using LC-MS. Samples were diluted with water and 10 mM TRIS pH 8.0 to 1 mg/mL and de-N-glycosylated overnight at 37°C using 2 μg of PNGase F (Roche) per 100 μg of protein. Samples were then reduced for 30 min at 37°C using 10 mM dithiothreitol (Pierce). The proteins were analyzed using Acquity system (Waters) and reverse ultra-high performance liquid chromatography (Waters) coupled to either a QToF Synapt G2 (Waters) or Orbitrap QExactive HFX (Thermo). For each chromatographic run 0.5– 2 μg of the protein was loaded onto Acquity C4 BEH column 1.7 µm, 2.1 mm x 50 mm (Waters). A linear gradient from 25% to 45% of acetonitrile with 0.1% formic acid and 0.01% of trifluoroacetic acid at a 150 μL/min was applied. Total runtime, including column wash and equilibration, was 22 min. Mass spectra were acquired at 500–4500 m/z range in the positive ion mode. Protein mass spectra were deconvoluted using either MassLynx or BiopharmaFinder software.
Cloning and construct design
Codon optimized gene inserts were designed and synthesized at GeneArt gene synthesis (Thermo Fischer Scientific). The pBAMF expression vector based on the pDest12.2 vector (Thermo Fisher) was generated via gene synthesis and sub-cloning to encode the origin of the viral DNA replication (OriP) sequence and the viral Epstein-Barr nuclear antigen 1 (EBNA1) protein under the control of the simian virus 40 (SV40) promoter. Gene inserts were cloned between the SbfI and NheI sites encoded in the modified vector.
pEP-Fc expression vector was generated by sub-cloning of gene fragments that encode CMV promoter, polyadenylation site, ampicillin resistance gene, bacterial origin of replication, and the origin of EBV replication (oriP) to facilitate expression of Fc-based secretory proteins in EBNA1-producing mammalian cells. Genes encoding the Fc-based proteins were synthesized and/or amplified by PCR and cloned using unique restriction enzyme sites NgoMIV and EcoR1 in frame with the modified immunoglobulin leader signal peptide to facilitate entry to the ER/Golgi and secretion.
Cell culture
Adherent CHO K1 and CHO K1 GALE KO cells were cultured in Ham’s F-12 nutrient mixture supplemented with 10% FCS, 8 mM L-GlutaMax (Invitrogen). Suspension CHO G22 cells were cultured in MedImmune-proprietaryCitation28 serum-free medium supplemented with 6 mM l‐glutamine and methionine sulfoxamine (MSX; Sigma‐Aldrich). HEK293F suspension cells and HEK293F GALE KO cells were cultured in FreeStyle™ 293 expression media (Thermo Fisher Scientific).
NS0 cells were cultured in EX-CELL NS0 Serum-Free Medium (Sigma). Adherent cells were incubated at 37°C, 80% humidity, 5% CO2. Suspension cells were incubated at 37°C, 80% humidity, 5% CO2, 140 RPM in shaker flasks.
Lentiviral transductions
Lentiviral construct pCDH-CMV-Fc-GALaXy-IRES-Puro – Fc protein with a C-terminal GALaXy tag (CD532A vector; System Biosciences) was co-transfected with pPackH1 lentiviral packaging mix (System Biosciences) into HEK293 T cells using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s protocol. After 48 h, viral particles were harvested. NS0, CHO K1, and CHO K1 GALE KO cells were infected with the Fc-GALaXy-expressing lentivirus in the presence of 8 μg/mL polybrene. Fc-expressing cell pools were selected using 5 µg/ml (NS0) and 10 µg/ml (CHO) of puromycin and used for subsequent protein expression studies.
Transfections and mammalian cell expression
All reagents were obtained from ThermoFisher unless otherwise stated. Transfections of adherent cells were performed at 80% confluency using Lipofecamine2000 as per the manufacturer’s protocol. HEK293F cell lines were seeded at 1 × 106 cells/ml and transfected with a final DNA concentration of 0.67 µg/ml using 293 Fectin transfection reagent. 293 Fectin:DNA complexes were prepared in OptiMEM at a 3:2 ratio prior to addition to the cells. Following transfection, cells were cultured at 37°C with rotation. Cells were fed by the addition of an equal volume of FreeStyle™ 293 expression media (Thermo Fisher Scientific) on day 3 of the incubation and harvested on day 6. For CHO G22 transfections, cells were seeded at a 4 × 106 cells/ml and transfected with a final DNA concentration of 1 µg/ml using linear polyethylenimine (PEI; Polysciences Europe). PEI:DNA complexes were prepared in 150 mM NaCl at a 5:1 ratio prior to addition to the cells. Following transfection, cells were cultured for up to 6 days at either 37°C or 34°C with the addition of MedImmune-proprietary nutrient supplement containing no GalNac or galactose. Cell culture media containing secretory proteins were harvested by centrifugation at 1500 g for 15 to 30 min followed by filtration using 0.2 µm filters.
Western blot analysis of total cell lysates
Cells were lysed in RIPA buffer containing cOmplete™ Protease Inhibitor Cocktail (Roche, Germany), and protein concentrations were determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Protein lysates were mixed with LDL-loading buffer and reducing agent (Invitrogen) 30 µg total protein sample per well was separated by SDS-PAGE, transferred onto a nitrocellulose membrane (Invitrogen), and probed with rabbit pAb UDP-glucose 4-epimerase (HPA007340, Prestige Antibodies®, Sigma-Aldrich) and mouse mAb α-tubulin (clone DM1A, Cell Signaling Technology). An IRDye® 800CW-labeled goat anti-rabbit and IRDye® 680RD-labeled donkey anti-mouse antibodies (both at 1:150,000) (LI-COR Biosciences) were used for detection, and blots were analyzed with the Odyssey imaging system (LI-COR Biosciences).
Protein purification and analysis
Harvested cell culture media containing secretory Fc-based proteins were purified by Affinity chromatography using MAbSelect SuReTM columns (GE Healthcare) and AKTA system (GE Healthcare). Proteins were eluted from the column using 0.1 M Glycine pH 2.7, neutralized with 10% volume 1 M Tris pH 9 and buffer exchanged to PBS using desalting columns PD-10 or dialysis. Yield of protein expression in culture medium, protein purity and efficiency of conjugation reactions were routinely assessed by SDS-PAGE using 4–12% NuPAGE Bis-Tris gels (ThermoFisher) in MES buffer and staining using InstantBlue (Expedeon).
GLP-1 cell-based assay
A cell-based cAMP accumulation assay was used to assess the activity of molecules at the GLP-1 receptor using a previously described CHO cell line expressing the human GLP‐1 receptor.Citation29 Dilutions of test molecules were prepared in duplicates in an assay buffer composed of HEPES-buffered saline solution; 25 mM HEPES, 0.5 mM IBMX and 0.1% bovine serum albumin, pH 7.4, using an acoustic liquid dispenser.Citation30 Cells were stimulated with different concentrations of compounds for 30 minutes, after which the cAMP accumulation was measured using the CisBio dynamic d2 cAMP HTRF assay kit (CisBio, Codolet, France) according to the manufacturer’s guidelines.
