
ABSTRACT
In contrast to natural antibodies that rely mainly on the heavy chain to establish contacts with their cognate antigen, we have developed a bispecific antibody format in which the light chain (LC) drives antigen binding and specificity. To better understand epitope-paratope interactions in this context, we determined the X-ray crystallographic structures of an antigen binding fragment (Fab) in complex with human CD47 and another Fab in complex with human PD-L1. These Fabs contain a κ-LC and a λ-LC, respectively, which are paired with an identical heavy chain (HC). The structural analysis of these complexes revealed the dominant contribution of the LCs to antigen binding, but also that the common HC provides some contacts in both CD47 and PD-L1 Fab complexes. The anti-CD47 Fab was affinity optimized by diversifying complementary-determining regions of the LC followed by phage display selections. Using homology modeling, the contributions of the amino acid modification to the affinity increase were analyzed. Our results demonstrate that, despite a less prominent role in natural antibodies, the LC can mediate high affinity binding to different antigens and neutralize their biological function. Importantly, Fabs containing a common variable heavy (VH) domain enable the generation of bispecific antibodies retaining a truly native structure, maximizing their therapeutic potential.
Introduction
Antibodies are a key component of the mammalian adaptive immune system used to fight pathogens. Beyond their biological importance, the exquisite specificity of antibodies has led to their widespread use as research tools, diagnostics, and therapeutic agents. Indeed, since the description of hybridoma fusion to generate the first mouse-derived monoclonal antibodies in 1975, the evolution of antibody generation and engineering technologies have led to the establishment of antibodies as one of the most successful and fast-growing classes of therapeutic modalities. Citation1–3
A natural IgG antibody is composed of four polypeptides, two copies of a heavy chain (HC) and two copies of a light chain (LC). The variable regions of the HC and LC each contain three hypervariable loops, named complementary-determining regions (CDRs), that contact the antigen and thus determine the specificity and affinity of the antibody. Antibody-antigen interfaces can vary significantly in shape and area (from 300 to 900 Å2), depending on the size and nature of the antigen. As the structure of more than a thousand intact IgGs or IgG fragments has been solved (www.rcsb.org), the structural features of CDRs and their contributions to antigen binding are well understood.Citation4–7 The CDR3 of HC and LC (H3 and L3, respectively) are located at the center of the paratope and invariably contribute to interaction with the antigen. In general, the binding is skewed toward the HC with CDRs H3, H2 and L3 providing most of the contact points.Citation8,Citation9 This is also due to the dominant role of H3, which is the most diverse CDR in length and sequence and is always involved in antigen contact.Citation10,Citation11 The high diversity of CDR H3 is created during B cell development by two recombination events linking V, D, and J regions, as well as nucleotide additions and deletions at the junctions of these segments.Citation12,Citation13
In the past two decades, many approaches and technologies to generate bispecific antibodies (bsAbs) capable of engaging two different antigens, or two different epitopes on the same antigen, have been developed.Citation14–16 In some cases, the VH and VL of both antibodies are combined into a single molecule by using linkers (e.g., single-chain variable fragment (scFv)) or reshaping interfaces to favor the correct chain pairing and thus maintain the original antibody specificities.Citation17 To simplify correct chain pairing, the use of antibodies that bear a common and invariable LC has emerged as a widespread strategy. In this case the binding is even more skewed toward the HC, as the LC is not diversified.Citation18–20
We previously described a different approach to generate native bsAbs by co-expressing a common HC and two different LCs: a kappa LC (κ-LC) and a lambda LC (λ-LC).Citation21 In this bsAb format, referred to as a κλ body, the LCs play two major functions. First, as the HC sequence is fixed and not diversified, the antigen binding and specificity are mainly driven by the LC. In addition, the presence of a κ-LC and a λ-LC in the bsAb allows for effective asymmetric purification of the bsAb. Unlike other bsAb formats, no linkers or mutations are introduced and thus κλ bodies retain a truly native human IgG structure, minimizing the potential for stability issues and immunogenicity. Using this platform, thousands of κλ bodies have been generated targeting a wide range of antigens. Our results demonstrate that LCs can effectively drive high affinity and specific binding, as well as provide potent inhibition for ligand – receptor interactions.Citation21–29 Several κλ bodies have been manufactured at large scale and two, TG-1801 (zeripatamig, NI-1701) and NI-1801, are currently undergoing Phase 1 clinical trials (NCT04806035 and NCT05403554, respectively). As such, κλ bodies represent an established clinical stage bsAb format.
To get more insight into the contribution of the LC and the HC to antigen binding in the context of κλ bodies, X-ray crystallography was conducted to resolve Fab-antigen complex structures. In the present study, we determined the structure of the K2 Fab in complex with human CD47 and the S79 Fab in complex with human PD-L1, which respectively contain a κ-LC and a λ-LC paired with the same common HC. The structural analysis of these complexes revealed the dominant contribution of the LCs to antigen binding, but also showed that some contacts are contributed by the common HC both in the context of CD47 and PD-L1. The structure of the K2 Fab:CD47 complex was then used for homology modeling of the K2-derived K38 Fab, which has higher affinity to CD47. This study provides structural evidence that, despite the naturally dominant role of the HC in antigen binding, antibody LCs can drive specific and high affinity antigen binding.
Materials and methods
Identification of anti-PD-L1 antibody arms
Phage display technology was used to identify the S79 anti-PD-L1 antibody. The phage libraries for selection and screening encode scFv that all share the same VH domain, based on the IGHV3–23 germline, and are solely diversified in the VL domain. For fixed VH library construction, human VH and VL germlines were synthesized according to the nucleotide sequence found in IMGT.Citation30 The VH was first cloned into the phagemid vector pNDS1. Then, the CDR1 and CDR2 of the VL genes were diversified using degenerate oligonucleotides containing DMT, RVK and NHT codons depending on the number of randomized positions. Diversified CDR L3 sequences (NNS, NHT and RVK codons) were introduced next by a Type IIS restriction cloning approach.Citation21 Recombinant pNDS1 was electroporated into Escherichia coli TG1 cells and phages expressing the scFv were produced. Aliquots from each scFv phage library (1012 pfu) were blocked with phosphate-buffered saline (PBS) containing 3% (w/v) skimmed milk and then deselected on streptavidin magnetic beads (Dynal). Deselected phages were incubated with 100 nM of recombinant biotinylated human PD-L1 extracellular domain (ACROBiosystems, #PD1-H82E5) captured on streptavidin magnetic beads for two hours at room temperature (RT) on a rotary mixer. The coated beads were captured using a magnetic stand followed by five washes with PBS/0.1% Tween 20 and two washes with PBS. Phages were eluted with 100 nM TEA for 30 minutes at room temperature on a rotary mixer and neutralized with Tris-HCl 1 M pH 7.4. These phages were reamplified after infecting TG1 bacteria cells and used in the next round of selection.
After three rounds of selections, individual infected TG1 clones were inoculated into a 96-deepwell plate containing 0.9 mL per well of 2×TYAG medium (2×TY medium containing 100 μg/mL ampicillin, 0.1% glucose) and grown at 37°C for 5–6 hours (240 rpm). To induce expression, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to give a final concentration of 0.02 mM. The plate was incubated overnight at 30°C with shaking at 240 rpm. After centrifugation, the pellets were resuspended in 150 µl TES buffer (50 mM Tris-HCl pH 8, 1 mM EDTA pH 8, 20% sucrose). A hypotonic shock was produced by adding 150 µL of diluted TES buffer (1:5 TES:H2O dilution) and incubation on ice for 30 minutes. After centrifugation, the supernatants were used for immediate testing.
