
ABSTRACT
The naïve human antibody repertoire has theoretical access to an estimated > 1015 antibodies. Identifying subsets of this prohibitively large space where therapeutically relevant antibodies may be found is useful for development of these agents. It was previously demonstrated that, despite the immense sequence space, different individuals can produce the same antibodies. It was also shown that therapeutic antibodies, which typically follow seemingly unnatural development processes, can arise independently naturally. To check for biases in how the sequence space is explored, we data mined public repositories to identify 220 bioprojects with a combined seven billion reads. Of these, we created a subset of human bioprojects that we make available as the AbNGS database (https://naturalantibody.com/ngs/). AbNGS contains 135 bioprojects with four billion productive human heavy variable region sequences and 385 million unique complementarity-determining region (CDR)-H3s. We find that 270,000 (0.07% of 385 million) unique CDR-H3s are highly public in that they occur in at least five of 135 bioprojects. Of 700 unique therapeutic CDR-H3, a total of 6% has direct matches in the small set of 270,000. This observation extends to a match between CDR-H3 and V-gene call as well. Thus, the subspace of shared (‘public’) CDR-H3s shows utility for serving as a starting point for therapeutic antibody design.
KEYWORDS:
Introduction
Antibodies are the largest group of biotherapeutics, with nearly 200 approved in at least one countryCitation1 (https://www.antibodysociety.org/antibody-therapeutics-product-data/) and comprising more than a quarter of new approvals by the US Food and Drug Administration in 2022.Citation2 These therapeutics derive from the successful harnessing of the natural process wherein an organism rapidly develops an antibody binder against a pathogen. The organism achieves this by a priori having a large number of naïve antibodies (~1011),Citation3 at least one of which ideally binds the pathogen and starts an immune response. Thereafter, in germinal centers, during the somatic hypermutation process, the organism explores an even larger (>1015) theoretically possible space of these molecules to develop an ever better binder. Understanding how this immense diversity is explored is crucial to our understanding of the immune system and developing better monoclonal antibody therapies.
The great diversity was proposed to be the result of accruing variability in response to pathogens, accumulated on an evolutionary scale by vertebrates.Citation4 However, this number is not plausible when taking into account the total number of epitopic sites that an organism would need to address within a reasonable time to mount an immune response.Citation5 It was noted that, despite theoretically great diversity, amino acid preferences in the complementarity-determining region (CDR)-H3s (the most variable loop) are biased.Citation6 The sequence space that is theoretically available to antibodies is ‘astronomical’, with some estimates placing it at as many as 1018 possible molecules.Citation5,Citation7,Citation8 With such large numbers, it appears implausible that an organism would explore all the possibilities, suggesting that a biased sampling of this space is more likely than purely random sampling.
The theoretically accessible sequence repertoire might indeed be theoretically ‘astronomical’ in size, but for practical reasons, there seems instead to be another, smaller, more manageable dimension that is being explored.Citation5 However, productive and nonproductive sequences appear to have similar profiles of somatic hypermutation, suggesting a stronger role in the random biochemical process rather than strategic selection.Citation9 To address this conundrum, theories of ‘minimal’ repertoires were proposed decades ago.Citation10 The structural dimension was proposed as one explanation, as there appears to be more conservation in structural configurations than there are sequences of antibodies.Citation11–13 Nevertheless, even at the most basic, sequence level, certain commonalities between independent repertoires are observed.
Despite the great variability available to repertoires, different individuals can evolve the same antibodies toward the same antigen challenge.Citation14 Commonality exists between repertoires of individuals as well, as documented by deep sequencing studies of threeCitation7 and ten individuals.Citation8 In both cases, the prevalence of clonotypes (same V-J assignment and CDR-H3 amino acid sequence) between different individuals was studied. In the Soto et al. study, 0.3% of heavy chain clonotypes were shared by all three individuals in the study (1–6% were shared between any two individuals). In the Briney et al. study, 0.022% of clonotypes were shared by all ten subjects in the study (on average 0.95% between any two individuals). Therefore, despite great diversity available to antibody repertoires, independent natural samples draw same/similar sequences, suggesting that the space is explored not randomly, but in a directed manner.
While antibodies are used as biotherapeutics, developing a biologic diverges from natural antibody processes. Monoclonal antibody harnessing originated in 1975 with Kohler and Milstein’s hybridoma technology, which involved fusing immunized mouse lymphocytes with myeloma cells. Yet, using mouse antibodies in therapy may trigger an immune response, generating anti-murine antibodies and reducing efficacy. Strategies such as chimerization, humanization, and use of fully human sequences aim to minimize this response.
Throughput limitations of hybridoma technology prompted the development of other selection methods, such as display systems producing fully human immunoglobulins that resemble adaptive immune response in vitro. Typically in this approach, antibody-binding domains such as antigen-binding fragments or single-chain variable fragments are presented on the outside of the cell wall or membrane. Both processes need to be followed by lead optimization.Citation15
Despite divergent development processes, therapeutic sequences can be found in naturally sourced repertoires.Citation16,Citation17 Position-specific scoring matrices developed on the basis of natural repertoires aligned with mutations selected for in therapeutics.Citation18 Therefore, despite the great diversity available to antibodies, a set of these developed by independent artificial methods appears to follow similar selection pressures.
Convergence between natural and therapeutic antibodies may facilitate drug discovery. Next-generation sequencing (NGS) repertoires are growing in numbers, providing an unprecedented sampling of the vast theoretical space available to antibodies. Drawing therapeutic molecules from this space is desirable, as human-origin antibodies can produce fewer downstream issues than non-human or display platforms.Citation19 Mapping the characteristics of the observed convergence between natural and therapeutic antibodies could potentially be used to limit the space of natural sequences that are used for drug discovery, accelerating the process.
Here, to interrogate the sampled natural antibody space, we automatically data-mined the public NGS repositories to create a heterogeneous set of naturally sourced B-cell receptors that is an order of magnitude larger than previously published datasets.Citation20–22 We used this dataset to identify commonalities between it and therapeutic antibodies.
Results
Large-scale data mining public next-generation sequencing repositories for B-cell receptor repertoires
We automatically mined the SRA repositories, to identify bioprojects associated with B-cell receptors (BCRs). For context, when conceived in 2018, the Observed Antibody Space (OAS), which has been a widely used resource for BCR NGS data,Citation23,Citation24 was created using manual identification of BCR-containing sequences. This resulted in 55 bioprojects, which was recently updated to 90 bioprojects.Citation20 Here, using an automatic identification pipeline requiring processing of more than 500,000 bioprojects, we identified 287 bioprojects with BCR data. Of these 287, 220 were earmarked for processing because they did not include single-cell data and had more than 10% of reads identifiable as immunoglobulin genes and associated metadata indicated that immunoglobulins were the main focus of the study.