Computer modeling
The Fc of trastuzumab was modeled in three different forms: 1) wt (trastuzumab without GALaXy tag), 2) with a GALaXy tag (amino acid sequence: PTAEPG) inserted in silico at position N380 without any linkers, and 3) with both a GALaXy tag and a linker (amino acid sequence: GGGGSGGGGSGPTAEPGGGGGSGGGGS) inserted in silico at position N380. The template used is the crystal structure of a full-length human IgGCitation31 (PDB ID: 1HZH). The modeling was generated using the Energy-based modeling panel from Schrödinger suite.Citation32,Citation33 The visualization and analysis of the model was done in Schrödinger and VMD.Citation34
FACS analysis
SKBR3 and NCI-N87 cells were maintained in RPMI supplemented with 10% FBS (Gibco). Prior to staining, cells were harvested using an Accutase® solution (Merck; as per manufacturer’s instructions), counted and a minimum of 2 × 105 cells were used for each FACS staining. Fc receptors were blocked for 10 mins using Human TruStain FcX™ (Fc Receptor Blocking Solution; Biolegend). Cells were resuspended in PBS and incubated with the indicated antibodies at 10 µg/ml for 30 mins on ice. Unstained cells and an irrelevant IgG control antibody were used as staining controls. Cells were washed three times after antibody incubation and re-suspended in cold PBS containing 1% v/v FBS for analysis using an LSRFortessa™ Flow Cytometer (BD Biosciences).
CLICK chemistry bio-conjugation
CLICK chemistry conjugation of DBCO-reagents (Click Chemistry Tools or in-house synthesis) to GalNaz-containing Fc-based proteins was carried out at a protein concentration of 0.5–5 mg/ml with a molar excess of DBCO-reagent to GalNaz between 3 and 10-fold. Protein samples were prepared in PBS, while DBCO-reagents were either dissolved in water or in dimethylsulfoxide (DMSO). Reactions were carried out at room temperature or at 25°C for 16–24 hours. Unreacted DBCO-reagents were removed by either a PD-10 desalting column or by affinity chromatography purification of the Fc-based bio-conjugate using MAbSelect SuReTM (GE Healthcare) column, followed by buffer exchange using PD-10.
Maleimide bio-conjugation
Conjugation reactions were performed at the protein concentration of 0.5 mg/ml. Prior to the conjugation reaction, the disulfide bonds were reduced, by addition of 40-fold molar excess of TCEP Bond Breaker 500 mM (Sigma) for 1 h at 37°C followed by re-buffering to PBS 1 mM ethylenediaminetetraacetic acid using a desalting column MidiTrap G-25. This was followed by oxidation at 25°C for 60 minutes with 20-fold molar excess of 100 mM dehydroascorbic acid in DMSO. The 20 mM maleimide- reagent in DMSO – DyLight488 Maleimide (Thermo Fisher Scientific) was added at the molar ratio of 10:1 (maleimide reagent: antibody) and the conjugation reaction was carried out for 2 hours at 25°C. The reaction was quenched for 15 min at 25°C with a 4-fold molar excess over the maleimide reagent using 100 mM N-acetyl cysteine. Bioconjugates were separated from unreacted fluorochrome label and buffer exchanged to PBS using PD-10 column
RESULTS
Discovery of an O-glycosylation motif from thymosin β-4
Thymosin β-4 (TB4) is a 43 amino acid intracellular cytoplasmic peptide that has been shown to play a role in the repair and regeneration of injured tissues,Citation35,Citation36 and therefore, has attracted attention as a therapeutic agent. The short serum half-life of TB4, however, limits its use as a therapeutic molecule. In order to extend the half-life of TB4, we expressed a variant of TB4 as a secreted fusion protein composed of the TB4 peptide linked to an Fc from human IgG1 (TB4-Fc) (, ). Liquid chromatography-mass spectrometry (LC-MS) analysis of the recombinant fusion protein unexpectedly showed two distinct species (). The first species was consistent with the expected reduced de-N-glycosylated mass of the TB4-Fc fusion protein of 30,838 Da, whilst the second species was 656 Da larger and consistent with addition of an O-glycan composed of one hexose, one N-acetylated-hexose and one sialic acid (NeuAc). Subsequent peptide mapping experiments (data not shown) confirmed the O-glycan addition and its location on Serine 30 (S30) within the TB4 peptide.
Table 1. Identification of a ‘minimal’ transferable motif for GalNAc O-glycosylation (GALaXy tag) in the context of GLP1-TB4 chimera – Fc fusions
Figure 1. Identification of an O-glycosylation motif in human thymosin beta 4 (TB4). (a) Schematic overview of the approach used to express TB4-fusion proteins with TB4 peptide sequence. Serine30 marked in red is a target for O-glycosylation. (b) LC-MS reduced mass analysis of de-glycosylated TB4-Fc fusion protein. Grey shaded region of the spectrum indicates non-glycosylated protein species (No O-glycans) The mass shift of 656 Da corresponds to addition of GalNac1Gal1NeuAc1. Peaks right-shifted from gray shading are O-glycosylated protein species (+O-glycans). (c) Mass spectrometry analysis of TB4 mutants that either abolish (A30 PLPAKET; top histogram) or promote full (T30 PLPTKET; bottom histogram) O-glycosylation of TB4 sequence. The expected mass shift of 656 Da (31,508 Da peak) corresponds to addition of GalNac1Gal1NeuAc1 and additional shift of 291 Da (31,799 Da peak) corresponds to addition of GalNac1Gal1NeuAc2 (d) Mass spectrometry analysis of GLP1-Fc (top panel) and GLP1Chimera1-Fc protein containing GLP1 peptide fused to a portion of TB4 T30 (bottom panel). Transposition of the mutant TB4 sequence (T30) into GLP1-Fc fusion protein resulted in an O-glycosylated protein. The non-glycosylated reduced protein with a theoretical mass of 31,720 Da was not detected. The mass shift of the 33,033 Da peak corresponds to the second O-glycosylation site (GalNac1Gal1) and the following peaks 33,324 Da and 33,615 Da correspond to addition of NeuAc molecules: GalNac1Gal1NeuAc1 and GalNac1Gal1NeuAc2, respectively. The symbolic representation of O-glycans is based of the most common O-glycan core-1. Yellow square = GalNAc, yellow circle = Gal, pink diamond = NeuAc
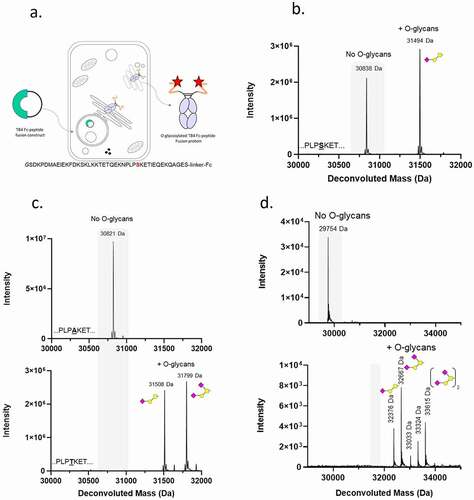
To confirm that S30 was the site of O-glycosylation, it was mutated to either an alanine (non-glycosylated residue) or threonine (commonly O-glycosylated residue) and the mutated proteins were expressed and purified. As expected, the substitution to alanine (TB4 A30 – Fc) completely abolished O-glycosylation of the recombinant protein. Substitution of S30 with threonine (TB4 T30–Fc) resulted in a protein in which 100% of the threonine residues at position 30 of the TB4 peptide were occupied with O-glycans. Two main species of 31,508 Da and 31,799 Da were observed, consistent with the masses of GalNAc-Gal-NeuAc or GalNAc-Gal-NeuAcx2, respectively (, ).
Generation of a widely applicable O-glycosylation tag
Our results showed TB4 T30 sequence could efficiently direct O-glycosylation in the context of the TB4 T30-Fc protein, which raised two key questions: 1) Could the motif be transferred to other recombinant proteins such that it would direct site-specific O-glycosylation, and 2) Could the motif be shortened and optimized to generate a discrete, widely applicable O-glycosylation tag?