Screening of scFv for binding to recombinant human PD-L1 was done in a homogenous assay using the CellInsightTM C×5High-Content Screening platform (ThermoFisher Scientific). The following reagents were mixed in each well of a 384-clear bottom well plate: 30 µL of a streptavidin polystyrene bead suspension (Polysciences; 3000 beads/well) coated with biotinylated recombinant human PD-L1 or a biotinylated irrelevant protein as a control, 60 µL of scFv periplasmic preparation, 10 µL of detection buffer (PBS containing human anti-c-myc antibody at 2 µg/mL; anti-human IgG Fc AlexaFluor® 647 diluted 1:500). After shaking at 600 rpm for 5 minutes and incubation at RT for 2 hours, the 384-well plate was read on the CellInsightTM platform. Clones expressing scFv giving a specific signal on human PD-L1 and not on the control protein were selected for sequencing.
The parental anti-PD-L1 antibody obtained, based on the IGLV2–14 germline, was further optimized by affinity maturation. Two new phage libraries were constructed based on the parental sequence. Mutations were only introduced in the three CDRs (based on Kabat definition and numbering) of the parental VL sequences, by blocks of 3 to 5 randomized residues at a time (Table S1). In the first library, NHT and RVK codons were introduced in positions 28, 29, 30, 31, 32 of the CDR1, and NHT/VNT codons in the positions 50, 51, 53 of the CDR2. The CDR3 sequence was kept unchanged. In the second library only the CDR3 was diversified using overlapping degenerated oligos containing blocks of 5 NNS codons and the CDR1 and CDR2 of the parental sequence were kept unchanged. Both libraries were used in selections as described above, the first round using 1 nM of biotinylated recombinant human PD-L1 (ACROBiosystems, #PD1-H82E5) and the second round using 0.1 nM of biotinylated recombinant cynomolgus PD-L1 (Sino Biological, #90251-C08H-B). After two rounds of selections, the plasmids of the outputs were used to create a third library, by recombining the CDR1 and CDR2 outputs of the first library with the CDR3 outputs of the second library. Three rounds of selections were performed with this third library using decreasing concentrations of biotinylated recombinant human PD-L1 (1, 0.1 and 0.01 nM).
The screening of individual clones was performed as described above for PD-L1 arm identification.
Identification of anti-CD47 antibody arms
The generation of the anti-CD47 K2 antibody was described previously in Fischer et al.Citation21 The K38 antibody was obtained after affinity maturation of K2. Briefly, a fixed VH phage display library was constructed by introducing diversity in the CDRs (Kabat definition and numbering) of the VL sequence of the K2 antibody, based on the IGKV1–39 germline. In the CDR1, positions 28, 30, 31, 32, 34, and in the CDR2, positions 50, 51, 53 were diversified using NHT/RVK codons (Table S1). The selections were performed as described above for anti-PD-L1 antibody arm identification with decreasing concentrations (10, 1, 0.1 nM) of biotinylated recombinant human CD47 protein (produced in-house). Finally, the screening of individual clones was performed as described above on biotinylated human CD47.
Bispecific antibody and fab fragment generation
Small-scale bsAb production and purification was previously described.Citation21
For the generation of S79 and K2 Fabs, an expression plasmid encoding the Fab was first generated for each Fab. Plasmids were transiently transfected in Expi293 cells by polyethyleneimine. Cells were grown in Expi293 culture medium for 7 days at 37°C, 8% CO2 and 125 rpm in an ISF1 Kuhner incubator. Supernatant was harvested and clarified by addition of diatomaceous earth and filtration at 0.22 μm (Sartorius). Fabs were then purified by affinity chromatography using CaptureSelect IgG-CH1 Affinity Matrix (ThermoFisher Scientific). Briefly, supernatants were incubated with the resin overnight (O/N) at 4°C and 15 rpm. Then, the resin was washed with PBS and transferred on an Amicon Pro column (Millipore) for elution steps with elution buffer (50 mM glycine pH 3.5). Elution fractions were pooled, dialyzed, and concentrated on Amicon 10 kDa centrifugal filter units (Millipore). Fabs were formulated in histidine 25 mM, NaCl 125 mM, pH 6.0.
Purified bsAbs and Fabs were analyzed by electrophoresis in denaturing and reducing or non-reducing conditions using the Bioanalyzer 2100 instrument with the Protein 80 kit, as described by the manufacturer (Agilent Technologies). Molecule integrity was assessed by isoelectric focusing (Cambrex pH 3–10 IsoGel agarose plates). Aggregate and fragment levels were determined by size exclusion chromatography – ultra-high-performance chromatography (SEC-UPLC) with an Acquity UPLC BEH SEC column, 200Å (Waters).
Bispecific antibody affinity measurements
Biolayer interferometry (BLI) (Octet RED96, Sartorius) was used at 30°C to determine the binding kinetics and affinity of S79 bsAb to human PD-L1 and K2 and K38 bsAbs to human CD47.
For S79, after hydration and a baseline step in kinetic buffer (Sartorius; PBS, 0.02% Tween20, 0.1% bovine serum albumin (BSA), 0.05% sodium azide), streptavidin biosensors (Sartorius) were loaded for 5 minutes with biotinylated human PD-L1 recombinant protein at 1 µg/mL (ACROBiosystems, #PD1-H82E5) in kinetic buffer. Then, biosensors were dipped into a serial dilution of S79 bsAb starting from 28.6 nM with a 2-fold dilution factor. Association and dissociation steps were monitored for 600 seconds and 900 seconds, respectively. The affinity was measured applying a 1:1 global fitting model on double referenced curves, on the full association and dissociation steps.
For K2, streptavidin biosensors were loaded for 5 minutes with biotinylated CD47 at 5 µg/mL in kinetic buffer. Loaded biosensors were transferred into a serial dilution of K2 bsAb starting from 166 nM with a 2-fold dilution factor. Association and dissociation steps were monitored for 60 seconds and 300 seconds, respectively. The affinity was measured applying a 1:1 global fitting model on double referenced curves, on the full association and dissociation steps.
Finally, for K38, after hydration and baseline steps, HIS1K biosensors (Sartorius) were loaded for 5 minutes with His-tagged human CD47 recombinant protein at 2.5 µg/mL (ACROBiosystems, #CD7-H82E9) in kinetic buffer. Then, biosensors were dipped into a serial dilution of K38 bsAb starting from 200 nM with a 2-fold dilution factor. Association and dissociation steps were monitored for 120 seconds and 300 seconds, respectively. The affinity was measured applying a 1:1 global fitting model on double referenced curves, on the full association step and on the first phase 100 seconds of the dissociation step.
ELISA binding assay
The ability of bsAbs to bind recombinant human PD-L1 or CD47 was assessed using a sandwich ELISA assay. A goat anti-human IgG Fc capture antibody (Jackson ImmunoResearch, #109-005-190) was coated at a concentration of 5 μg/mL in PBS O/N at 4°C in a 96-well black MaxiSorp plate (Nunc) and discarded before blocking buffer (PBS, BSA 2%, Tween 20 0.05%) was added for one hour at RT. After a washing step with wash buffer (PBS, Tween 20 0.05%), bsAbs were captured on plates at a fixed concentration for one hour at RT, and, after a second washing step, increasing concentrations of biotinylated human PD-L1 (ACROBiosystems, #PD1-H82E5) or human CD47 (produced in-house) proteins were added and the plates incubated for one hour at RT. Plates were washed three times and incubated with Streptavidin-HRP (Jackson ImmunoResearch, #016-030-084, dilution 1:15’000 in blocking buffer) for one hour at RT followed by a washing step. Finally, plates were read using a fluorescence reader (Biotek Synergy HT) after incubation with Amplex UltraRed detection solution (Invitrogen) for 20 minutes at RT in the dark.