The 220 bioprojects contributed a total of 7 billion sequences that were processed into IMGT-aligned amino acid sequences, allowing for CDR delineation. We processed the data using a single uniform pipeline, creating the AB-NGS database (). By contrast, previously in OAS, the raw sequences were processed using IgBLASTCitation25 to convert nucleotides to amino acid sequences. The amino acid sequences were IMGT numbered and germlines identified using ANARCI.Citation26 Both pieces of software used IMGTCitation27 as the underlying germline database. It is known, however, that annotation results depend on the underlying germline database and piece of annotation software used in the process.Citation28 For this reason, we adapted best practices from IgBLAST, ANARCI, and MMSeqsCitation29 programs to create a single pipeline that filters and numbers nucleotide sequences in a single pass, using the VDJBase germline databaseCitation30 (see Methods).
Table 1. Statistics of the number of heavy-chain sequences processed to make the AB-NGS database. We stratified our datasets by the organism and whether the sequence was productive (no stop codons, no truncation, see Methods for more details). The multiple repeats within the bioproject were taken as identical variable region sequences on the nucleotide level to increase the stringency of including only the confident sequences.
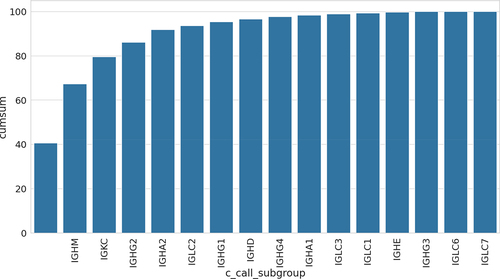
The processing of 7 billion sequences resulted in 6 billion productive sequences (). The processing allowed us to identify the CDR regions, closest germlines and, wherever available, the isotype (). The gene usage across our datasets places some of the most common genes as having the largest coverage,Citation7,Citation8 so our annotation pipeline recapitulates the data distribution from its constituent experiments.Citation7,Citation8,Citation14 In some projects, isotype was provided in metadata, but we performed sequence level annotation by aligning constant region of each sequence to C genes database, obtained from NCBI. For a considerable number of sequences, it was impossible to identify the isotype. The most common constant region identification was IGHM. In second place is IGK, with IGHG2 only in third position. Altogether, IGG isotype is typically associated with an active immune response whereas IGM is associated with the naïve repertoire. The constant region identification suggests a predominantly naïve makeup of our database, though with the caveat that in multiple cases identification of the constant region was not possible. This observation, however, is further confirmed by metadata analysis.
Figure 1. Annotated C regions as cumulative plot. We automatically identified the constant region, wherever it was possible. On the basis of the heavy C region call, the isotype can be inferred. For most of the sequences it was not possible to infer the C region (the blank leftmost column).
For each bioproject, we collected and cleaned its associated metadata. The 220 bioprojects were associated with more than 11,000 biosamples, that in turn are linked to author-submitted metadata, such as organism, B-cell type, tissue, and disease. This data format is not uniform, requiring substantial manual processing to achieve normalization across all 11,000 samples. For each sample, where present, we extracted the organism, B-cell type, tissue, and disease and their per-sequence distribution is given in .
Figure 2. Metadata landscape of our database. a. Author-annotated organism distribution of sequences in our dataset. b. Source of B-cells in our dataset. c. Type of B-cells in our dataset. d. Author-annotated disease present in the dataset.
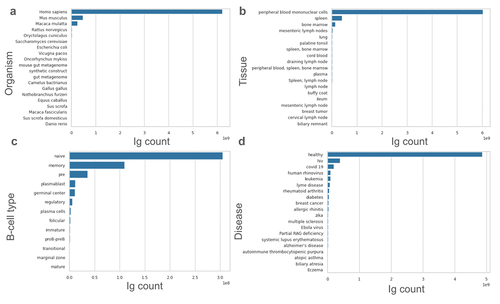
The BCR sequences in our database cover a range of organisms, with the most common being human, followed by mouse (). This reflects the therapeutic focus of sequencing efforts that address humans, wherein mice are a typical model organism in immunization campaigns. Other model organisms such as macaques, rats and rabbits are also represented. The presence of camelids (Vicugna pacos, Camelus bactrianus) indicates some studies that target the naturally occurring heavy-chain only antibodies, or VHHs, marketed therapeutically as nanobodies.Citation31,Citation32
Most of the sequences in our database come from peripheral blood mononuclear cells, followed by spleen and bone marrow (). Most of the sequences come from naïve B-cells, with memory B-cells in the second place (). The majority of sequences in our database come from individuals who were not mounting an immune response to any disease (). Nevertheless, in a large proportion of cases, the disease status information was not available.
Altogether, sequence and metadata analysis indicates that the contents of our database are largely human, naïve, and not responding to any immune challenge. We believe that it is the biggest compilation of publicly available human BCR sequencing data to date. By contrast, the current version of OAS (Oct 2023), held 2.2 billion redundant human antibody sequences from 70 bioprojects, contrasted to our 5.3 billion from 135 bioprojects (). We thus consider the dataset to be well placed to study commonalities to constrain the great antibody diversity for therapeutic discovery.
Highly public CDR-H3s account for 0.07% of all CDR-H3s and 0.31% of confident CDR-H3s
Drawing from observations that natural antibodies can be sampled by different individuals and therapeutic antibodies can arise naturally, we hypothesized that there is a reduced set of ‘shared’ antibodies within which therapeutic varieties are more likely to be found.
Taking advantage of the large set of independent human studies, we quantified the extent to which the human natural antibody sequence space is sampled in the same way, by checking how many of the sequences are ‘shared’ or ‘public’. To avoid confounding factors such as gene usage and disease states, we focused only on the CDR-H3 region, as the most diverse and instrumental element of an antibody for ease of analysis and interpretation.
We took bioproject as the quantum of an independent sample, as many studies differ in variables such as number of individuals and sequencing regimens. Therefore, any commonalities that result do so despite the diversity in our datasets. To further stratify the diversity, we divided the dataset into the Confident and All sequences. The Confident set was composed of sequences that had to occur at least twice in the source bioproject. This further ensured that the same CDR-H3s observed across different bioprojects did not occur as a result of technical artifacts, such as sequencing errors.