To address the first question, a portion of the TB4 sequence (amino acids 25–43) encompassing the T30 O-glycosylation site was fused to a glucagon-like peptide-1 (GLP1) variant to generate a series of chimeric molecules (). The GLP1 peptide, similarly to the TB4 peptide, can be expressed as an N-terminal Fc fusion in mammalian cells, but unlike TB4-Fc, GLP1-Fc is not O-glycosylated (, top panel). LC-MS evaluation of the recombinant GLP1Chimera 1-Fc (TB4 aa 25–43) showed that the transfer of amino acids 25–43 of TB4 T30 led to complete O-glycosylation, demonstrating the transferability of the TB4-derived O-glycosylation motif ( and ).
To define the minimum, optimal sequence for this transferable O-glycosylation motif, we performed sequential deletional and mutational analysis of the peptide sequence as chimeric peptide-Fc proteins () starting with C-terminal truncations of the peptide contained in GLP1Chimera 1-Fc (TB4 aa 25–43) to create GLP1Chimera 2-Fc (TB4 aa 25–38) bearing amino acids 25 to 38 of TB4 T30. All subsequent truncations and mutations of the TB4 T30 sequence were performed in the context of the same linker as in GLP1Chimera 2 -Fc (). The first four chimeric proteins, GLP1Chimera 1-Fc (TB4 aa 25–43) to GLP1Chimera 4-Fc (TB4 aa 25–33), retained high levels of O-glycosylation (>90%), while further truncations of the region from either the C-terminus or N-terminus reduced total O-glycosylation level to 71% for GLP1Chimera 5 -Fc (TB4 aa 25–32), 60% for GLP1Chimera 6-Fc (TB4 aa 27–32) and 0% for GLP1Chimera 7-Fc (TB4 aa 29–32). Introducing an additional proline, which can act as an O-glycosylation promoting amino acid, at the end of the GLP1Chimera 6 peptide at position +3 in relation to T30 (GLP1Chimera 9-Fc) restored the high O-glycosylation levels that were previously observed for chimeras bearing a longer motif. Further to this observation, we noted that removing a positive charge from the position adjacent to T30 and substituting lysine 31 for alanine increased O-glycosylation in GLP1Chimera 8-Fc to 93% when compared to GLP1Chimera 6-Fc (TB4 aa 27–32) at 60%. The combination of these two changes (K31A and +3P) rescued the O-glycosylation deficient phenotype of GLP1Chimera 7-Fc (TB4 aa 29–32), giving rise to a fully O-glycosylated variant GLP1Chimera 11-Fc bearing a minimal 5 amino acid tag (PTAEP) attached to the C-terminus of the GLP1 sequence.
Table 2. Optimization of the GALaXy tag and assessment of the impact of adjacent residues on the levels of secondary O-glycosylation
Secondary O-glycan addition at threonine and serine residues adjacent to the primary O-glycosylation site are commonly observed in O-glycoproteins. shows the effect of truncations (chimeras 1 to 7) and mutations of residues adjacent to T30 (chimeras 8 to 13) on inducing secondary O-glycosylation events in a context of a linker containing both serine and threonine. Among all the variants analyzed, the one containing a PTAEP motif followed by the linker GSTASSGSGSATGGGGAA (GLP1Chimera 11-Fc) provided the strongest signal for promoting multiple O-glycosylation events, with 100% of molecules containing 3 O-glycans, which could be linked to T30 and either of the two additional serines and threonines in the linker. Changing the terminal proline of the PTAEP motif to alanine (PTAEA; GLP1Chimera 12 – Fc) shifted the O-glycosylation pattern in favor of a single O-glycosylation site. From this series of experiments, we determined the O-glycosylation motif contained within GLP1Chimera 11-Fc, PTAEP to be the minimal peptide motif required for high occupancy and reproducible O-glycosylation ( and ).
Next, we proceeded to test if this short peptide could be used as a tag fused to the C-terminus of an Fc without a GLP1 peptide or the linker, to provide greater utility in a context of a monoclonal antibody (mAb). We hypothesized that, in the absence of additional adjacent serine or threonine residues, only the primary T30 site would become O-glycosylated. As shown in , a fusion of the PTAEP motif, referred to from now on as the GALaXy tag, to the C-terminus of the CH3 domain, resulted in an efficient fully glycosylated protein containing a single O-glycosylation site.
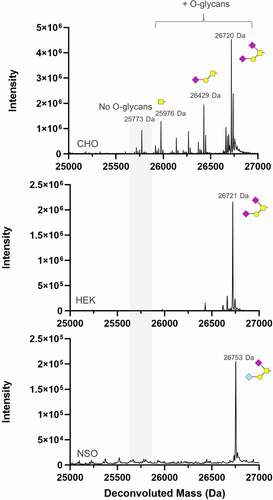
Figure 2. Assessment of GALaXy tag O-glycosylation in common expression hosts. An Fc protein containing a C-terminal GALaXy tag fused to human IgG1 Fc as a polypeptide GAGPTAEPG (Fc-GALaXy) was expressed in three commonly used expression cell lines: Chinese hamster ovary CHO G22, human embryonic kidney HEK293F and a mouse myeloma NS0 cell line. LC-MS reduced mass analysis shows de-N-glycosylated Fc-GALaXy proteins produced in CHO (top), HEK (middle) and NS0 (bottom) cells lines. The gray shaded region of the spectrum indicates non-glycosylated protein species (No O-glycans). Peaks right-shifted from gray shading are O-glycosylated protein species. The symbolic representation of O-glycans is based of the most common O-glycan core-1; yellow square = GalNAc, yellow circle = Gal, pink diamond = NeuAc, light blue diamond = NeuGc
Expression of GALaXy tag in mammalian cell lines
Having defined and optimized our transferable GALaXy tag, we next sought to access the general applicability and functionality of the GALaXy tag in a variety of expression hosts commonly used for manufacturing antibodies and protein-based molecules.
Fc molecules with a C-terminal GALaXy tag (Fc-GALaXy) were expressed in either Chinese hamster ovary (CHO) or human embryonic kidney (HEK) 293 F cells transiently transfected with plasmids, or mouse NS0 cells transduced with lentivirus (). LC-MS analysis demonstrated that transient expression in CHO cells yielded Fc proteins with glycoforms consistent with the addition of either GalNAc (Tn), GalNAc1Gal1, GalNAc1Gal1NeuAc1, or GalNAc1Gal1NeuAc2. The dominant O-glycan attached to the PTAEP motif was GalNAc1Gal1NeuAc2. Only small amounts (<10%) of CHO-produced protein remained O-glycan free. Mouse NS0 cells also yielded highly O-glycosylated Fc protein containing O-glycans differing from the CHO-expressed material due to the presence of additional Gal-alpha-Gal or glycolylneuraminic acid (Neu5Gs)Citation37–38 glycoforms. The mass shift observed for the NS0 O-glycan is consistent with either GalNAc1Gal1(alpha-Gal)2-NeuAc or GalNAc1Gal1NeuAc-Neu5Gs, but the structure of the glycan has not been confirmed. The glycoforms produced in HEK293F cells showed only two species: GalNAc1Gal1NeuAc and GalNAc1Gal1NeuAc2. The differences in the glycosylation patterns observed between proteins expressed by the three different species were as expected and have been demonstrated previously.
Controlling O-glycosylation via metabolic engineering of expression cell lines using CRISPR genome editing
Having developed a simple 5 amino acid tag for site-specific O-glycosylation, we next sought to investigate a method to control the timing and nature of the sugars added to the tag. The endogenous synthesis pathway of the O-glycan precursor, UDP-GalNAc, is governed by the enzyme GALE and we hypothesized that the deletion of endogenous GALE by CRISPR-mediated genome engineering would render the cells dependent on exogenous sugars from the salvage pathway to perform O-glycosylation (). To assess this, we generated GALE KOs in both CHO K1 and HEK293F cells using guide RNAs designed to target exon 2 of the GALE gene of CHO and HEK cells. Clonal CRISPR-edited cells from both CHO K1 and HEK293F cells were isolated via fluorescence-activated cell sorting (FACS), and GALE KO was confirmed by Western blot ( and C, respectively). Transfection of the adherent CHO K1 cells with plasmid DNA is inefficient; therefore, to achieve high levels of protein expression, a pool of CHO K1 GALE KO cells overexpressing an Fc-GALaXy protein was generated via lentiviral transduction and subsequent selection with puromycin. These cells were then adapted to serum-free media supplemented with either GalNAc (the natural sugar substrate used by the salvage pathway) or Ac4GalNAz (a cell-permeable azido-containing derivative of GalNAc). As a control, media without sugar supplementation was used (). In the absence of supplements, the Fc-GALaXy proteins produced in CHO K1 GALE KO cells were free of O-glycans as expected, while upon supplementation with either GalNAc or GalNAz the parental pattern of complex branched O-glycosylation was restored ( and ).