A clinical batch of anti-PD-L1 atezolizumab (TecentriqTM, FarmaMondo) was used as a reference in PD-L1 binding assays.
Cell-based binding assay
HT-1080 tumor cells (ATCC; CCL-121), that endogenously express PD-L1 and CD47, were exposed to IFNγ (50 ng/mL) for 24 hours to enhance PD-L1 expression. 3 × 105 cells/well in a 96-well conical-bottom plate (Nunc) were incubated with serial dilutions of bispecific antibodies in fluorescence-activated cell sorting (FACS) buffer (PBS, BSA 2%) for 15 minutes at 4°C. After a washing step, cells were resuspended in FACS buffer and incubated with AF647-conjugated F(ab’)2 Fragment goat anti-human IgG, Fcγ fragment specific (Jackson ImmunoResearch, #109-606-170, dilution 1:200) in the dark at 4°C for 15 minutes. After washing, cells were resuspended in 200 μL FACS buffer with SYTOX Green Dead Cell Stain (ThermoFisher Scientific, dilution 1:5000) per well and analyzed on a CytoFLEX S flow cytometer (Beckman Coulter). Gating was performed on the viable cells and mean fluorescence intensity (MFI) was determined for each concentration of antibody tested.
Cell-based blocking assays
To assess PD-1/PD-L1 blocking activity, PD-L1-transfected CHO cells (CHO-PD-L1, negative for human CD47) or HT-1080 tumor cells, pre-activated with IFNγ for 24 hours (50 ng/mL), were incubated with CellTrace Violet dye (ThermoFisher Scientific) for 20 minutes at 37°C and diluted in PBS-BSA 2% before distribution in 384-well clear-bottom black polystyrene microplates (Corning). Serial dilutions of bsAbs were incubated for 1 hour at RT after a short centrifugation step. As detection reagent, a mix of 0.1 µg/mL of human PD-1-moFc protein (ACROBiosystems, #PD1-H5255) for CHO-PD-L1 cells, or 0.2 µg/mL of high-affinity human PD-1-moFc protein (produced in-house, based on the high affinity consensus PD-1 variant published in Maute et al.Citation31 for HT-1080 cells and AF647-conjugated goat anti-mouse IgG, Fcγ fragment specific (Jackson ImmunoResearch, #115-605-071, dilution 1:5000) was added for 3 hours at RT before reading. A clinical batch of anti-PD-L1 atezolizumab (TecentriqTM, FarmaMondo) was used as a reference for PD-1/PD-L1 blockade.
To evaluate CD47/SIRPα blocking activity, NALM-6 tumor cells (ATCC; CRL-3273, negative for PD-L1) or HT-1080 cells, pre-activated with IFNγ for 24 hours (50 ng/mL), were incubated with CellTrace Violet dye for 20 minutes at 37°C and diluted in PBS-BSA 2% before distribution in 384-well clear-bottom black polystyrene microplates. Serial dilutions of antibodies were incubated for 1 hour at RT after a short centrifugation step. As detection reagent, a mix of 0.05 µg/mL of human SIRPα-moFc protein (produced in-house) and AF647-conjugated goat anti-mouse IgG, Fcγ fragment specific (Jackson ImmunoResearch, #115-605-205, dilution 1:5000) was added for 3 hours at RT.
Finally, the plates were read using the CellInsightTM C×5High Content Screening Platform (ThermoFisher Scientific).
Expression and purification of extracellular domain of human CD47 and IgV domain of PD-L1 for crystallography study
The extracellular domain (ECD) of human CD47 gene (Accession number Q08722, residues 19–141, C33G) with a C-terminal 6-histidine tag was cloned in the pTT5 vector and expressed and purified as described previously.Citation32 Briefly, human CD47 was produced through transient expression in HEK293 cell line as secreted protein in the presence of kifunensine. The protein was purified by nickel affinity chromatography using a HisTrap excel column (Cytiva) followed by a size exclusion chromatography (SEC) step on a Superdex 75 column (Cytiva) in buffer composed of 10 mM HEPES, 140 mM NaCl, pH 7.4. The purified CD47 protein was then deglycosylated using endoglycosidase Hf and cleaned through a final polishing SEC step.
The IgV domain of human PD-L1 protein (Accession number Q9NZQ7, residues 18–133) with a C-terminal 6-histidine tag was cloned into the vector pET21b and expressed and purified as described previously.Citation33 Briefly, the plasmid was transformed into E. coli BL21 (DE3) bacteria and expressed as inclusion bodies. Bacteria were cultured at 37°C in LB, induced with 0.5 mM IPTG at OD600 = 0.7, and incubated for 5 hours at 37°C. After a centrifugation step, bacteria were resuspended in lysis buffer (20 mM Tris-HCl pH 7.4, 1% Triton X-100, 20 mM EDTA, 2 mM PMSF, 0.25 mg/mL lysozyme, and DNase) and lysed by sonication. Inclusion bodies were collected by centrifugation, washed 3 times in 20 mM Tris-HCl pH 7.4, 1% Triton X-100, and 20 mM EDTA and resolubilized in dissolving buffer (20 mM Tris-HCl pH 7.4, 6 M GuHCl, 10 mM DTT). PD-L1 IgV domain was refolded by drop-wise dilution with gentle stirring in buffer consisting of 0.1 M Tris-HCl pH 8.0, 1 M L-Arginine, 2 mM EDTA, 0.2 mM oxidized glutathione and 0.25 mM reduced glutathione. The protein was concentrated, subsequently dialyzed against 10 mM Tris-HCl pH 8.0 and 20 mM NaCl and clarified by centrifugation before loading on HisTrap FF columns (Cytiva) pre-equilibrated with buffer A (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 25 mM Imidazole) and elution with a gradient of buffer B (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 500 mM Imidazole). Fractions from immobilized metal ion affinity chromatography (IMAC) purification containing the protein of interest were finally combined, concentrated, and loaded onto a Superdex 200 column using a SEC buffer composed of 10 mM Tris-HCl pH 8.0, 20 mM NaCl for a final purification step.
K2 Fab:CD47 and S79 Fab:PD-L1 complex formation and purification
To generate crystals of K2 Fab with deglycosylated human CD47-His tag, the two proteins were mixed at a molar ratio of 1:1.2, respectively. The complex was purified by SEC on a Superdex 200 column and concentrated at 15.5 mg/mL in 10 mM HEPES pH 7.4, 140 mM NaCl.
To obtain crystals of S79 Fab with IgV domain of human PD-L1, both proteins were mixed at a molar ratio of 1:1.5, respectively. The complex was purified by SEC on a Superdex 75 column and concentrated at 10.5 mg/mL in 10 mM Tris pH 8.0, 20 mM NaCl.
Crystallization, data collection and crystal structure determination of the K2 Fab:CD47 complex
The crystal structure of the complex between human CD47 ECD and K2 in Fab format has been deposited in the protein data bank with PDB ID 8RP8.