To test for ‘public sequences’, we counted how often the same CDR-H3 occurred among different naturally sourced NGS studies. Our dataset includes data from studies that are known for their depth of sequencing (e.g., Soto et al., Briney et al.,Citation7,Citation8 which could potentially skew any statistics. All but one bioproject of 135 contributed CDR-H3s to the public datasets, suggesting that no single dataset is a biasing factor. Altogether, we note that when bioproject redundancy of just two is imposed, it captures a greater number of CDR-H3s (1.58% for All and 4.17% for Confident) than for more stringent redundancies (). Furthermore, the difference between public CDR-H3s from All or Confident datasets becomes blurred with higher redundancies (). This suggests that, at bioproject redundancy of just two, chance occurrences can be observed. The percentage of public CDR-H3s appears to drop asymptotically with the higher stringencies, appearing to stabilize in the range of one promile (0.07% for redundancy 5 or 0.01% at redundancy 9). The corresponding figure for the confident CDR-H3s is 0.31 at redundancy 5 and 0.05 at redundancy 9. Altogether, the public CDR-H3 levels are within the same range that the public clones (that also included the V call) reported between individuals within the Briney et al. and Soto et al. studiesCitation7,Citation8 (though direct comparison between the numbers is tenuous). Nevertheless, here, we report public sequence occurrences that are drawn from much larger and more heterogeneous samples than either of the two studies.
Table 2. Number of public CDR-H3s by the number of independent bioprojects they occurred in. The ‘Confident’ CDR-H3 dataset is a collection of CDR-H3s from sequences that occurred at least once within each bioproject. By contrast the ‘All’ dataset includes all the sequences without such constraints. Within Confident and All datasets, we noted how many separate bioprojects each CDR-H3 occurred in. The table shows the number of CDR-H3s in each dataset with CDR-H3s occurrences equal to or greater than specified bioproject redundancy. The percentages in both columns refer to different totals, in case of All CDR-H3s it is 385,878,857 sequences and in case of Confident 85,688,290.
Any occurrences of shared CDR-H3s between samples could either be attributed to biological convergence or the presence of common features that increase likelihood of multiple occurrences. The biological convergence is hard to define and difficult to check. Features driving higher probability of occurring between different studies are likely CDR length and sequence composition. To check for both, we plotted the CDR-H3 lengths of All CDR-H3s and the public datasets (). The results shows unequivocally that the public CDR-H3s are generally shorter. One would expect increasingly more common CDR-H3s to be increasingly shorter. Nonetheless, increasing the stringency of the number of CDR-H3 occurrences does not reduce the CDR-H3 length. To assess CDR-H3 composition, which we expressed as positional diversity, we plotted the positional entropy for a range of most common CDR-H3 lengths (). The results show that All CDR-H3s have slightly higher diversity (entropy) than the public datasets. Nevertheless, the diversity is not reduced radically if samples of, comparatively small, 700 are drawn from the public datasets.
Figure 3. Length distribution of the CDR-H3s across the entire dataset and public datasets. The solid black line shows the length distribution in the entire dataset of 385 m sequences. The public datasets are divided into the Confident ones (dashed lines, subscript ‘r’ in legend) and the ones not requiring redundancy (solid lines, subscript ‘all’ in legend). The numeric subscripts in public datasets indicate the across bioproject redundancy required, e.g., public_all_3, has all the CDR-H3s that were observed across at least three bioprojects.
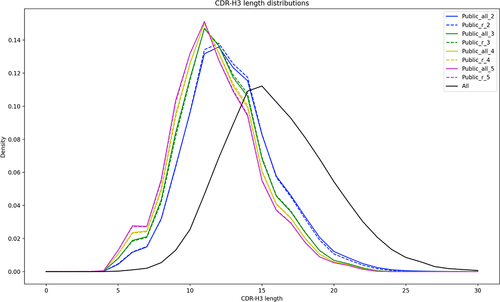
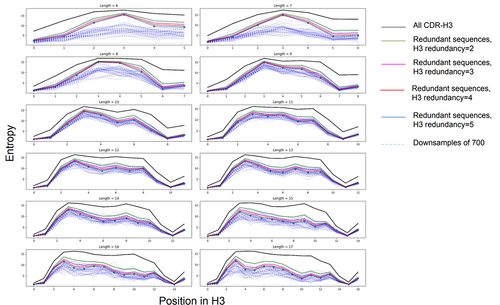
Figure 4. Diversity of the CDR-H3s in the public datasets. Here we show the Confident (redundant) dataset diversity against the background of all the CDR-H3s (‘All’ dataset, black line). Diversity was calculated as amino acid frequency entropy calculated for each position on length-matched CDR-H3s. The entropy is lower in the in the N-terminal region because of higher conservation (e.g., the AR motif). Each panel shows specific CDR-H3 lengths according to the IMGT definition. As expected, downsampling each public dataset to 700 (the number of therapeutic CDR-H3s we have) and recalculating the entropy reduces the diversity further.
Therefore, our results indicate that public CDR-H3s bear features making them more likely to be observed across multiple datasets: they are shorter and less diverse. We checked whether such a collection of CDR-H3s with common features indeed has any strategic immunological diversity bias that can be used therapeutically or whether it is purely a statistical coincidence.
Public CDR-H3s define a reduced set of clonotypes closely reflecting antibodies derived from therapeutic programs
We have previously noted that very good sequence identity matches within the NGS space can be found for some therapeutic CDR-H3s.Citation16,Citation17 Though our previous studies indicated that the public NGS for therapeutic sequences can be mined, they did not shed light on patterns in the convergence across naturally sourced and artificially developed molecules. Any immune repertoire selection biases, especially those shared with the therapeutic space, could be made clinical actionable by biasing selection in that direction. To check whether the set of public CDR-H3s, that is a priori ‘shared’ and radically constrains the diversity space, bears any therapeutic actionables, we checked to what extent it overlaps with therapeutic CDR-H3s.
We identified 700 CDR-H3s that are unique within the set of INN-drawn 827 therapeutics, at various stages of development.Citation16 We next checked the length matched Hamming distance from each of the 700 therapeutic CDR-H3s to one of our ca. 385 m CDR-H3s. For instance, a Hamming distance between sequences POTATOS and TOMATOS is two as they differ in that many length-matched aligned characters. We performed the CDR-H3 matching for the Confident and All datasets and for five CDR-H3 public stringencies (e.g., redundancy 3 indicates that CDR-H3s occurred in at least three different bioprojects).