Figure 3. Developing an on-demand O-glycosylated protein expression system. (a) Schematic of the pathways of galactose and GalNAc metabolism. Disruption of GALE enzyme via CRISPR (indicated) disrupts endogenous synthesis pathway. (b, c) Western blot analysis of GALE expression (lower bands) in CHO K1 (b) or HEK293F (c) parental cells (Wt) or CRISPR GALE KO (KO) cells. Alpha-tubulin (upper bands) is included as a loading control. (d) LC-MS analysis of Fc-GALaXy proteins expressed in CHO K1 GALE KO cells in the absence of sugar supplementation (top histogram), supplemented with 100 µM GalNAc (middle histogram) or supplemented with 100 µM GalNAz (bottom histogram). (e) LC-MS analysis of Fc-GALaXy proteins expressed in HEK293F GALE KO cells without GalNAc supplementation (top histogram) or with 100 µM GalNAc (bottom histogram). A mass shift of 203 Da corresponds to addition of GalNAc1 (f) LC-MS analysis of Fc-GALaXy proteins expressed in HEK293F GALE KO cells in the absence of sugar supplementation (top histogram) or with 100 µM GalNAz (middle histogram). The bottom histogram shows the mass spectrometry analysis of the GalNaz-labeled Fc-GALaXy protein after its conjugation with DBCO-AlexaFluor488 by CLICK chemistry. (g) LC-MS analysis of Fc-GALaXy protein containing the multi-tag fused to CH3 -GAGPTAEPGSTASSGSGSATGGGGA (permits multiple O-glycosylations within the same tag) expressed in HEK293F GALE KO cells in the absence of sugar supplementation (top histogram) or with 100 µM GalNAc (bottom histogram). Numbers on peaks indicate the number of GalNAc molecules added. (h) LC-MS analysis of the heavy chain of IgG-GALaXy proteins expressed in HEK293F GALE KO cells in the absence of sugar supplementation (top histogram), with 100 µM GalNAc only (middle histogram) or with 100 µM GalNAc and galactose (bottom histogram). For all LC-MS analysis samples are reduced and de-N-glycosylated. Grey shaded region of the spectrum indicates non-glycosylated protein species (No O-glycans). Peaks right-shifted from gray shading are O-glycosylated protein species (plus O-glycans). The symbolic representation of O-glycans is based of the most common O-glycan core-1. Yellow square = GalNAc, yellow circle = Gal, pink diamond = NeuAc, red square = GalNAz
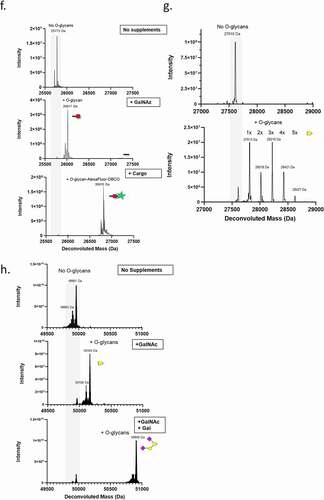
In contrast to CHO K1 GALE KO cells, the knockout of GALE in HEK293F () resulted in cell clones that, upon supplementation with GalNAc, incorporated only a single GalNAc monosaccharide (). This was unexpected as the parental cells produced analogous proteins with extended O-glycan structures incorporated at the tag site (). Interestingly, when the HEK293F GALE KO cells were supplemented with GalNAz, the azido-modified sugar was also incorporated into the tag as a single GalNAz monosaccharide (). This observation raises the possibility of generating homogeneously labeled proteins containing azido functional groups that would provide a chemical handle for subsequent downstream reactions.
To demonstrate this, the Fc-GALaXy GalNAz protein produced from HEK293F GALE KO cells was subjected to a CLICK chemistry reaction with DBCO-functionalized AlexaFluor488 (AF488), and the product was assessed by LC-MS analysis. Conjugation efficiency was assessed by the appearance of a peak with a mass of 26,810 Da, consistent with a reduced de-N-glycosylated Fc-GALaXy-GalNAz-DBCO-AF488 product. As can be seen from , the conjugation of AF488 to the Fc protein was highly efficient, yielding almost complete conversion to a homogeneous bioconjugate.
To investigate if the single GalNAc addition could be applied to multiple O-glycosylation events, we compared Fc proteins containing a C-terminal GALaXy tag to a version extended by a linker containing additional potential O-glycosylation sites termed the ‘multi-tag’ PTAEPGSTASSGSGSATGGGGA (). As expected, protein expressed in media supplemented with GalNAc exclusively contained monosaccharide additions. When using this multi-tag version, we also observed the addition of Tn antigens at up to 5 separate glycosylation sites with no complex sugars or branching present at any of the glycosylation sites ().
One explanation for the absence of more complex O-glycans from the recombinant proteins produced in HEK293F GALE KO is an insufficient substrate supply. As shown in , the dominant type of O-glycans observed in HEK293F contains GalNAc as well as galactose, which are both dependent on the GALE enzyme for conversion to O-glycan building blocks (Figure S1 details O-glycan building blocks). The HEK293F GALE KO cells generated in this study are grown in defined media lacking both of these sugars. To test whether supplying both sugars would restore the observed parental cell-line O-glycosylation pattern, we transfected HEK293F GALE KO cells with a construct encoding an IgG molecule with a GALaXy tag at the C-terminus of the heavy chain (IgG-GALaXy) and expressed this protein in the presence of GalNAc alone (100 µM final), a combination of GalNAc and galactose (100 µM final of each) or with no supplements. As demonstrated by LC-MS analysis, in the absence of supplemented sugars the IgG-GALaXy heavy chain lacked O-glycans () and upon addition of GalNAc, the heavy chain was labeled with a single GalNAc moiety (Tn antigen) as expected. However, when the IgG-GALaXy protein was expressed in the presence of both GalNAc and galactose, the parental O-glycosylation phenotype was completely restored, with the heavy chain exhibiting O-glycans consistent with the glycoforms seen on GALaXy-tagged proteins expressed in the parental HEK293F cells ().
