
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
T-cell engaging (TCE) bispecific antibodies are potent drugs that trigger the immune system to eliminate cancer cells, but administration can be accompanied by toxic side effects that limit dosing. TCEs function by binding to cell surface receptors on T cells, frequently CD3, with one arm of the bispecific antibody while the other arm binds to cell surface antigens on cancer cells. On-target, off-tumor toxicity can arise when the target antigen is also present on healthy cells. The toxicity of TCEs may be ameliorated through the use of pro-drug forms of the TCE, which are not fully functional until recruited to the tumor microenvironment. This can be accomplished by masking the anti-CD3 arm of the TCE with an autoinhibitory motif that is released by tumor-enriched proteases. Here, we solve the crystal structure of the antigen-binding fragment of a novel anti-CD3 antibody, E10, in complex with its epitope from CD3 and use this information to engineer a masked form of the antibody that can activate by the tumor-enriched protease matrix metalloproteinase 2 (MMP-2). We demonstrate with binding experiments and in vitro T-cell activation and killing assays that our designed prodrug TCE is capable of tumor-selective T-cell activity that is dependent upon MMP-2. Furthermore, we demonstrate that a similar masking strategy can be used to create a pro-drug form of the frequently used anti-CD3 antibody SP34. This study showcases an approach to developing immune-modulating therapeutics that prioritizes safety and has the potential to advance cancer immunotherapy treatment strategies.
Introduction
Cancer immunotherapies that engage the adaptive immune response to destroy tumors have made substantial progress, particularly in extending the survival of patients with life-threatening malignancies, in the past decade.Citation1–3 T cells have a central role in the clinical success of cancer immunotherapies because they are capable of robust tumor cell killing and can signal other immune cells to assist in tumor destruction.Citation4 One subset of immunotherapies, known as T-cell engager (TCE) bispecific antibodies are engineered antibody drugs that are capable of directing T cells to kill tumors. TCEs work by binding surface receptors on T cells and tumor cells to create a physical link between the two cells that triggers T cell activation and T cell-mediated tumor cell killing.
The first TCE granted a marketing approval was blinatumomab, which the Food and Drug Administration approved in 2014 for the treatment of B-cell precursor acute lymphoblastic leukemia (ALL), initiating the development and expansion of additional TCEs to treat hematological cancers, solid tumors and other clinical targets.Citation5 To date, three additional TCEs, glofitamab, tebentafusp, and teclistamab, have been approved by FDA for the treatment of diffuse large B-cell lymphoma (DLBCL), uveal melanoma, and multiple myeloma (MM), respectively.Citation6–8
Potent T cell-activating therapies, including TCEs, are often accompanied by the serious toxicity of cytokine release syndrome (CRS).Citation9 CRS is a systemic hyperinflammatory response caused by high levels of pro-inflammatory cytokines released from TCE-activated T cells and remains a dose-limiting safety liability for patients.Citation10 Patients with CRS experience symptoms that vary in severity, from mild fever and rash to life-threatening organ failure. While patients with mild CRS can be treated and supported, intervention and termination of treatment is required for patients with severe CRS, limiting the therapeutic window of TCEs.Citation11 We set out to engineer a safer TCE prodrug that prevents CRS by relying on proteolytic conditional activation of the TCE by a tumor-enriched protease, matrix metalloproteinase 2 (MMP-2), to activate T cells locally at tumor microenvironments and not within peripheral tissues. Recent work co-opting MMP-2 for tumor-specific activation of immune checkpoint inhibitors and cytokines demonstrate the potential for using tumor-dysregulated proteases to improve targeted delivery of immune-modulating therapeutics for cancer that are toxic systemically, but curative when delivered locally to the tumor.Citation12–16
Here, we report the crystal structure of a novel anti-human/cynomolgus monkey (cyno) CD3ε antibody antigen-binding fragment (Fab), E10, in complex with a CD3εγ fusion protein and use information from this structure to design an MMP-2 activatable prodrug T-cell engager (pro-TCE). The pro-TCE consists of a full-length, Fc-silenced IgG1Citation17 bispecific antibody with one arm of the antibody, derived from E10, targeting CD3 on T cells and the other arm targeting a tumor-associated antigen. The CD3-targeting antibody arm has a masking domain that engages the antibody complementarity-determining regions (CDRs) so that binding by the prodrug to T cells is weakened while the tumor-targeting arm remains functional to accumulate the pro-TCE at tumor antigen-expressing tissues. The masking domain is connected to the anti-CD3 arm via a flexible linker incorporating an MMP-2 cut site. Cleavage of the linker with MMP-2 releases the mask, restoring affinity for CD3. Design of the mask was informed by the crystal structure of E10 in complex with a CD3εγ fusion protein. Only the N-terminal 7 amino acids (PCA-DGNEEM, where PCA is a pyroglutamate) of the cyno CD3εγ-histag protein were resolved within the crystal structure, suggesting the N-terminus represents the entirety of the epitope. In the structure, the CD3ε peptide N-terminal pyroglutamate is buried deep in a groove formed between the VH and VL domains of the antibody (). This result led us to hypothesize that the CD3 peptide could be linked to the N terminus of E10 in a manner that allows it to bind and mask the CDR region of E10.