We plotted the best Hamming distance from our 700 therapeutics to all our varieties of public datasets and the results are shown in . For a non-trivial proportion of therapeutic CDR-H3s, we find perfect matches at all levels of public CDR-H3 stringency. For Confident public sets at bioproject redundancy of 2, we find perfect matches for a total of 9% of all CDR-H3s. Increasing the redundancy to 5, decreases the amount of public CDR-H3s from 3,577,323 to 271,147 (7% of 3,577,323). However, perfect therapeutic matches at bioproject stringency 2 at 9% decreases to only 6%. Moreover, the 271,147 CDR-H3s from Confident dataset at redundancy 5 have 1 Hamming distance and 2 Hamming distance matches for 10% and 15% of therapeutic CDR-H3s, respectively. Therefore, the relatively small number of 271,147 public CDR-H3s cover ca 30% of all therapeutic CDR-H3s within two substitutions. Similar good correspondences between public and therapeutic CDR-H3s are noted for all bioproject redundancies regardless of whether CDR-H3s are from All or Confident datasets (). Comparatively, if we take the entire dataset of 385 m CDR-H3s, it covers 67% of all therapeutics up to two substitutions, despite being ca. 1420 larger in size than the public dataset with 271,147 CDR-H3s. Increasing the bioproject redundancy, radically reduces the number of CDR-H3s in a public dataset, and does not proportionally reduce the number of therapeutic matches. This suggests that there might be a non-trivial convergence between public and therapeutic datasets.
Table 3. Number of therapeutic CDR-H3s within a specific Hamming distance from a dataset. We created a range of reference datasets of CDR-H3s that we compare to the 700 therapeutic CDR-H3s using Hamming distance. As references, we created variants of the Confident and All datasets akin to , where the CDR-H3s were only constrained to those that occurred in a sufficient number of bioprojects (given by bioproject redundancy column). We also created randomizations of each such dataset either by randomizing the CDRs in the NGS dataset (the -D subscript) or the 700 therapeutic CDR-H3s we compared to (−T subscript). For instance, Public All-2 is the subset of CDR-H3s from the ‘All’ dataset, constrained to CDR-H3s occurring in at least two bioprojects and compared to the original unmodified 700 therapeutic CDR-H3s. Random All T-2 is the same dataset, but the 700 therapeutics CDR-H3s are randomized. In Random All D-2, only the original NGS dataset is randomized and therapeutic CDR-H3s are left unmodified. The CDR-H3s are compared by length-matching and hamming distance at five levels, with 0 Hamming distance indicating identity and 4, indicating four differences. Additionally we compared the therapeutic contrast with germline CDR-H3s, in original form from IMGT and applying our randomization scheme to the same germline CDR-H3s.
A non-trivial element that could be contributing to the overlap we observe is the germline nature of the public and therapeutic datasets. It could be the case that the public CDR-H3s are in fact not very diversified away from the germline. To carry out the germline test, we created a set of artificial germline CDR-H3s by combining all possible human V, D and J amino acid segments found in the IMGT database.Citation6 We did not account for V(DD)J fusionsCitation33 because of their low frequency measured in peripheral blood. An alternative approach would be to use a simulation framework such as immuneSIM, SoNNIa,Citation34,Citation35 though we opted for IMGT CDR-H3s because we consider these unencumbered by mutations, representing a germline ground truth.
To check germline-therapeutic overlap, we compared the germline IMGT CDR-H3s and for each therapeutic CDR-H3, calculated the smallest length-matched Hamming distance. The results are shown in . Only at a Hamming distance of 2 do we start to observe any overlap, suggesting that trivial germline nature is not the driving factor in the convergence we see between public and therapeutic CDR-H3s.
Another factor that could be contributing to the therapeutic overlap with the public database is a bias inherent to the discovery procedure of a given therapeutic. We hypothesized that therapeutics derived from murine sources are less likely to be found as good matches in our highly public dataset of 271,147 CDR-H3s, as opposed to human platforms. Of our 700 therapeutic CDR-H3s, we identified 226 CDR-H3s whose origin can be labeled as human and 312 that can be labeled as humanized. There were some chimeric, murine, and antibodies of other sequence source, but their numbers were much smaller with respect to these two groups so we considered a contrast between the human/humanized as the best way forward. A total of 20 human therapeutic CDR-H3s found direct matches in our highly public dataset as opposed to 19 humanized ones. We used Fisher’s exact test to check whether there is a statistically significant difference between observing direct match within a human or humanized dataset. The test statistic was not significant at a strict significance level of 0.01. Therefore, the development platform or organism source did not appear to bestow features that separated the CDR-H3s between those that are frequently found in our highly public dataset.
Since we established previously that public CDR-H3 datasets have features that increase the likelihood of occurrences among datasets, we checked whether the good correspondence between therapeutic and public datasets is due to chance. We devised a conservative randomization scheme for each CDR-H3 that kept two N-terminal residues and three C-terminal residues constant (as these positions are typically less diverse,Citation36 see ), whilst uniformly randomizing the rest. Using this scheme, the shorter CDR-length distribution is already factored in. Since we compare the 700-member CDR-H3 therapeutic set versus public datasets of varying sizes, we introduced two dataset randomization schemes, therapeutic-sided and public-sided. In the therapeutic-sided test, we randomize the therapeutic dataset and compare it against the undisturbed reference dataset (e.g., public dataset). In the public-sided randomization, we modify only the reference dataset leaving the therapeutic CDR-H3s undisturbed. We performed ten separate runs in each case of therapeutic-sided and public-sided randomizations, with the results shown in . Despite quite lenient length-matched randomization scheme, it is clear that randomizations consistently produce much worse matches to therapeutics than undisturbed public datasets. For instance, whereas perfect matches for public datasets are in the range of 6–10% for all therapeutics, they do not go beyond 1–2% for the randomization experiments. It appears that randomizing the public dataset rather than therapeutics produces slightly better results. Though public CDR-H3s have features that are common among different datasets, there is a nontrivial signal driving therapeutic-public dataset convergence that cannot be explained by shorter lengths and random diversity alone.