The best diffracting crystal was obtained from sitting drops formed by 160 nL of 15.5 mg/mL K2 Fab:CD47 complex, 120 nL of precipitant solution, and 40 nL of microcrystal seed solution. The precipitant solution consisted of 20% w/v PEG 8000 and 100 mM sodium citrate pH 5.0, and the micro-crystal stabilization solution contained 20% w/v PEG 3350, 50 mM NaCl, 150 mM sodium D,L-malate pH 7.2, and 10 mM sodium HEPES pH 7.4. The crystallization drops were set up using a Mosquito crystallization robot (SPT Labtech) and were left to equilibrate against 40 μL precipitant solution at 20°C. The crystals appeared within 24 hours and grew to full size in a week. The crystals were cryo-protected in a drop of precipitant solution supplemented with 16% v/v PEG 400 and 8% v/v glycerol prior to flash-cooling in liquid nitrogen.
Data were collected to 2.0 Å resolution at beamline BioMAX, MAX IV Laboratory, Lund, Sweden, at 100 K and λ = 0.9763 Å. The beamline was equipped with a DECTRIS Eiger 16 M hybrid-pixel detector. 3600 images were collected with an oscillation range of 0.10° per image, and the data were processed using the XDSAPP pipeline,Citation34 which includes the software XDSCitation35 and Pointless.Citation36
An initial 3D structure was obtained by the molecular replacement method using PhaserCitation37 and two published structures of human CD47 and a Fab protein, with the CDR loops removed, as search models (PDB IDs 5TZU and 6UTA, respectively). The initial structure refinement was carried out in Refmac5Citation38 and model-building was performed with Coot.Citation39 The final stage of the refinement was carried out in Buster.Citation40 Non-crystallographic local symmetry restraints between the two independent Fab complexes were used during the refinement. The summary of data collection and refinement statistics is displayed in . The interactions between the different subunits of the K2 Fab:CD47 complex were analyzed using QtPISA, without including the hetero atoms.
Table 1. Data collection and refinement statistics for the K2 Fab:CD47 and the S79 Fab:PD-L1 crystal structures. aNumbers in parentheses are corresponding values for the highest resolution shell. bValues given by Molprobity.Citation41
Crystallization, data collection and crystal structure determination for S79 Fab:PD-L1 complex
The crystal structure of the complex between human PD-L1 IgV ECD and S79 in Fab format has been deposited in the protein data bank with PDB ID 8RPB.
Crystallization drops were formed by 100 nL of 5.2 mg/mL S79 Fab:PD-L1 complex and 100 nL of reservoir solution consisting of 0.1 M Tris-HCl pH 8.0 and 1.6 M Li2SO4. The crystals were cryo-protected in reservoir solution supplemented with 20% v/v glycerol prior to flash-cooling in liquid nitrogen.
Data were collected to 2.79 Å resolution at beamline BioMAX, MAX IV Laboratory, Lund, Sweden, at 100 K and λ = 0.9763 Å. The beamline was equipped with a DECTRIS Eiger 16 M hybrid-pixel detector. 3600 images were collected with an oscillation range of 0.10° per image. The data was processed using EDNACitation42 and the software XDS.Citation35
PD-L1 IgV domain/S79 Fab structure was determined by molecular replacement using Phaser. The search models were the crystal structure of PD-L1 (PDB ID 3FN3) with the IgC domain removed and a Fab (PDB ID 5VEB) with the CDR loops removed. The structure was initially refined using Refmac5 and model building was carried out in Coot. The final stage of the refinement was carried out in Buster. The higher resolution structures with PDB IDs 5JDS (chain A), 6GHG (chain A) and 3TNN (chain B) were used as target structures for the refinement of PD-L1, S79 Fab HC, and S79 Fab LC, respectively. The summary of data collection and refinement statistics is displayed in . The interactions between different subunits of the S79 Fab:PD-L1 complex were analyzed using QtPISA, without including the hetero atoms.
Modelling of K38 Fab:CD47 interactions
Knowing that the HC is the same for both Fabs, a comparative homology model of the LC of the K38 Fab was designed based on the structure of the LC of the K2 Fab.
Residues 1 to 214 of K38 LC were built using the Prime module from the Schrödinger computational chemistry suite of programs (Schrödinger, L.L.C., Portland, OR, USA). The sequence identity was 97% with the chain B of the K2 Fab crystal structure and rotamers from conserved residues were retained. The C-terminal cysteine residue (C215) was not built, as this residue is not visible in the K2 Fab crystal structure. No gaps existed so no extensive refinement was necessary. Only a short energy minimization of the side chains for V28, A30, N31, W32, G51, H53 and S67 was performed after inserting the K38 LC into the crystal structure of the K2 Fab:CD47 complex. The PDB file of the complex was prepared using the Protein Preparation Wizard in the Schrödinger suite, where hydrogens were added to all chains prior to the minimization. Minimization was performed using the MacroModel module from the Schrödinger suite and the OPLS4 force field.Citation43
Results
Identification of Fabs with identical heavy chain for bispecific κλ body assembly
In this study we focused on 3 Fabs that bind specifically to different antigens while sharing an identical VH. Two Fabs, K2 and K38, contain a κ-LC and bind human CD47, whereas the S79 Fab contains a λ-LC and binds to human PD-L1. These antibodies were isolated from phage display libraries containing a fixed VH combined with diversified LCs. The general approach that allowed the generation of the bispecific κλ body platform, as well as the isolation of the low affinity anti-CD47 arm K2, was previously described in details.Citation21 The higher affinity K38 antibody was derived from K2 via affinity optimization, by diversification of the CDR L1 and CDR L2, followed by phage display selections under stringent conditions.
The same fixed VH libraries were used to identify high-affinity neutralizing anti-PD-L1 antibodies. After initial screening and characterization, two consecutive optimization campaigns performed on the selected candidate led to the isolation of the high-affinity S79 anti-PD-L1 antibody, as described in the methods section. Affinity measurement confirmed the high affinity of S79 for human PD-L1 (KD = 0.5 nM) (Figure S1A and S1D) and the improved affinity of the K38 arm for human CD47 (KD = 13.6 nM) (Figure S1B and S1D), which represents a more than 20-fold increase over the low-affinity anti-CD47 K2 arm (KD = 315 nM) (Figure S1C and S1D).
Generation of κλ bodies mediating either tumor selective CD47/SIRPα inhibition or simultaneous PD-1/PD-L1 and CD47/SIRPα inhibition
CD47 is an innate immune checkpoint receptor and attractive target in immuno-oncology, but has proven to be challenging in a therapeutic setting due to its ubiquitous expression.Citation44–46 For that reason, we developed bispecific antibody approaches to improve tumor targeting and overcome safety risks.
A first mode of action that we have previously described, and which is currently evaluated in clinical trials, is the selective blockade of the CD47/SIRPα axis on tumor cells leading to increased killing via phagocytosis.Citation21–26,Citation29 To achieve this goal, the K2/TAA bispecific κλ bodies combine the low-affinity anti-CD47 K2 Fab arm with high affinity Fab arms targeting tumor-associated antigens (TAA) and carry a hIgG1 Fc that triggers effector functions (). The significant difference in affinities between the TAA targeting Fab arm (sub- or low nM range), such as CD19 or mesothelin, and the CD47-blocking Fab arm K2 (KD = 315 nM) is key to support the intended mode of action.Citation22–24 Importantly, low affinity circumvents hematological toxicities linked to the ubiquitous expression of CD47, as the κλ bodies only minimally engage cells that do not express the TAA, such as erythrocytes and platelets. In contrast, effective neutralization of CD47 and enhanced phagocytosis of TAA-expressing cells is achieved via avidity provided by co-engagement of the two Fab arms on the cell surface.Citation22,Citation23
Figure 1. Schematic representation of bispecific antibodies used for structural studies.