Positional dependency of the GALaXy tag
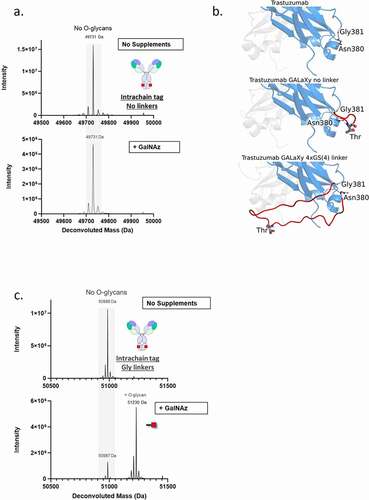
To test the effect of the position and accessibility of the GALaXy tag on the efficiency of O-glycosylation, we compared IgGs with either a C-terminal or intrachain GALaXy tag (incorporated into the CH3 domain at position N380). While the C-terminal tag facilitated efficient O-glycosylation (), no labeling was observed when the GALaXy tag was placed at the intrachain position of the IgG molecule (). The lack of labeling of the GALaXy tag when buried within the structure of the CH3 domain implied that accessibility of the GalNAc-T enzymes at the site could have been restricted. In silico modeling of the Fc CH3 domain with and without the GALaXy tag demonstrated that the threonine residue, which is the target for O-glycosylation within the tag sequence, lies in close proximity to the Fc backbone, which potentially hinders the accessibility of cellular glycosyltransferases to the site. We reasoned that the addition of a flexible linker flanking the tag could lead to higher sugar incorporation rates by increasing accessibility to the threonine residue (). To test this, we generated an IgG molecule with a CH3 GALaXy tag flanked on either side by an 11–12 amino acid linker (composed of glycines and serines or poly-glycine). The addition of flexible linkers significantly improves labeling efficiencies, yielding proteins that were ~82% O-glycosylated (), indicating that accessibility to the O-glycosylation site is critical to achieve high sugar incorporation rates (see supplemental Figure S2 for sugar structures).
Figure 4. Effect of tag accessibility on O-glycosylation efficiency. (b). Model of the structure of the CH3 domain of human IgG1 containing no tag (top), GALaXy tag with no linkers (middle) and GALaXy tag with G4S linker (bottom). Tag insertions were modeled between Asn380 and Gly381. The threonine residue representing the site of O-glycosylation is marked. In the absence of a linker the tag threonine residue lies in close proximity to the Fc backbone. The addition of a flexible linker makes the threonine more exposed and accessible for glycosylation reactions. LC-MS analysis was performed on IgG proteins expressed in HEK293 F GALE KO cells, containing a GALaXy tag inserted within CH3 domain without a linker(a) or flanked by 11 and 12 amino acid glycine linkers (c), where following medium supplementation a mass shift of 243 Da was observed corresponding to incorporation of GalNAz Proteins were expressed either in the absence of sugar supplementation (top histograms) or with 100 µM GalNAz (bottom histograms). For all LC-MS analysis only heavy chain is shown. Grey shaded region of the spectrum indicate non-glycosylated protein species (No O-glycans). Peaks right-shifted from gray shading are O-glycosylated protein species (plus O-glycans). GalNAz is represented by the red square
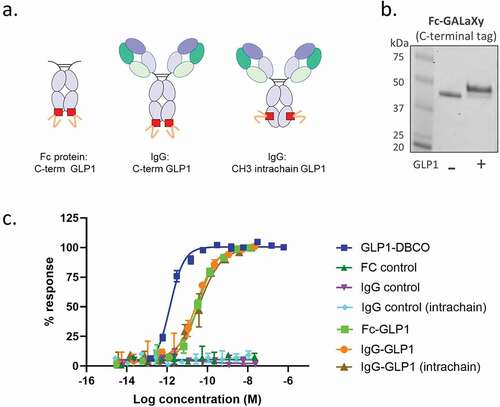
Having observed the importance of tag positioning on sugar incorporation, we next sought to understand whether positioning would affect the activity of payloads attached at these sites. A DBCO-functionalized GLP-1 mimetic peptide was conjugated to either a human Fc-GALaXy (C-terminal GALaXy tag) or a human IgG-GALaXy (C-terminal and intrachain plus linker; see ) generated using the HEK293F GALE KO cells with GalNAz supplementation. The efficient CLICK chemistry conjugation of the peptide to the Fc could be visualized by SDS-PAGE where a higher molecular weight product, consistent with the size of an Fc-peptide conjugate, could be observed (). Peptide conjugates were tested for GLP1 receptor agonism in a cell-based cAMP accumulation assay using cells overexpressing the GLP1 receptor. All bioconjugates retained agonistic activity and induced maximal increases in cAMP levels comparable to unconjugated peptide controls (). The rightward shift in the dose response curves observed for conjugated molecules, indicating an increased EC50, was expected and has been observed before for peptide-conjugates of this class.
Figure 5. Peptides conjugated via modified O-glycans at different positions retain bioactivity. (a) Schematics of GLP1 peptide-conjugates for use in GLP1 cell-based bioactivity assays. (b) SDS-PAGE analysis of Fc-GALaXy before (- GLP1) and after (+ GLP1) CLICK chemistry conjugation with DBCO-modified GLP1 peptide. (c) Representative concentration‐response curves for either GLP1-DBCO, GALaXy proteins (Fc control, IgG control, IgG control intrachain) or GALaXy conjugates – either Fc-GLP1 or IgG-GLP1 (either C-terminal or intrachain conjugation) in cAMP accumulation assays in CHO cell line expressing human GLP‐1 receptors. Plotted values are mean (±SEM) from duplicate analysis fitted with 4‐parameter logistic fit. Data shown representative of n ≥ 3 experiments
Using the GALaXy tag for site-specific conjugation
Having demonstrated that the GALaXy system is a simple and efficient conjugation platform for recombinantly expressed secretory proteins, we next looked to expand the capabilities of the technology. Combining the CLICK chemistry-based GALaXy system approach with maleimide-based conjugation would provide a facile way to generate site-specifically labeled, dual-payload proteins, thereby increasing the potential for generating molecules with novel and/or improved functionalities. To test this approach, we generated an anti-human epidermal growth factor receptor 2 (Her2) IgG molecule based on trastuzumab containing both a free cysteine inserted at position 239 in CH2 domain and a C-terminal GALaXy tag (IgG-Cys-GALaXy). The IgG-Cys-GALaXy protein was expressed in GalNAz-supplemented HEK293F GALE KO cells and LC-MS analysis confirmed a high-level of GalNAz incorporation (). Maleimide conjugation to cysteines requires partial reduction of the molecule prior to the reaction between the thiol and maleimide. If carried out first, a partial reduction of the IgG-Cys-GALaXy-GalNaz would reduce the azido group via the Staudinger reaction, preventing the subsequent CLICK chemistry conjugation (ref. Citation39 and data not shown). Therefore, we proceeded with the reaction in a stepwise manner whereby the CLICK reaction was performed first followed by the partial reduction of the molecule with TCEP and conjugation of the maleimide-payload in the second step. The payloads chosen for this proof-of-principle experiment were a DBCO-AlexaFluor647 dye and a maleimide-DyLight488 dye, to be conjugated at the GALaXy site and Cys 239, respectively (). Conjugation efficiency was monitored at each step by LC-MS to assess the degree of payload incorporation into the IgG heavy chain. After the CLICK reaction, a 51,336 Da species that was consistent with the mass of an IgG-Cys-GALaXy-AF647 conjugate with one DBCO-AF647 per heavy chain was observed (). The subsequent conjugation of the maleimide-functionalized DyLight488 dye resulted in the generation of a 52,113 Da species consistent with the mass of an IgG-Cys-(DyLight488)GALaXy-AF647 dual conjugate with one AF647 and one Dylight488 molecule per heavy chain (). As can be seen from , the conjugation reactions resulted in almost complete conversion to a site-specifically labeled, dual dye-conjugated molecule, demonstrating the compatibility of the GALaXy system with maleimide-based conjugation approaches.