Though most of the study focused on CDR-H3 alone to avoid the many confounding factors such as isotypes and germlines, selection of antibody-based therapeutics does not hinge on this loop alone. Whilst constructing phage display libraries, a known binder or a framework with known good biophysical properties, such as trastuzumab, might be used.Citation37 For this reason, we also checked whether the public-therapeutic correspondence disappears when information beyond CDR-H3 is introduced, such as the clonotype,Citation7,Citation8 that encompasses information of the entire variable region. For each public CDR-H3 noticed in a public dataset, we also noted its gene call, either V only or in combination with the J-gene. A match between a therapeutic clonotype was called when the CDR-H3 matched to a specific extent (e.g., Hamming distance 2) and there was a match in gene calls.
The best matches to therapeutic clonotype (V or V+J call and CDR-H3 Hamming distance) within a public dataset are shown in . For the Confident dataset at bioproject stringency 5, we found perfect CDR-H3 matches for 6% of therapeutic CDR-H3s compared to 5% when clonotype information is required. At bioproject stringency 5, public CDR-H3s were within 2 substitutions of ca. 30% of all therapeutic CDR-H3s, but this decreases to only ca 27% when clonotype information is required. A similar high correspondence between clonotype () and CDR-H3 () only matches is seen for other bioproject stringencies. This indicates that correspondence between public and therapeutic molecules is not only constrained to CDR-H3s but also to clonotype, which is already information that can be used for drug discovery, e.g., for library design or virtual screening. The set of public CDR-H3s with their assigned clonotypes is much smaller than the number of possible antibodies, but appears to closely reflect diversity of known antibodies discovered via therapeutic programs.
Table 4. Clonotype-level matches of public H3s to therapeutics. As in , for All and Confident datasets, we constrained them to only those CDR-H3s that occurred in a number of bioprojects specified by the Bioproject redundancy column. The CDR-H3 Hamming distance between a reference and therapeutic is calculated only if their V or V+J calls match between the NGS sequence and a therapeutic sequence.
Discussion
Therapeutic antibody discovery hinges on our ability to translate the binding diversity of these molecules into developable therapeutics. The task is daunting due to the vast sequence space of approximately 1018 possible (natural) molecules. In that space, a molecule needs to be found with the right combination of suitable target-binding and manufacturability properties. Antibodies are naturally bestowed with target-binding properties, but not manufacturability. ‘Developable’ antibodies appear to nevertheless form a subset of all available natural antibodies.Citation38
Identifying potentially therapeutic antibodies in this vast space can be facilitated by a combination of an ‘oracle’Citation39 calling binding (or specificity) and developability properties or by reducing the space to more viable molecules. Traditional discovery methods such as display protocols have the benefit of an oracle in the form of a process that filters out binders.Citation40,Citation41 Display methods have no way of calling developable antibodies, which can lead to multiple downstream issues.Citation19 A workaround these methods are using is in fact constraining the initial libraries (or search space) to sequences that are more developable, e.g., without any liabilities.Citation41
Computational binding oracles have been proposed.Citation37,Citation42,Citation43 Opposed to display approaches that ‘constrain’ the space a priori because of their low-throughput nature, computational methods promise to enable exploration of an almost unconstrained set of antibodies. In theory, by being able to perform binding and developability checks quickly computationally, all the combinatorial combinations of sequences could be checked. In practice, there is currently no universal ‘binding oracle’ available, with published approaches relying on a priori deep mutational scans against individual antigens to train the oracles. Developability filtering approaches, though becoming standard, still do not fully reflect experimental performance.Citation44
Both experimental and computational approaches could benefit from ‘constraining’ the search space to only antibodies that a priori have a higher chance of becoming a therapeutic. Here, we tackled this task by setting out an approach to mapping the ‘common diversity’ shared across natural and therapeutic antibodies.
Studying this diversity poses a multi-dimensional challenge even on the sequence level. For example, different elements, such as frameworks and CDRs, all associated with different functions (frameworks as scaffoldsCitation45 or intrinsic features (e.g., canonical classes of CDRs,Citation46 are included within a single variable region. Such considerations become exacerbated with other dimensions, such as structuralCitation47 or embeddings from transformers.Citation48,Citation49 To allow straight-forward analysis and interpretation of our results, we used only the IMGT-derived CDR-H3 sequence. We thus did not account for all the higher-dimensional considerations, even the clonotype, which is a limitation of the study.
Our analysis shows substantial overlap between public CDR-H3s and the therapeutic ones that is not due to statistical prevalence of shared features alone. As little as ca. 270k CDR-H3s are within two substitutions of 30% of all known therapeutic antibodies and 6% are perfect matches. The finding was possible via gross oversimplification of the antibody space to just CDR-H3, but is actually reflected by extending it to clonotypes where public CDR-H3s are observed. Therefore, combining the 270,000 CDR-H3s and the common therapeutic frameworks IGHV3–23, for instance, in the form of a display library, should statistically cover a great number of possible therapeutics.
The biological significance/cause of this observation is beyond the scope of this study (e.g., similar to the GRAB motifs.Citation50 It is plausible to assume that despite great theoretical diversity, antibodies in fact sample a smaller space,Citation5 just by more constrained means, such as conserved structure.Citation51 Our study appears to show that there are biases in which repertoires are constructed, which should be reflected in somatic hypermutation trajectories whenever these can be reasonably reconstructed.Citation52–54 We showed that there does not appear to be statistical significance between humanized or human CDR-H3s being found in the highly public dataset. Therefore, the convergences across immunoglobulins could go beyond therapeutic/natural, but between different organisms as well. Thorough investigation of organism-specific diversity, including murine as well as single-domain antibodies,Citation31 is needed to reveal more actionable observations that constrain the search space for developable therapeutics.
The public-therapeutic overlap we note has an immediate actionable for ‘constraining’ the search space for either experimental or computational approaches. Naive display libraries could be biased toward only the public clones/CDR-H3s to increase the chances of molecules hitting the therapeutic, though unlikely in practice as display library providers need to guarantee ‘uniqueness of their sequences’. With poor performance of generalistic virtual antibody screening methods, constraining their search space to sequences with higher probability of being ‘therapeutics’, could fill the predictive inadequacy gap and make them usable in practice.
We hope that the findings and the data associated with the study will help in antibody engineering efforts.
Materials and methods
Sequence processing
We implemented an internal antibody numbering pipeline to avoid issues with versioning of germline databases and those caused by using different pieces of software at nucleotide and amino acid annotation level. As the human germline reference, we used the freely available germlines from VDJBase.Citation30
The software operates similarly to IgBLAST,Citation25 with a major difference of using the MMseqs229 software instead of BLAST,Citation55 for efficient sequence alignment.