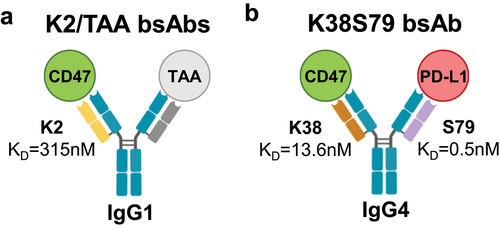
To support a second mode of action relying on independent and simultaneous inhibition of the CD47/SIRPα and PD-1/PD-L1 checkpoints, we generated the K38S79 κλ body incorporating the K38 anti-CD47 and S79 anti-PD-L1 arms and carrying an IgG4 isotype that has reduced Fc-mediated effector functions to mitigate CD47-related toxicities ().
The S79, K2 and K38 Fab arms were reformatted to express and purify the K2/TAA and K38S79 κλ bodies (Figures S2A, S2C and S2D) that were characterized for binding to their recombinant targets (). Using a sandwich ELISA format to avoid avidity, S79 showed a binding profile similar to that of atezolizumab, an approved anti-PD-L1 therapeutic antibody that has an affinity for PD-L1 close to S79Citation47 (). The same assay format was used to evaluate the binding of K2 and K38 to human CD47 and demonstrated the higher binding capacity of K38 compared to K2, consistent with their respective affinities ( and S1D). Then, the PD-1/PD-L1 or CD47/SIRPα blocking properties of the 3 arms were assessed in a cell-based assay. In line with their binding properties, S79 and K38 showed efficient blockade of PD-1/PD-L1 () and CD47/SIRPα interactions (), respectively, while the low-affinity K2 arm triggered only partial SIRPα blockade at high concentration ().
Figure 2. Functional characterization of anti-PD-L1 S79 and anti-CD47 K2 and K38 antibody arms used for structural studies.
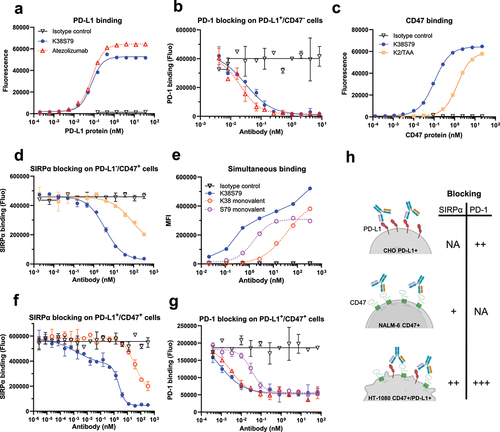
Subsequently, we evaluated the binding and blocking properties of the anti-CD47×PD-L1 K38S79 κλ body using HT-1080 tumor cells that express both human CD47 and PD-L1. Compared to monovalent K38 and S79 controls, K38S79 bsAb showed increased binding (), CD47/SIRPα blockade () and PD-1/PD-L1 blockade () enhanced by simultaneous engagement of CD47 and PD-L1 on the cell surface. Overall, these results show that K38S79 can independently block both CD47/SIRPα and PD-1/PD-L1 interactions and that its activity is enhanced by avidity following co-engagement of both targets, as illustrated in .
Then, to gain insights into the κλ body LC contribution to antigen binding, we selected for structural studies the above-described K2 and S79 arms. The 2 Fabs were successfully expressed, produced, and purified with good quality attributes in terms of purity and aggregate level (Figure S2B, S2C and S2D).
Crystallization and structure determination of K2 Fab:CD47 complex
The K2 Fab:CD47 complex was purified (Figure S3), crystallized, and the structure was determined to a 2.0 Å resolution () with a high-quality electron density map for both K2 (Figure S4A) and CD47 (Figure S4B). The structure contains two complexes in the asymmetric unit and includes HC residues 1–131 and 137–217, LC residues 1–214 and CD47 residues 1–118.
Figure 3. Structural basis for blocking the CD47/SIRPα interaction by the anti-CD47 K2 fab.
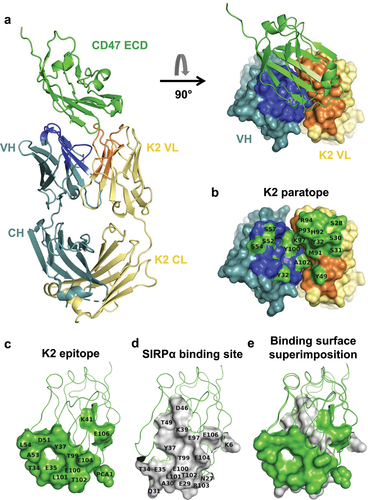
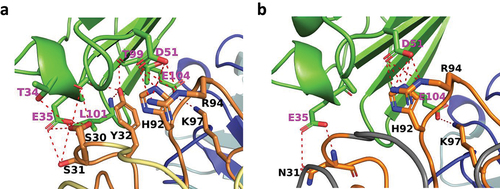
The average solvent-accessible areas excluded in the pairwise interactions between CD47 and the K2 Fab were calculated by the software Areaimol of the CCP4 software packageCitation49 to 662.7 Å2 and 671.3 Å2 for the two complexes, in the asymmetric unit. Residues of CD47 with atoms within 4.0 Å of either the K2 Fab HC or LC were defined as epitope residues () and, likewise, residues of the K2 Fab with atoms within 4.0 Å of CD47 protein defined as paratope residues (). The paratope and epitope interfaces of the K2 Fab:CD47 complex are depicted in , respectively. The interfaces between CD47 and the K2 Fab LC and HC were also analyzed by QtPISA and hydrogen bonds and salt-bridges expected to be present in the interaction were reported in Supplementary Table S2. The analysis clearly shows that the major interaction contribution between CD47 and the K2 Fab comes from the LC and to a lesser extent from the HC, as illustrated by and .
Table 2. Contact residues forming the epitope of K2/K38 and S79 on CD47 and PD-L1, respectively.
Table 3. Contact residues within each chain forming the paratope of K2, K38 and S79 fabs. Residues are displayed according to the PDB numbering described in Supplementary Table S1. CDRs are delimited according to contact definition.Citation50,Citation51
Several CD47 residues that have been shown to make contacts with SIRPα are part of the K2 paratope, providing structural evidence for the neutralizing properties of the K2 arm (). These residues include T34, E35, Y37, T99, E100, L101, T102, E104, E106.The residues L101 and T102 are part of the FG loop of CD47, which is a key determinant of the interaction with SIRPα and is often targeted by neutralizing anti-CD47 antibodies.Citation48,Citation52,Citation53
Crystallization and structure determination of S79 Fab:PD-L1 complex
After purification of the S79 Fab:PD-L1 complex (Figure S5), we obtained crystals that diffracted to a 2.79 Å resolution with good electron density for both S79 and PD-L1 IgV (Figure S6A and S6B). The structure of S79 Fab:PD-L1 contains one complex in the asymmetric unit and includes residues 1–218 for the Fab HC, 1–215 for the Fab LC and 18–130 for the IgV domain of PD-L1 ().
Figure 4. Structural basis for blocking the PD-1/PD-L1 interaction by the anti-PD-L1 S79 fab.