Figure 6. Combination of azido-functionalized O-glycoproteins with maleimide chemistry allows efficient dual payload conjugation. (a) Overview of dual labeling workflow using CLICK and maleimide chemistries. (b) LC-MS analysis of IgG-Cys-GALaXy proteins expressed in HEK293 F GALE KO cells supplemented with 100 µM GalNAz (top histogram), after conjugation of a DBCO-AlexaFluor647 dye via CLICK chemistry (middle histogram) and a dual labeled IgG containing both AlexaFluor647 and DyLight488 dyes conjugated using CLICK chemistry followed by maleimide-thiol chemistry (bottom histogram). (c) Representative FACS histograms showing the expression Her2 on SKBR3 (top histograms) and NCI-N87 (bottom histograms) breast cancer cell lines. Histograms show fluorescence quantification of AlexaFluor647 (APC) and Dylight488 (FITC) from staining using a single (Trast-AF647) or dual-dye (Trast-AF647-Dy488) labeled anti-Her2 trastuzumab antibody. An irrelevant antibody (NIP228, AstraZeneca) is used for staining controls. GalNAz is represented by the red square, AlexaFluor647 = red star and DyLight488 = green star
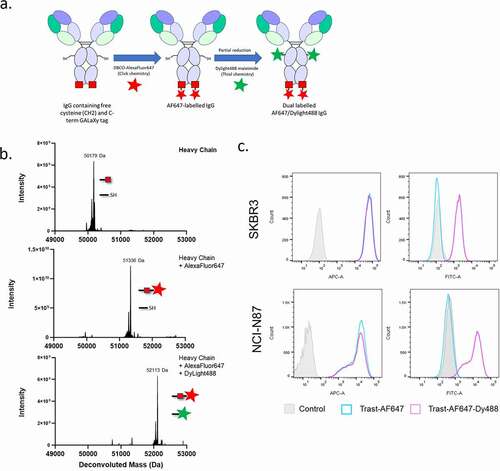
To ensure that the two-step conjugation did not compromise protein function, we tested the binding of dual dye-labeled trastuzumab to the Her2-positive breast cancer cell lines SKBR3 and NCI-N87 by FACS (). Conjugation of the two dyes did not affect the binding of this IgG to Her2, as demonstrated by a strong IgG fluorophore signal in both the APC (AF647) and FITC (Dylight488) channels and a corresponding rightward shift in fluorescence intensities in stained cells ().
DISCUSSION
The complexities of the endogenous O-glycosylation pathways, combined with the lack of an O-glycosylation consensus sequence, have limited its use for glycoengineering approaches. Here, we have identified a simple 5 amino acid, widely applicable O-glycosylation tag (GALaXy tag) that can be used to produce site-specific and uniformly O-glycosylated recombinant proteins. When expressed in a CRISPR-engineered HEK293F GALE KO cell line with metabolically defined media, ‘conjugation-ready’ recombinant proteins that are also compatible with multiple conjugation chemistries can be readily made.
Our serendipitous finding that a short peptide sequence from TB4 could efficiently direct site-specific O-glycosylation correlates well with the findings of previous researchers investigating the O-glycosylation of other short peptides. In fact, the idea of a transferable O-glycosylation domain was proposed over 30 years ago by Conradt et al.,Citation40 who mutated the natural O-glycosylation motif in interleukin-2 (IL-2) to include multiple prolines downstream from the target threonine, which resulted in more efficient O-glycosylation. Their concept, however, was only tested in the context of an IL-2 sequence,Citation41 failing to identify a consensus transferable sequence for O-glycosylation, and was not further developed. The idea of an O-glycosylation consensus sequence was later explored by Yoshida et al.,Citation42 who devised an in vitro assay using peptides from human erythropoietin (Epo), a known O-glycosylation substrate, to identify short sequence motifs in the primary peptide structure that were required to produce high levels of Epo O-glycosylation. This in vitro-based approach used recombinantly expressed bovine colostrum O-GalNAC-T to glycosylate Epo-derived peptides and marked a step forward in the understanding that O-glycosylation sites can be represented by discrete, short amino acid motifs. However, as their experiments were limited to Epo-derived peptides evaluated in vitro, it is difficult to assess how representative these are of motifs functional in cells from different species where the proteins would be subjected to the full complement of cellular GalNAC-Ts. We have shown that the GALaXy tag is active in the most commonly used protein expression systems (CHO, mouse and human) and thus represents a functional, generally applicable, mammalian O-glycosylation motif.
Examples of tags that bestow O-glycosylation sites on proteins have been previously described, e.g., roTag, but these have not been fully characterized and provide lesser utility.Citation16 These approaches do not provide a method to control O-glycosylation, such as our HEK293F GALE KO expression cell line, and it is likely that a heterogenous mix of glycoforms would be generated using these tags in non-engineered cells.
We observed that the levels and heterogeneity of O-glycosylation varied widely between cell backgrounds (). These differences most likely reflect the expression profile of the resident GalNAC-T enzymes and their substrate specificities for the tag.Citation43 However, it should be noted that the mechanism that determines the exact O-glycosylation site occupancy is poorly understood. Interestingly, we noted in this study, as others have found previously,Citation42–44 that amino acids from position −3 to +3 relative to the glycosylation site were especially important and that sequences rich in serines or threonines with a proline in position −1 or −3 and +3 favored O-glycosylation (, and Table S1).
While the GALaXy tag proved transferable and functional across several proteins, the tag alone would only allow the user to specify the location of glycosylation and not permit the control of the type or complexity of the glycans added. Control of sugar addition to generate an ‘on demand’ system required engineering of the endogenous O-glycosylation pathways of the host cell. Previous work had shown that mutant CHO IdID cells (deficient in GALE expression)Citation22,Citation45 HeLa GALE −/−,Citation25 and HEK293T GALE KOCitation24 cells were viable and grew at comparable rates to parental cells,Citation24 which is consistent with our findings for HEK293F GALE KO cells (data not shown). This demonstrates that GALE enzyme activity, or indeed UDP-GalNAc, does not appear to be critical for cell proliferation, which is in agreement with previous reports in the field.Citation22,Citation24,Citation45 However, the complete lack of extended O-glycans as a result of GALE knockout in HEK293F was unexpected, as the activity of the GALE enzyme was reported to be limited to catalyzing two distinct reactions, i.e., the epimerization of UDP-glucose to UDP-galactose, and the epimerization of UDP-N-acetylglucosamine to UDP-N-acetylgalactosamine.
A direct impact of GALE KO on the extension of the initial glycan chain and further branching was not anticipated. However, most HEK293 O-glycans contain a subtype structure consisting of galactose in a β1–3 linkage to GalNAc called core 1.Citation19 Elongation of core 1 produces complex, branched, mature O-glycans, and, therefore, we reasoned that, without a supply of galactose from the cell culture media, the formation of core 1, and hence longer O-glycans, was compromised, thus explaining our single GalNAc/GalNAz addition in HEK293F GALE KO cells. This phenomenon appears to be a characteristic of the culture conditions of the HEK293F GALE KO cells, as GALaXy-tagged proteins produced in CHO K1 GALE KO cells exhibited O-glycans with extended chain lengths.
The presence of complex O-glycans in CHO K1 GALE KO cells is consistent with previous findings showing that ldlD CHO cells could scavenge sugar precursors from serum-supplemented media to restore O-glycosylation patterns observed in wild-type (wt) cells.Citation46 Indeed, using our HEK293F GALE KO system, we could prove that the lack of galactose was responsible for the lack of O-glycan chain extension. Expression of the recombinant proteins in cells supplemented with both GalNAc and galactose restored the O-glycosylation pattern characteristic for a non-modified cell line. These findings were in agreement with previous observations that found that galactose supplementation could rescue the maturation of N- and O-glycans under conditions of sugar starvation.Citation47 In contrast, however, a recent publication by Brossard et al. showed, using GALE KO cell lines, that galactose alone was able to restore both N- and O-glycosylation,Citation25 which requires further investigation. An implication of this work is that either simple O-glycans (containing Tn antigen only) or complex O-glycans can also be generated in HEK293F GALE KO on demand simply by switching medium supplementation from GalNAc (or its derivatives) to GalNAc and galactose.