The processing pipeline consists of following steps:
Prepare V, J, and C genes alignments. V gene alignments are the most time consuming, as they are performed on a full germline database as well as both forward and reverse complemented queries. From this result, we can determine a locus as well as a reading frame of the query sequence, which is used to narrow the number of possible germlines during J and C alignments. Next, the already aligned query sequence fragment (for V gene) is masked, and only the rest is taken into consideration during J alignment. This process is repeated for the C gene.
Join alignment results into AIRRCitation56 rearrangement format. Besides germline ids and aligned query sequence fragments, information such as V and J frames, region offsets as well as several useful flags (presence of stop codon, presence of frameshift or other defects that might render the immunoglobulin nonfunctional) are calculated and added to the results.
Number sequences with IMGT schema. We again use MMSeqs2 to calculate pairwise alignments between translated query sequence and gapped germline sequence. Resulting CIGAR string is converted to a state vector and then used directly as input to a modified version of ANARCI,Citation26 without the IMGT database and HMMER. This allows for bypassing the default HMMER alignment, which is computationally expensive.
The annotated alignment uses custom validation rules. This produces a collection of flags, part of which are meant to extend the alignment quality checks (absence of empty regions, conserved amino acid presence) and various technical sanity checks, such as are region offsets correct, are there no negative offsets, is CDR3 in junction.
Add IMGT numbering and validations to alignment data. Results in extended AIRR format.
Summarizing, there are nine extra columns in our extended AIRR format:
study_accession
run_accession
cluster_size
v_frame
j_frame
imgt_residue_mapping
positional_imgt_mapping
exc – optional, exception from IMGT numbering
additional_validation_flags – from step 4 described above
As one can notice, the alignment is performed twice: 1) By precisely aligning query nucleotide sequences to multiple ungapped germline sequences; and 2) By aligning translated query to gapped (using IMGT schema) amino acids sequences of corresponding genes. The second alignment, however, is much less time consuming, as we only need to align to a single, previously selected germline.
MMseqs2 performs this alignment in the following steps:
Frame Generation – generating all possible reading frames (forward and reverse complemented).
Prefiltering – the secret to MMseqs2 performance lies in this step. Using k-mers generated for query and target sequences, the software finds a matching subset and rearranges it into a diagonal. Next, top n (5 in our case) genes sharing the longest diagonal with query sequence are selected. Thanks to that, computational-heavy pairwise alignment is performed on a significantly reduced gene dataset.
Pairwise alignment – a fast implementation (SIMD) of Smith-Waterman algorithm is executed for query-target pairs that were selected during prefiltering.
Choosing best – assign the germline with the lowest E-value. It is defined as the number of expected alignments with similar scores, between a target and a set of random sequences. This way we are taking into consideration also the quality of alignment, which raw Smith-Waterman scores do not provide.
Automatic identification of B-cell receptor-containing bioprojects
The sequencing datasets are abstracted into bioprojects, which consolidate sequencing runs within one study (e.g., scientific publication) into SRA files. To identify novel bioprojects containing antibodies, we analyzed SRA files from ca. 500,000 bioprojects. For each SRA, we applied our antibody numbering pipeline, which returns an empty value for a sequence if a sequence cannot be reliably matched to a germline sequence. Because of the data volume, the pipeline was implemented in Amazon AWS Batch to perform the calculations faster, via parallelization.
We only considered SRA files where more than 10% of sequences were identified as antibodies. This is a very low threshold that was designed to increase the coverage, as SRA files with only 10% antibodies are not considered to be of good quality. A total of 287 bioprojects were identified with more than 10% antibody content. Each of the bioprojects was further manually checked that the subject of the study was BCR sequencing and were not single-cell. This brought the number to 220 high-quality bioprojects.
Metadata processing pipeline
Bioprojects are associated with biosample data that might contain a range of biological parameters relating to sequencing runs such as age and disease. The attributes are not consistent between bioprojects and thus we created a normalized attribute set where attributes available in each biosample were transferred. In the first instance, biosample metadata were automatically downloaded for our 220 bioprojects, resulting in 11,000 biosamples. Each of the biosamples was then inspected manually to fix the correspondence between attributes available in the biosample and our normalized attribute set.
Disclosure statement
PD, DC, JK, TS, BJ, SW, TG, IJ, WB, and KK are current or former employees of NaturalAntibody that commercializes computational solutions for the therapeutic antibody discovery industry.
Data availability statement
The AB-NGS dataset is available for nonprofit organizations for noncommercial use via following instructions on our site: https://naturalantibody.com/ngs/.
Additional information
Funding
References
- Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. Mabs-austin. 2023;15(1):2153410. doi:10.1080/19420862.2022.2153410.
- Mullard A. 2022 FDA approvals. Nat Rev Drug Discov. 2023;22(2):83–12. doi:10.1038/d41573-023-00001-3.
- Boyd SD, Joshi SA, Crowe JE Jr., Boraschi D, Rappuoli R. High-throughput DNA sequencing analysis of antibody repertoires. Microbiol Spectr. 2014;2(5). doi:10.1128/microbiolspec.AID-0017-2014.
- Marchalonis JJ, Adelman MK, Schluter SF, Ramsland PA. The antibody repertoire in evolution: chance, selection, and continuity. Dev Comp Immunol. 2006;30(1–2):223–47. doi:10.1016/j.dci.2005.06.011.
- Rees AR. Understanding the human antibody repertoire. Mabs-austin. 2020;12(1):1729683. doi:10.1080/19420862.2020.1729683.
- Khass M, Vale AM, Burrows PD, Schroeder HW Jr. The sequences encoded by immunoglobulin diversity (DH) gene segments play key roles in controlling B-cell development, antigen-binding site diversity, and antibody production. Immunological Reviews. 2018;284(1):106–19. doi:10.1111/imr.12669.
- Soto C, Bombardi RG, Branchizio A, Kose N, Matta P, Sevy AM, Sinkovits RS, Gilchuk P, Finn JA, Crowe JE Jr. High frequency of shared clonotypes in human B cell receptor repertoires. Nature. 2019;566(7744):398–402. doi:10.1038/s41586-019-0934-8.
- Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 2019;566(7744):393–97. doi:10.1038/s41586-019-0879-y.
- Lupo C, Spisak N, Walczak AM, Mora T, Yates AJ. Learning the statistics and landscape of somatic mutation-induced insertions and deletions in antibodies. PLOS Comput Biol. 2022;18(6):e1010167. doi:10.1371/journal.pcbi.1010167.