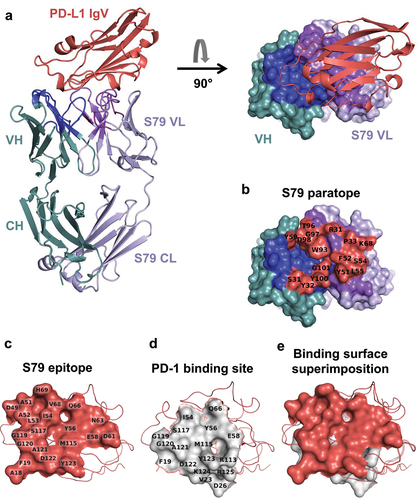
The average buried surface area calculated for the complex was 860.3 Å2. Residues of IgV_PD-L1 with atoms within 4.0 Å of either the HC or LC were defined as epitope residues ( and ) and residues of the S79 Fab with atoms within 4.0 Å of the IgV_PD-L1 molecule were defined as paratope residues ( and ). All contact residues fall within the CDRs except for K68, which is in the framework 3 region of the S79 λ-LC.
The QtPISA tool was also run on this complex to analyze the interactions between the different chains. From all results generated, the interface parameters are reported in Supplementary Table S2. Similarly to the K2 Fab:CD47 complex, the analysis clearly shows that the LC, in this context as well, contributes more to the interaction with PD-L1 than the HC, as illustrated by and .
PD-L1 residues I54, Y56, Q66, M115, S117, A121, D122 and Y123, that lie in the center of the S79 epitope, are also key contact residues for interaction with PD-1 (). In addition, the S79 epitope is similar and overlapping with those of atezolizumab, avelumab and durvalumab, three clinically approved neutralizing anti-PD-L1 monoclonal antibodies.Citation54–57
Structural comparison of the common VH in different contexts
We compared the structures of the K2 and S79 Fabs in complex with their antigens by superimposing the protein backbone of their common VH domains (Figure S7). Interestingly, and in contrast to CDR H1 and CDR H2, the CDR H3 loops do not overlap. This demonstrates that, despite its short length and identical sequence, the CDR H3 can adopt different conformations in different contexts, such as pairing with a different LC and/or in complex with a different antigen.
Modelling K38 Fab:CD47 interaction/homology model
The K38 Fab has approximately 20-fold higher affinity toward CD47 compared to the K2 Fab, which is mainly driven by a slower off-rate (Figure S1D). In order to understand the structural differences between K2 and K38 that lead to this increase in affinity, a homology model of the LC of K38 Fab was generated using the Prime module in the Schrödinger software. The crystal structure of the K2 Fab:CD47 complex was used as template for the model.
The epitope of CD47 in the K38 complex is identical to the one in the K2 complex (). A few intermolecular hydrogen bonds are lost due to the change of amino acid types in CDR1 and CDR2 of K38 VL (, , Table S1 and S2). Mainly the change of Y32 into a tryptophan removes the hydrogen bonds to E35 and L101 in CD47. Furthermore, the change of S30 into an alanine removes the hydrogen bond to T34. No new intermolecular hydrogen bonds are formed in the complex. However, due to the change of S31 into an asparagine, new hydrogen bonds are formed with the backbone carbonyl of A50 and the side chain of S52 in the K38 CDR L2 loop, while the hydrogen bond to S67 is retained. This likely stabilizes the whole CDR L1 loop that makes most of the interactions with CD47. In this CDR loop, W32 makes more extensive van der Waals interactions with residues in the CD47 epitope compared to the previous tyrosine, especially with the side chain of L101 in CD47. In addition, replacement of S30 into an amino acid with smaller side chain like alanine creates more space for the large tryptophan side chain. W32 residue most likely also makes a stronger π-stacking with H92 in the CDR L3 loop, stabilizing the intermolecular hydrogen bonds between H92 and R94 in the CDR L3 loop and D51 in CD47. Furthermore, the replacement of S28 by a valine also adds some hydrophobic interactions with L54 in CD47. Finally, the replacement of S53 by histidine stabilizes the CDR L2 loop by π-π stacking of H53 with Y49.
Figure 5. Modelling of the binding interface to CD47 of the K2-derived, affinity-enhanced, K38 fab.
Discussion
During the process of antibody repertoire generation by the immune system of mammals, the variable regions of the heavy chain are more diversified than the LC variable regions. The high sequence and length diversity of the CDR H3 correlates with its central role in antigen binding and to a clear bias toward heavy chain contribution to the antibody paratope. This is observed in most antibody-antigen interactions that have been studied via structural approaches. Using antibody libraries containing a fixed heavy chain antibody and diversified LC repertoires, we isolated thousands of antibodies that are specific for a wide variety of soluble or cell surface antigens. In this study, the structural analysis of such antibodies provides a better understanding of how the LCs can drive specificity against different antigens while paired with the same HC.
The buried surface areas in the complexes between the Fabs and CD47 or PD-L1 (670 Å2 and 860 Å2, respectively) fall within the normal range that is observed for antibodies binding to proteins. An important finding is that even if most interactions are provided by residues located in the LC CDRs, the heavy chain also contributes to the binding in both Fab-Ag complexes. Thus, the hypothesis that the binding could have been solely mediated by the VL, mimicking a single domain-antigen interaction, could clearly be excluded. Interestingly, two tyrosines, Y32 and Y100, located in CDR H1 and H3 respectively, make contact both with CD47 and PD-L1. In addition, several serine residues in CDR H1 and H2 are also involved in antigen contact with these two antigens. It is interesting to note that tyrosines can mediate different types of interactions and have been shown to be intrinsically well suited to mediate a molecular recognition of different antigens and are often enriched in naturally occurring paratopes.Citation58 It can be anticipated that these residues are able to promiscuously contribute to binding to different antigens.Citation59 The versatility of these residues was demonstrated as antibodies specific for different antigens could be readily isolated from libraries with drastically restricted CDR diversity, as they contained only serine and tyrosine residues.Citation60
It is important to highlight that the fixed HC we used contains a VH3–23 variable domain with germline CDR H1 and H2 sequences and a short CDR H3 of 7 amino acids length. Thus, the heavy chain residues involved in antigen contact are naturally occurring and were not specifically engineered. The short H3 was deliberately chosen in the library design to create space at the center of the antibody combining site and allow for contribution of the LC CDRs.Citation21 The superimposition of the two K2 and S79 VH domains showed that, in the context of CD47 and PD-L1, the CDR H3 loop can adopt different conformations, suggesting a potential induced fit mechanism. Elucidating the structure of the two free Fabs would allow confirmation of that hypothesis.Citation61
The canonical structure of an antibody paratope composed of 6 CDRs that can all contribute to antigen binding has been revisited by the discovery and characterization of antibodies based on a single variable domain, such as VHH found in camelids and VNARs present in sharks.Citation62,Citation63 These single domain antibodies demonstrate that 3 CDRs are sufficient to achieve high specificity and affinity antigen binding. Our approach to generate fixed heavy chain antibodies further challenges the central and dominant role of the heavy chain, as it appears that, by displacing the focus of CDR diversification from the heavy chain to the LC, the main interaction and binding energy can be mediated by the LC. It is therefore reasonable to speculate that if the immune system had evolved a different antibody gene diversification mechanism, for instance with V-D-J recombination occurring in the LC rather than the heavy chain locus, the LC would play a more prominent role in antigen recognition in natural antibody repertoires.