In developing our controlled O-glycosylation system, we aimed to generate a platform that could produce recombinant secreted proteins at significant levels and yield a homogeneous product. Standard protein expression platforms use cells that can grow in suspension cultures at high density and with high viability rates. CHO IdID and HEK293T GALE KO cells, which grow as adherent cultures, are not best suited to high level recombinant protein expression and, in terms of controlling O-glycosylation, require the use of lipo-depleted fetal bovine serum (FBS)Citation24 to ensure they are free from sugars that could be used in the salvage pathway, which would otherwise confound metabolic labeling. Therefore, we used the HEK293F cell line as a host because it is more amenable to high-yielding recombinant protein expression and grows in defined serum-free media in the absence of galactose or GalNAc. Moreover, our findings that O-glycoproteins recombinantly expressed in HEK293F GALE KO cells supplemented with GalNAc in the absence of galactose contained only a single GalNAc were critical to the development of this expression platform. We have shown that this feature, combined with the promiscuity of the salvage pathway and GalNAC-T enzymes, allowed the metabolic labeling of recombinant proteins with azido-functionalized GalNAc (GalNAz) and thus produced a system whereby homogenously labeled, conjugation-ready proteins could be easily produced. To our knowledge, this is the first description of generating a site-specific O-glycosylation system by combining an O-glycosylation tag and a metabolically engineered cell line.
Additional factors needed to be considered when using the GALaXy system, such as: 1) multiple GalNAc additions to adjacent serine or threonine residues proximal to the initial glycosylation site (as observed for the multi-tag), and 2) varying labeling efficiencies dependent on tag location and expression conditions. With regard to multiple O-glycan additions, even though in the absence of galactose the O-glycans added to the tag were not extended or branched, we observed multiple individual GalNAc additions (up to 5 per Fc chain) when the multi-tag was used. The multi-tag provides additional serine and threonine substrate residues proximal to the initiating O-glycosylation site and thus takes advantage of the substrate specificities of individual GalNAC-T enzymes. A subset of these enzymes have a preference for proteins that have at least one O-glycan already added.Citation43 However, as can be seen from our data (, ), the presence of these additional potential glycosylation sites does not guarantee that all serines and threonines proximal to the tag will be O-glycosylated and thus lead to varying O-glycan incorporation rates. A potential solution to this issue could be to concatenate multiple GALaXy tags (PTAEP) and thereby achieve homogenous multi-site O-glycan incorporation. This work is the subject of further experiments in our lab.
Tag location, as expected, was also found to have a substantial impact on the efficiency of labeling. In agreement with our findings, Grabenhorst et al.Citation41 demonstrated that the efficiency of O-glycosylation of baculovirus-expressed IL-2 mutants in Sf21 insect cells depended on the positioning of an ‘artificial O-glycosylation domain’, GGKAPTPPPK at either C-terminus or in a loop region of IL-2. However, it has to be noted that a labeling efficiency of over 80% was never achieved and a mix of GalNAc and Gal(β1- 3)GalNAc was observed. These effects are most likely protein and insect cell-specific. In our HEK-based system, the most efficient incorporation of GalNAc or GalNAz to Fc-based proteins was achieved when the GALaXy tag was positioned at the C-terminus of the Fc. This positioning of the tag is also the most convenient for labeling or conjugating payload to a mAb without interfering with binding or function.
The GalNAc-Ts accessibility to the O-glycosylation tag plays a role in the efficiency of labeling, as demonstrated by the variable rates of sugar incorporation into the intrachain CH3 tags, which were highly dependent on the flexible linkers flanking the tags (). O-glycosylation occurs late in the secretory pathway when the protein is usually already fully folded. The CH3 domain of an antibody adopts a compact and stable conformation during folding, which is obligatory for the structure and dimerization of the heavy chains,Citation48,Citation49 and which could impair access to a buried tag in the absence of additional flexible linkers. Work is ongoing within our group to optimize linker lengths with the aim of obtaining improved O-glycan occupancy at all sites.
GALaXy technology provides an easy-to-use platform for generating proteins for bioconjugation using CLICK chemistry. Other methods that have been used to produce proteins for CLICK-chemistry conjugation include codon-reassignment and incorporation of unnatural amino acids,Citation50,Citation51Citation52 post production by in vitro chemoenzymatic glycan remodeling,52 or using bifunctional cross-linkers such as maleimide-azido or maleimide-alkyne, including maleimide-DBCO for copper-free CLICK chemistry. While copper-free CLICK-chemistry conjugation has been well validated and widely used, the methods for generating CLICK-chemistry-ready homogeneous proteins often require the use of complex purification steps. The use of the GALaXy system streamlines the production process, as it fits into standard recombinant protein expression workflows and obviates the need for additional processing steps post-purification, thereby simplifying the generation of CLICK-chemistry “ready” proteins for subsequent payload conjugation. We would envisage the GALaXy system as a platform that could greatly simplify the production of recombinant proteins destined for conjugation. It would ultimately minimize downstream handling and losses associated with the processing of bioconjugates while providing a simple to use and flexible approach to generating bioconjugates through O-glycan engineering.
Abbreviations
Supplemental Material
Download Zip (188.3 KB)Disclosure statement
This work was funded by AstraZeneca.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Lim SI. Site-specific bioconjugation and self-assembly technologies for multi-functional biologics: on the road to the clinic. Drug Discov Today. 2020;25(1):168–17. doi:10.1016/j.drudis.2019.10.002.
- van Berkel SS, van Delft FL. Enzymatic strategies for (near) clinical development of antibody-drug conjugates. Drug Discov Today Technol. 2018;30:3–10. doi:10.1016/j.ddtec.2018.09.005.
- Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol Ther Nucleic Acids. 2017;6:116–32. doi:10.1016/j.omtn.2016.12.003.
- Cavaco M, Castanho M, Neves V. Peptibodies: an elegant solution for a long-standing problem. Pept Sci. 2018;110(1):e23095.
- Lang K, Chin JW. Bioorthogonal reactions for labeling proteins. ACS Chem Biol. 2014;9(1):16–20. doi:10.1021/cb4009292.
- Nguyen TA, Cigler M, Lang K. Expanding the genetic code to study protein-protein interactions. Angew Chem Int Ed Engl. 2018;57:14350–61. doi:10.1002/anie.201805869.
- Neumann H, Slusarczyk AL, Chin JW. De novo generation of mutually orthogonal aminoacyl-tRNA synthetase/tRNA pairs. J Am Chem Soc. 2010;132:2142–44. doi:10.1021/ja9068722.
- Agatemor C, Buettner MJ, Ariss R, Muthiah K, Saeui CT, Yarema KJ. Exploiting metabolic glycoengineering to advance healthcare. Nat Rev Chem. 2019;3(10):605–20. doi:10.1038/s41570-019-0126-y.
- Chen Z, Cole PA. Synthetic approaches to protein phosphorylation. Curr Opin Chem Biol. 2015;28:115–22. doi:10.1016/j.cbpa.2015.07.001.
- Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z, Lin H. Protein lipidation: occurrence, mechanisms, biological functions, and enabling technologies. Chem Rev. 2018;118(3):919–88. doi:10.1021/acs.chemrev.6b00750.
- Zhang P, Woen S, Wang T, Liau B, Zhao S, Chen C. Challenges of glycosylation analysis and control: an integrated approach to producing optimal and consistent therapeutic drugs. Drug Discov Today. 2016;21:740–65.
- Byrne G, O’Rourke SM, Alexander DL, Yu B, Doran RC, Wright M. CRISPR/Cas9 gene editing for the creation of an MGAT1-deficient CHO cell line to control HIV-1 vaccine glycosylation. PLoS Biol. 2018;16:e2005817. doi:10.1371/journal.pbio.2005817.
- Anyaogu DC, Mortensen UH. Manipulating the glycosylation pathway in bacterial and lower eukaryotes for production of therapeutic proteins. Curr Opin Biotechnol. 2015;36:122–28. doi:10.1016/j.copbio.2015.08.012.
- Pan C, Sun P, Liu B, Liang H, Peng Z, Dong Y. Biosynthesis of conjugate vaccines using an O-linked glycosylation system. mBio. 2016;7:e00443–16. doi:10.1128/mBio.00443-16.