- Perelson AS, Oster GF. Theoretical studies of clonal selection: minimal antibody repertoire size and reliability of self-non-self discrimination. J Theor Biol. 1979;81(4):645–70. doi:10.1016/0022-5193(79)90275-3.
- Krawczyk K, Kelm S, Kovaltsuk A, Galson JD, Kelly D, Trück J, Regep C, Leem J, Wong WK, Nowak J. et al. Structurally mapping antibody repertoires. Front Immunol. 2018;9:1698. doi:10.3389/fimmu.2018.01698.
- Raybould MIJ, Marks C, Kovaltsuk A, Lewis AP, Shi J, Deane CM, Dunbrack RL. Public baseline and shared response structures support the theory of antibody repertoire functional commonality. PLOS Comput Biol. 2021;17(3):e1008781. doi:10.1371/journal.pcbi.1008781.
- Kovaltsuk A, Raybould MIJ, Wong WK, Marks C, Kelm S, Snowden J, Trück J, Deane CM, Ofran Y. Structural diversity of B-cell receptor repertoires along the B-cell differentiation axis in humans and mice. PLOS Comput Biol. 2020;16(2):e1007636. doi:10.1371/journal.pcbi.1007636.
- Galson JD, Trück J, Fowler A, Clutterbuck EA, Münz M, Cerundolo V, Reinhard C, van der Most R, Pollard AJ, Lunter G. et al. Analysis of B cell repertoire dynamics following hepatitis B vaccination in humans, and enrichment of vaccine-specific antibody sequences. EBioMedicine. 2015;2(12):2070–79. doi:10.1016/j.ebiom.2015.11.034.
- Khetan R, Curtis R, Deane CM, Hadsund JT, Kar U, Krawczyk K, Kuroda D, Robinson SA, Sormanni P, Tsumoto K. et al. Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. Mabs-austin. 2022;14(1):2020082. doi:10.1080/19420862.2021.2020082.
- Młokosiewicz J, Deszyński P, Wilman W, Jaszczyszyn I, Ganesan R, Kovaltsuk A, Leem J, Galson JD, Krawczyk K, Alkan C. AbDiver: a tool to explore the natural antibody landscape to aid therapeutic design. Bioinformatics. 2022;38(9):2628–30. doi:10.1093/bioinformatics/btac151.
- Krawczyk K, Raybould MIJ, Kovaltsuk A, Deane CM. Looking for therapeutic antibodies in next-generation sequencing repositories. Mabs-austin. 2019;11(7):1197–205. doi:10.1080/19420862.2019.1633884.
- Petersen BM, Ulmer SA, Rhodes ER, Gutierrez-Gonzalez MF, Dekosky BJ, Sprenger KG, Whitehead TA. Regulatory approved monoclonal antibodies contain framework mutations predicted from human antibody repertoires. Front Immunol. 2021;12:728694. doi:10.3389/fimmu.2021.728694.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y. et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci USA. 2017;114(5):944–49. doi:10.1073/pnas.1616408114.
- Olsen TH, Boyles F, Deane CM. Observed antibody space: a diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. 2022;31(1):141–46. doi:10.1002/pro.4205.
- Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, Krawczyk K. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J Immunol. 2018;201(8):2502–09. doi:10.4049/jimmunol.1800708.
- Corrie BD, Marthandan N, Zimonja B, Jaglale J, Zhou Y, Barr E, Knoetze N, Breden FMW, Christley S, Scott JK. et al. iReceptor: a platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories [Internet]. Immunol Rev. 2018;284(1):24–41. doi:10.1111/imr.12666.
- Norman RA, Ambrosetti F, Bonvin AMJJ, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Briefings Bioinf. 2020;21(5):1549–67. doi:10.1093/bib/bbz095.
- Wilman W, Wróbel S, Bielska W, Deszynski P, Dudzic P, Jaszczyszyn I, Kaniewski J, Młokosiewicz J, Rouyan A, Satława T. et al. Machine-designed biotherapeutics: opportunities, feasibility and advantages of deep learning in computational antibody discovery. Brief Bioinform [Internet]. 2022;23(4). doi:10.1093/bib/bbac267.
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41(W1):W34–40. doi:10.1093/nar/gkt382.
- Dunbar J, Deane CM. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics. 2016;32(2):298–300. doi:10.1093/bioinformatics/btv552.
- Lefranc MP, Giudicelli V, Ginestoux C, Bodmer J, Müller W, Bontrop R, Lemaitre M, Malik A, Barbié V, Chaume D. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 1999;27(1):209–12. doi:10.1093/nar/27.1.209.
- Smakaj E, Babrak L, Ohlin M, Shugay M, Briney B, Tosoni D, Galli C, Grobelsek V, D’Angelo I, Olson B. et al. Benchmarking immunoinformatic tools for the analysis of antibody repertoire sequences. Bioinformatics. 2020;36(6):1731–39. doi:10.1093/bioinformatics/btz845.
- Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35(11):1026–28. doi:10.1038/nbt.3988.
- Omer A, Shemesh O, Peres A, Polak P, Shepherd AJ, Watson CT, Boyd SD, Collins AM, Lees W, Yaari G. VDJbase: an adaptive immune receptor genotype and haplotype database. Nucleic Acids Res. 2020;48(D1):D1051–6. doi:10.1093/nar/gkz872.
- Deszyński P, Młokosiewicz J, Volanakis A, Jaszczyszyn I, Castellana N, Bonissone S, Ganesan R, Krawczyk K. INDI—integrated nanobody database for immunoinformatics. Nucleic Acids Res. 2022;50(D1):D1273–81. doi:10.1093/nar/gkab1021.
- Jovčevska I, Muyldermans S. The therapeutic potential of nanobodies. BioDrugs. 2020;34(1):11–26. doi:10.1007/s40259-019-00392-z.
- Briney BS, Willis JR, Hicar MD, Thomas JW 2nd, Crowe JE Jr. Frequency and genetic characterization of V(DD)J recombinants in the human peripheral blood antibody repertoire. Immunology. 2012;137(1):56–64. doi:10.1111/j.1365-2567.2012.03605.x.
- Weber CR, Akbar R, Yermanos A, Pavlović M, Snapkov I, Sandve GK, Reddy ST, Greiff V, Schwartz R. immuneSIM: tunable multi-feature simulation of B- and T-cell receptor repertoires for immunoinformatics benchmarking. Bioinformatics. 2020;36(11):3594–96. doi:10.1093/bioinformatics/btaa158.