From a drug development perspective, antibodies containing a fixed HC can be used for the straightforward assembly of bispecific antibodies containing a κ-LC in one Fab arm and a λ-LC in the other Fab arm. These κλ bodies have the unique characteristic of being devoid of any sequence alteration that are used in other bispecific formats.Citation17 Retaining a native human IgG structure is beneficial from a stability and development standpoint, but might also be favorable when administered to patients as the appearance of anti-drug antibodies is emerging as a growing concern in the field of therapeutic bispecific antibodies.Citation64,Citation65
List of abbreviations
| BLI: | = | BioLayer Interferometry |
| BSA: | = | Bovine Serum Albumin |
| bsAb: | = | Bispecific Antibody |
| CDRs: | = | Complementarity-Determining Regions |
| CH: | = | Constant Heavy |
| ECD: | = | ExtraCellular Domain |
| ELISA: | = | Enzyme-Linked Immunosorbent Assay |
| Fab: | = | Antigen binding Fragment |
| FACS: | = | Fluorescence-Activated Cell Sorting |
| Fc: | = | Fragment crystallizable |
| FR: | = | Framework |
| HC: | = | Heavy Chain |
| IgG: | = | Immunoglobulin G |
| IgV: | = | Immunoglobulin Variable |
| IMAC: | = | Immobilized Metal ion Affinity Chromatography |
| IPTG: | = | Isopropyl β-D-1-thiogalactopyranoside |
| κ-LC: | = | kappa Light Chain |
| LB: | = | Luria-Bertani |
| LC: | = | Light Chain |
| λ-LC: | = | lambda Light Chain |
| MFI: | = | Mean Fluorescence Intensity |
| O/N: | = | Overnight |
| PBS: | = | Phosphate-Buffered Saline |
| rpm: | = | rotations per minute |
| RT: | = | Room Temperature |
| scFv: | = | single-chain Fragment variable |
| SEC-UPLC: | = | Size Exclusion Chromatography – Ultra-high Performance Chromatography |
| SIRPα: | = | Signal Regulatory Protein alpha |
| TAA: | = | Tumor-Associated Antigen |
| VH: | = | Variable Heavy |
| VHH: | = | Variable Heavy domain of Heavy chain |
| VL: | = | Variable Light |
| VNAR: | = | Variable New Antigen Receptor |
Author contributions
P.M., G.M., M.L., V.K., N.R., F.G., U.R. and X.C. contributed to the conception and design of the experiments and interpretation of the data. P.M., J.B., N.B., S.C., T.B., M.B., M.L., G.M. and V.K. developed assays, performed experiments, acquired data and/or analyzed data. P.M., X.C., M.L., V.K., G.M. and N.F. wrote the manuscript and N.F., F.G., U.R. and N.R. reviewed and/or revised the manuscript. All authors read and approved the final manuscript.
Availability of data and materials
All the data relevant to the study results are included in the main figures of the article or available in the additional files. The X-ray crystallography data of K2 Fab:CD47 and S79 Fab:PD-L1 complexes that support the findings of this study have been deposited in the Protein Database (www.rcsb.org) with the accession codes 8RP8 and 8RPB, respectively.
Supplemental Material
Download Zip (589.6 KB)Acknowledgments
We acknowledge MAX IV Laboratory for time on Beamline BioMAX under Proposal 20180356 and 20220012. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. For technical support and discussions during data collection we would like to thank Dr. Ana Gonzales and Dr. Jie Nan.
We also thank Dr. Anders Svensson and Dr. Björn Walse from SARomics Biostructures for their help in structure refinement, modelling, and helpful discussions.
Disclosure statement
All authors are current employees of Light Chain Bioscience/Novimmune SA or SARomics Biostructures AB.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2024.2362432
Additional information
Funding
References
- Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021;20(7):491–14. doi:10.1038/d41573-021-00079-7.
- Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. Mabs-austin. 2023;15(1):2153410. doi:10.1080/19420862.2022.2153410.
- Antibody therapeutics approved or in regulatory review in the EU or US. The antibody society. https://www.antibodysociety.org/resources/approved-antibodies/.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–42. doi:10.1093/nar/28.1.235.
- Kunik V, Peters B, Ofran Y, Baker B. Structural consensus among antibodies defines the antigen binding site. PLOS Comput Biol. 2012;8(2):e1002388. doi:10.1371/journal.pcbi.1002388.
- Reis PBPS, Barletta GP, Gagliardi L, Fortuna S, Soler MA, Rocchia W. Antibody-antigen binding interface analysis in the big data era. Front Mol Biosci. 2022;9. doi:10.3389/fmolb.2022.945808.
- Luo S, Su Y, Peng X, Wang S, Peng J, Ma J. Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures. 2022; doi:10.1101/2022.07.10.499510.
- Xu JL, Davis MM. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity. 2000;13(1):37–45. doi:10.1016/S1074-7613(00)00006-6.
- Tsuchiya Y, Mizuguchi K. The diversity of H 3 loops determines the antigen-binding tendencies of antibody CDR loops. Protein Sci. 2016;25(4):815–25. doi:10.1002/pro.2874.
- Barrios Y, Jirholt P, Ohlin M. Length of the antibody heavy chain complementarity determining region 3 as a specificity-determining factor. J Mol Recognit. 2004;17(4):332–38. doi:10.1002/jmr.679.
- Ippolito GC, Schelonka RL, Zemlin M, Ivanov II, Kobayashi R, Zemlin C, Gartland GL, Nitschke L, Pelkonen J, Fujihashi K, et al. Forced usage of positively charged amino acids in immunoglobulin CDR-H3 impairs B cell development and antibody production. J Exp Med. 2006;203(6):1567–78. doi:10.1084/jem.20052217.
- Cobb RM, Oestreich KJ, Osipovich OA, Oltz EM. Accessibility control of V(D)J recombination. Adv Immunol. 2006;91:45–109.
- Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109(2):S45–55. doi:10.1016/S0092-8674(02)00675-X.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng Des Sel. 1996;9(7):617–21. doi:10.1093/protein/9.7.617.
- Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. Mabs-austin. 2016;8(6):1010–20. doi:10.1080/19420862.2016.1197457.
- Sandeep Shinde SH, Shinde SH, Pande AH. Polyspecificity - an emerging trend in the development of clinical antibodies. Mol Immunol. 2023;155:175–83. doi:10.1016/j.molimm.2023.02.005.
- Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18(8):585–608. doi:10.1038/s41573-019-0028-1.
- Brinkmann U, Kontermann RE. Bispecific antibodies. Science. 2021;372(6545):916–17. doi:10.1126/science.abg1209.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. Mabs-austin. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Kontermann RE. Dual targeting strategies with bispecific antibodies. Mabs-austin. 2012;4(2):182–97. doi:10.4161/mabs.4.2.19000.
- Fischer N, Elson G, Magistrelli G, Dheilly E, Fouque N, Laurendon A, Gueneau F, Ravn U, Depoisier J-F, Moine V, et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat Commun. 2015;6(1):6113. doi:10.1038/ncomms7113.
- Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, Cons L, Calloud S, Majocchi S, Nelson R, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol Ther. 2017;25(2):523–33. doi:10.1016/j.ymthe.2016.11.006.
- Ferlin W, Masternak K, Shang L. Selective CD47 targeting with a bispecific antibody. Cancer Immunol Immunother. 2021;70(4):1161–62. doi:10.1007/s00262-020-02812-4.
- Buatois V, Johnson Z, Salgado-Pires S, Papaioannou A, Hatterer E, Chauchet X, Richard F, Barba L, Daubeuf B, Cons L, et al. Preclinical development of a bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B cell lymphoma and leukemia. Mol Cancer Ther. 2018;17(8):1739–51. doi:10.1158/1535-7163.MCT-17-1095.