- Lodish HBA, Zipursky SL. Molecular Cell Biology. 4th edition. Section 17.7, Protein Glycosylation in the ER and Golgi Complex. New York: W. H. Freeman; 2000.
- Petris G, Bestagno M, Arnoldi F, Burrone OR. New tags for recombinant protein detection and O-glycosylation reporters. PLoS One. 2014;9:e96700. doi:10.1371/journal.pone.0096700.
- Varki A, Etzler ME, Cummings RD, Esko JD. Discovery and Classification of Glycan-Binding Proteins. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, editors. Essentials of Glycobiology. Cold Spring Harbor (NY); 2009. Chapter 26.
- Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346–66.
- Brockhausen I, Stanley P. O-GalNAc Glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, editors. Essentials of Glycobiology. New York: Cold Spring Harbo; 2015. p. 113–23.
- Clausen H, Bennett EP. A family of UDP-GalNAc: polypeptide N-acetylgalactosaminyl-transferases control the initiation of mucin-type O-linked glycosylation. Glycobiology. 1996;6:635–46. doi:10.1093/glycob/6.6.635.
- Frey PA. The leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 1996;10(4):461–70. doi:10.1096/fasebj.10.4.8647345.
- Kingsley DM, Kozarsky KF, Hobbie L, Krieger M. Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase deficient mutant. Cell. 1986;44:749–59. doi:10.1016/0092-8674(86)90841-X.
- Matzuk MM, Krieger M, Corless CL, Boime I. Effects of preventing O-glycosylation on the secretion of human chorionic gonadotropin in Chinese hamster ovary cells. Proc Natl Acad Sci U S A. 1987;84:6354–58. doi:10.1073/pnas.84.18.6354.
- Termini JM, Silver ZA, Connor B, Antonopoulos A, Haslam SM, Dell A. HEK293T cell lines defective for O-linked glycosylation. PLoS One. 2017;12:e0179949. doi:10.1371/journal.pone.0179949.
- Broussard A, Florwick A, Desbiens C, Nischan N, Robertson C, Guan Z. Human UDP-galactose 4ʹ-epimerase (GALE) is required for cell-surface glycome structure and function. J Biol Chem. 2020;295:1225–39. doi:10.1016/S0021-9258(17)49882-6.
- Boyce M, Carrico IS, Ganguli AS, Yu SH, Hangauer MJ, Hubbard SC. Metabolic cross-talk allows labeling of O-linked beta-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc Natl Acad Sci U S A. 2011;108:3141–46. doi:10.1073/pnas.1010045108.
- Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc Natl Acad Sci U S A. 2003;100(25):14846–51. doi:10.1073/pnas.2335201100.
- Daramola O, Stevenson J, Dean G, Hatton D, Pettman G, Holmes W. A high-yielding CHO transient system: coexpression of genes encoding EBNA-1 and GS enhances transient protein expression. Biotechnol Prog. 2014;30(1):132–41. doi:10.1002/btpr.1809.
- Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes Metab. 2016;18:1176–90. doi:10.1111/dom.12735.
- Naylor J, Rossi A, Hornigold DC. Acoustic dispensing preserves the potency of therapeutic peptides throughout the entire drug discovery workflow. J Lab Autom. 2016;21(1):90–96. doi:10.1177/2211068215587915.
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293:1155–59. doi:10.1126/science.1061692.
- Mp J, DL P, Cs R, TJ D, Honig B, De S. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–67. doi:10.1002/prot.10613.
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320(3):597–608. doi:10.1016/S0022-2836(02)00470-9.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(33–8):27–28. doi:10.1016/0263-7855(96)00018-5.
- Goldstein AL, Hannappel E, Sosne G, Kleinman HK. Thymosin beta4: a multi-functional regenerative peptide. Basic properties and clinical applications. Expert Opin Biol Ther. 2012;12:37–51. doi:10.1517/14712598.2012.634793.
- Pipes GT, Yang J. Cardioprotection by thymosin beta 4. Vitam Horm. 2016;102:209–26.
- Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358:1109–17. doi:10.1056/NEJMoa074943.
- Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol. 2010;28(8):863–67. doi:10.1038/nbt.1651.
- Liu S, Edgar KJ. Staudinger reactions for selective functionalization of polysaccharides: a review. Biomacromolecules. 2015;16(9):2556–71. doi:10.1021/acs.biomac.5b00855.
- Conradt HS, Nimtz M, Dittmar KE, Lindenmaier W, Hoppe J, Hauser H. Expression of human interleukin-2 in recombinant baby hamster kidney, Ltk-, and Chinese hamster ovary cells. Structure of O-linked carbohydrate chains and their location within the polypeptide. J Biol Chem. 1989;264:17368–73. doi:10.1016/S0021-9258(18)71502-0.
- Grabenhorst E, Hofer B, Nimtz M, Jager V, Conradt HS. Biosynthesis and secretion of human interleukin 2 glycoprotein variants from baculovirus-infected Sf21 cells. Characterization of polypeptides and posttranslational modifications. Eur J Biochem. 1993;215(1):189–97. doi:10.1111/j.1432-1033.1993.tb18022.x.
- Yoshida A, Suzuki M, Ikenaga H, Takeuchi M. Discovery of the shortest sequence motif for high level mucin-type O-glycosylation. J Biol Chem. 1997;272(27):16884–88. doi:10.1074/jbc.272.27.16884.
- Choi J, Wagner LJS, Timmermans S, Malaker SA, Schumann B, Gray MA. Engineering orthogonal polypeptide GalNAc-transferase and UDP-sugar pairs. J Am Chem Soc. 2019;141(34):13442–53. doi:10.1021/jacs.9b04695.
- Gerken TA, Tep C, Rarick J. Role of peptide sequence and neighboring residue glycosylation on the substrate specificity of the uridine 5ʹ-diphosphate-alpha-N-acetylgalactosamine: polypeptideN-acetylgalactosaminyl transferases T1 and T2: kinetic modeling of the porcine and canine submaxillary gland mucin tandem repeats. Biochemistry. 2004;43:9888–900.
- Schulz JM, Ross KL, Malmstrom K, Krieger M, Fridovich-Keil JL. Mediators of galactose sensitivity in UDP-galactose 4ʹ-epimerase-impaired mammalian cells. J Biol Chem. 2005;280:13493–502. doi:10.1074/jbc.M414045200.
- Krieger M, Reddy P, Kozarsky K, Kingsley D, Hobbie L, Penman M. Analysis of the synthesis, intracellular sorting, and function of glycoproteins using a mammalian cell mutant with reversible glycosylation defects. Methods Cell Biol. 1989;32:57–84.
- Sasaoka N, Imamura H, Kakizuka A. A trace amount of Galactose, a major component of milk sugar, allows maturation of Glycoproteins during sugar starvation. iScience. 2018;10:211–21. doi:10.1016/j.isci.2018.11.035.
- Dall’Acqua W, Simon AL, Mulkerrin MG, Carter P. Contribution of domain interface residues to the stability of antibody CH3 domain homodimers. Biochemistry. 1998;37:9266–73. doi:10.1021/bi980270i.
- Feige MJ, Hendershot LM, Buchner J. How antibodies fold. Trends Biochem Sci. 2010;35(4):189–98. doi:10.1016/j.tibs.2009.11.005.
- Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A. 2012;109:16101–06. doi:10.1073/pnas.1211023109.
- Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug Chem. 2014;25:351–61. doi:10.1021/bc400490z.
- van Geel R, Wijdeven MA, Heesbeen R, Verkade JM, Wasiel AA, van Berkel SS. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-Glycan of native mAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug Chem. 2015;26:2233–42. doi:10.1021/acs.bioconjchem.5b00224.