- Isacchini G, Walczak AM, Mora T, Nourmohammad A. Deep generative selection models of T and B cell receptor repertoires with soNnia. Proc Natl Acad Sci USA. 2021;118(14):e2023141118. doi:10.1073/pnas.2023141118.
- Ostrovsky-Berman M, Frankel B, Polak P, Yaari G. Immune2vec: Embedding B/T Cell Receptor Sequences in ℝ N using natural language processing. Front Immunol. 2021;12:680687. doi:10.3389/fimmu.2021.680687.
- Mason DM, Friedensohn S, Weber CR, Jordi C, Wagner B, Meng SM, Ehling RA, Bonati L, Dahinden J, Gainza P. et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat Biomed Eng. 2021;5(6):600–12. doi:10.1038/s41551-021-00699-9.
- Bashour H, Smorodina E, Pariset M, Zhong J, Akbar R, Chernigovskaya M, Quý KL, Snapkov I, Rawat P, Krawczyk K. et al. Cartography of the developability landscapes of native and human-engineered antibodies [Internet]. bioRxiv. 2023 [accessed 2023 Nov 9]; 2023.10.26.563958. doi:10.1101/2023.10.26.563958v1.
- Akbar R, Robert PA, Weber CR, Widrich M, Frank R, Pavlović M, Scheffer L, Chernigovskaya M, Snapkov I, Slabodkin A. et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. Mabs-austin. 2022;14(1):2031482.
- Erasmus MF, Ferrara F, D’Angelo S, Spector L, Leal-Lopes C, Teixeira AA, Sørensen J, Nagpal S, Perea-Schmittle K, Choudhary A. et al. Insights into next generation sequencing guided antibody selection strategies. Sci Rep. 2023;13(1):18370. doi:10.1038/s41598-023-45538-w.
- Teixeira AAR, D’Angelo S, Erasmus MF, Leal-Lopes C, Ferrara F, Spector LP, Naranjo L, Molina E, Max T, DeAguero A. et al. Simultaneous affinity maturation and developability enhancement using natural liability-free CDRs. Mabs-austin. 2022;14(1):2115200. doi:10.1080/19420862.2022.2115200.
- Friedensohn S, Neumeier D, Khan TA, Csepregi L, Parola C, de Vries ARG, Erlach L, Mason DM, Reddy ST. Convergent selection in antibody repertoires is revealed by deep learning [Internet]. bioRxiv. 2020 [cited 2023 Jul 11];2020.02.25.965673. doi:10.1101/2020.02.25.965673v1.
- Lim YW, Adler AS, Johnson DS. Predicting antibody binders and generating synthetic antibodies using deep learning. Mabs-austin. 2022;14(1):2069075. doi:10.1080/19420862.2022.2069075.
- Jain T, Boland T, Vásquez M. Identifying developability risks for clinical progression of antibodies using high-throughput in vitro and in silico approaches. Mabs-austin. 2023;15(1):2200540. doi:10.1080/19420862.2023.2200540.
- Saerens D, Pellis M, Loris R, Pardon E, Dumoulin M, Matagne A, Wyns L, Muyldermans S, Conrath K. Identification of a universal VHH framework to graft non-canonical antigen-binding loops of camel single-domain antibodies. J Mol Biol. 2005;352(3):597–607. doi:10.1016/j.jmb.2005.07.038.
- Kelow S, Faezov B, Xu Q, Parker M, Adolf-Bryfogle J, Dunbrack RL. A penultimate classification of canonical antibody CDR conformations [Internet]. bioRxiv. 2022 [cited 2022 Nov 24]. 2022.10.12.511988. doi:10.1101/2022.10.12.511988.
- Jaszczyszyn I, Bielska W, Gawlowski T, Dudzic P, Satława T, Kończak J, Wilman W, Janusz B, Wróbel S, Chomicz D. et al. Structural modeling of antibody variable regions using deep learning—progress and perspectives on drug discovery. Front Mol Biosci. 2023;10. doi:10.3389/fmolb.2023.1214424.
- Leem J, Mitchell LS, Farmery JHR, Barton J, Galson JD. Deciphering the language of antibodies using self-supervised learning. Patterns Prejudice. 2022;3(7):100513. doi:10.1016/j.patter.2022.100513.
- Ruffolo JA, Gray JJ, Sulam J. Deciphering antibody affinity maturation with language models and weakly supervised learning [Internet]. arXiv [q-bio.BM]. 2021. http://arxiv.org/abs/2112.07782.
- Shrock EL, Timms RT, Kula T, Mena EL, West AP Jr, Guo R, Lee I-H, Cohen AA, McKay LGA, Bi C. et al. Germline-encoded amino acid–binding motifs drive immunodominant public antibody responses. Science. 2023;380(6640):eadc9498. doi:10.1126/science.adc9498.
- Chomicz D, Kończak J, Wróbel S, Satława T, Dudzic P, Janusz B, Tarkowski M, Deszyński P, Gawłowski T, Kostyn A. et al. Benchmarking antibody clustering methods using sequence, structural, and machine learning similarity measures for antibody discovery applications. Front Mol Biosci. 2024;11:11. doi:10.3389/fmolb.2024.1352508.
- Corcoran MM, Phad GE, Vázquez Bernat N, Stahl-Hennig C, Sumida N, Persson MAA, Martin M, Karlsson Hedestam GB. Production of individualized V gene databases reveals high levels of immunoglobulin genetic diversity. Nat Commun. 2016;7(1):13642. doi:10.1038/ncomms13642.
- Ralph DK, Matsen FA, Buhler J. Per-sample immunoglobulin germline inference from B cell receptor deep sequencing data. PLoS Comput Biol. 2019 4th. 15(7):e1007133. doi:10.1371/journal.pcbi.1007133.
- Nouri N, Kleinstein SH. A spectral clustering-based method for identifying clones from high-throughput B cell repertoire sequencing data. Bioinformatics. 2018;34(13):i341–9. doi:10.1093/bioinformatics/bty235.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi:10.1016/S0022-2836(05)80360-2.
- Christley S, Aguiar A, Blanck G, Breden F, Bukhari SAC, Busse CE, Jaglale J, Harikrishnan SL, Laserson U, Peters B. et al. The ADC API: a web API for the programmatic query of the AIRR data commons. Front Big Data. 2020;3:22. doi:10.3389/fdata.2020.00022.