- Hatterer E, Chauchet X, Richard F, Barba L, Moine V, Chatel L, Broyer L, Pontini G, Bautzova T, Juan F, et al. Targeting a membrane-proximal epitope on mesothelin increases the tumoricidal activity of a bispecific antibody blocking CD47 on mesothelin-positive tumors. Mabs-austin. 2020;12(1):1739408. doi:10.1080/19420862.2020.1739408.
- Chauchet X, Cons L, Chatel L, Daubeuf B, Didelot G, Moine V, Chollet D, Malinge P, Pontini G, Masternak K, et al. CD47xCD19 bispecific antibody triggers recruitment and activation of innate immune effector cells in a B-cell lymphoma xenograft model. Exp Hematol Oncol. 2022;11(1):26. doi:10.1186/s40164-022-00279-w.
- Gueneau F, Ravn U, Fischer N. Round optimization for improved discovery of native bispecific antibodies. Methods. 2019;154:51–59. doi:10.1016/j.ymeth.2018.11.009.
- Seckinger A, Majocchi S, Moine V, Nouveau L, Ngoc H, Daubeuf B, Ravn U, Pleche N, Calloud S, Broyer L, et al. Development and characterization of NILK-2301, a novel CEACAM5xCD3 κλ bispecific antibody for immunotherapy of CEACAM5-expressing cancers. J Hematol Oncol. 2023;16(1):117. doi:10.1186/s13045-023-01516-3.
- Seckinger A, Buatois V, Moine V, Daubeuf B, Richard F, Chatel L, Viandier A, Bosson N, Rousset E, Masternak K, et al. Targeting CEACAM5-positive solid tumors using NILK-2401, a novel CEACAM5xCD47 κλ bispecific antibody. Front Immunol. 2024;15:1378813. doi:10.3389/fimmu.2024.1378813.
- Lefranc MP, Giudicelli V, Ginestoux C, Bodmer J, Muller W, Bontrop R, Lemaitre M, Malik A, Barbie V, Chaume D, et al. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 1999;27(1):209–12. doi:10.1093/nar/27.1.209.
- Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG, Kimura R, Tsai JM, Manglik A, Kruse AC, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci USA. 2015;112(47):E6506–14. doi:10.1073/pnas.1519623112.
- Pietsch EC, Dong J, Cardoso R, Zhang X, Chin D, Hawkins R, Dinh T, Zhou M, Strake B, Feng P-H, et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J. 2017;7(2):e536–536. doi:10.1038/bcj.2017.7.
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23(12):2341–48. doi:10.1016/j.str.2015.09.010.
- Sparta KM, Krug M, Heinemann U, Mueller U, Weiss MS. Xdsapp2.0. J Appl Crystallogr. 2016;49(3):1085–92. doi:10.1107/S1600576716004416.
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(2):125–32. doi:10.1107/S0907444909047337.
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(1):72–82. doi:10.1107/S0907444905036693.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(4):658–74. doi:10.1107/S0021889807021206.
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(4):355–67. doi:10.1107/S0907444911001314.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66(4):486–501. doi:10.1107/S0907444910007493.
- BUSTER Documentation: Usage. https://www.globalphasing.com/buster/manual/autobuster/manual/autoBUSTER4.html.
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server):W375–83. doi:10.1093/nar/gkm216.
- Incardona M-F, Bourenkov GP, Levik K, Pieritz RA, Popov AN, Svensson O. EDNA: a framework for plugin-based applications applied to X-ray experiment online data analysis. J Synchrotron Radiat. 2009;16(6):872–79. doi:10.1107/S0909049509036681.
- Lu C, Wu C, Ghoreishi D, Chen W, Wang L, Damm W, Ross GA, Dahlgren MK, Russell E, Von Bargen CD, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291–300. doi:10.1021/acs.jctc.1c00302.
- Bouwstra R, van Meerten T, Bremer E. CD47‐SIRPα blocking‐based immunotherapy: current and prospective therapeutic strategies. Clin Transl Med. 2022;12(8):e943. doi:10.1002/ctm2.943.
- van Duijn A, Van der Burg SH, Scheeren FA. CD47/SIRPα axis: bridging innate and adaptive immunity. J Immunother Cancer. 2022;10(7):e004589. doi:10.1136/jitc-2022-004589.
- Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα immune checkpoint. Immunity. 2020;52(5):742–52. doi:10.1016/j.immuni.2020.04.011.
- Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–67. doi:10.1038/nature14011.
- Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol Cell. 2008;31(2):266–77. doi:10.1016/j.molcel.2008.05.026.
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(4):235–42. doi:10.1107/S0907444910045749.
- MacCallum RM, Martin AC, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol. 1996;262(5):732–45. doi:10.1006/jmbi.1996.0548.
- bioinf.org.uk - Prof. Andrew C.R. Martin’s group at UCL. http://www.bioinf.org.uk/abs/info.html.
- Huang B, Bai Z, Ye X, Zhou C, Xie X, Zhong Y, Lin K, Ma L. Structural analysis and binding sites of inhibitors targeting the CD47/SIRPα interaction in anticancer therapy. Comput Struct Biotechnol J. 2021;19:5494–503. doi:10.1016/j.csbj.2021.09.036.
- Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, Volkmer AK, Volkmer J-P, Liu J, Lim JS, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest. 2016;126(7):2610–20. doi:10.1172/JCI81603.
- Liu K, Tan S, Chai Y, Chen D, Song H, Zhang CWH, Shi Y, Liu J, Tan W, Lyu J, et al. Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res. 2017;27(1):151–53. doi:10.1038/cr.2016.102.
- Tan S, Liu K, Chai Y, Zhang CWH, Gao S, Gao GF, Qi J. Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell. 2018;9(1):135–39. doi:10.1007/s13238-017-0412-8.
- Zhang F, Qi X, Wang X, Wei D, Wu J, Feng L, Cai H, Wang Y, Zeng N, Xu T, et al. Structural basis of the therapeutic anti-PD-L1 antibody atezolizumab. Oncotarget. 2017;8(52):90215–24. doi:10.18632/oncotarget.21652.
- Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G, Holak TA. Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Struct. 2017;25(8):1163–74. doi:10.1016/j.str.2017.06.011.
- Mejias-Gomez O, Madsen AV, Skovgaard K, Pedersen LE, Morth JP, Jenkins TP, Kristensen P, Goletz S. A window into the human immune system: comprehensive characterization of the complexity of antibody complementary-determining regions in functional antibodies. Mabs-austin. 2023;15(1):2268255. doi:10.1080/19420862.2023.2268255.
- Koide S, Sidhu SS. The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem Biol. 2009;4(5):325–34. doi:10.1021/cb800314v.
- Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J Mol Biol. 2005;348(5):1153–62. doi:10.1016/j.jmb.2005.03.041.
- Stanfield RL, Wilson IA. Antigen-induced conformational changes in antibodies: a problem for structural prediction and design. Trends Biotechnol. 1994;12(7):275–79. doi:10.1016/0167-7799(94)90139-2.
- Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82(1):775–97. doi:10.1146/annurev-biochem-063011-092449.
- Matz H, Munir D, Logue J, Dooley H. The immunoglobulins of cartilaginous fishes. Dev Comp Immunol. 2021;115:103873. doi:10.1016/j.dci.2020.103873.
- Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT, Szpurka AM, Copeland V, Hodi FS, Rottey S, et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res. 2021;27(10):2773–81. doi:10.1158/1078-0432.CCR-20-3716.
- Zhou Y, Penny HL, Kroenke MA, Bautista B, Hainline K, Chea LS, Parnes J, Mytych DT. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J Immunother Cancer. 2022;10(4):e004225. doi:10.1136/jitc-2021-004225.