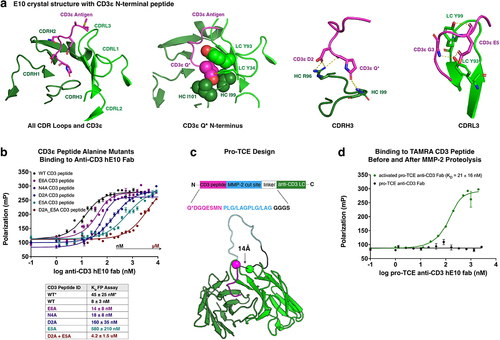
Figure 1. Crystal structure of E10 antibody with CD3ε peptide (PDB 8VY4) informs design of pro-TCE. (a) Top-down view of the isolated CDR loops (green) from anti-CD3 antibody E10 in contact with the CD3ε peptide antigen (pink). Key binding interaction features are highlighted from left to right respectively: N-terminal pyroglutamate, CDRH3 contacts with CD3ε, and CDRL3 contacts with CD3ε. (b) Fluorescence polarization binding data with WT and alanine mutant CD3ε peptides with anti-CD3 hE10 fab. The table below shows two KD values for WT. “WT*” represents our best estimate for KD and associated error from all replicates, while “WT*” and all other KD values in the table are the fitted values and errors to the fit from the specific experiment shown in the figure. (c) PyRosetta Model of designed pro-TCE anti-CD3 antibody fragment and sequence of CD3ε peptide (pink) prepended with MMP-2 cleavage sites (blue). (d) Fluorescence polarization binding data with pro-TCE antibody fragment before and after MMP-2 proteolysis. KD with reported standard deviation of the mean is shown for two replicate measurements. *Q undergoes cyclization to pyroglutamic acid (Pyr) under physiological conditions.
We investigated pro-TCE binding to soluble CD3 and in vitro cellular T cell activity and cytokine release using Jurkat T cells and human primary T cells as effector T cells with targeted killing of human tumor cells expressing HER2 target antigen to demonstrate before and after proteolysis with MMP-2 protease. After confirming that our designed pro-TCE can selectively control CD3 binding and T cell activation dependent upon proteolysis, we characterized the release of pro-inflammatory cytokines IFN-γ and IL-2 from human T cells in the presence of target antigen-expressing cancer cells. Our results demonstrate selective pro-TCE activation only in the presence of MMP-2 with successful removal of the masking domain. Additionally, we show that the pro-TCE masking strategy works for different anti-CD3 antibody clones and may broadly be used to trigger T cell activation. Overall, we demonstrate a rational engineering approach for conditional activation of TCE bispecific antibodies designed to evoke a higher therapeutic response in a tumor environment to reduce off-target toxicity.
Results
Structure of anti-CD3 mAb clone E10 bound to CD3ε peptide
The E10 mAb was generated by immunizing rabbits with recombinant human/cyno CD3εγ-Fc in a process described previously.Citation18 Briefly, the mAb was cloned via RT-PCR after single-cell B cell sorting against the human and cyno antigens. To reveal how the rabbit mAb E10 binds to CD3ε, we solved the crystal structure of E10’s Fab in complex with cyno CD3εγ-histag protein. The structure resolved only the N-terminal 7 amino acids (PCA-DGNEEM, where PCA is a pyroglutamate) of the cyno CD3εγ-histag protein, suggesting the N-terminus of CD3ε is representative of the entire epitope. These 7 N-terminal residues are identical in sequence between human and cyno CD3εγ. The pyroglutamate at the N-terminus of the CD3 peptide buries deep into a cleft formed between the Fab VH and VL domains where it forms several hydrophobic and polar interactions. Hydrophobic interactions are made between the methylene groups on the pyroglutamate and Ile97 on the heavy chain (HC) and Tyr93 and Ile101 from the light chain (LC). Hydrogen bonds are formed between the side-chain carbonyl of the pyroglutamate and the backbone amide groups of HC-Ile99 and HC-Gly100. The backbone carbonyl of the pyroglutamate also forms a hydrogen bond with HC-Arg96 (). CD3ε peptide residue Asp2 further stabilizes the interaction by hydrogen bonding with HC-Arg96. Hydrogen bonds are also made between the CD3 residues Gly3 and Glu5 with LC-Tyr93 and LC-Tyr99 ().
Affinity measurements with CD3ε variants
To confirm insights gained from the E10 crystal structure, we used fluorescent polarization (FP) measurements to determine the binding affinity of the Fab domain of the antibody for various CD3ε peptide variants.Citation19,Citation20 In these and all further experiments, we used a humanized version of E10, referred to as hE10. E10 was humanized by direct CDR1/CDR2/CDR3 grafts of the VH and Vkappa, respectively, into VH1–46 and Vk-A26 human germline sequences, respectively. Using a TAMRA-labeled WT CD3ε peptide (sequence PCA-DGNEEM), we determined an equilibrium dissociation constant (KD) of 45 ± 25 nM for the interaction (). Consistent with the crystal structure, mutating Asp2 or Glu5 to alanine, both of which form hydrogen bonds to the antibody CDR loops, reduces binding affinity with a KD of 160 ± 35 nM or 580 ± 210 nM, respectively. A double mutant peptide, containing both Asp2 and Glu5 alanine mutations, further reduces binding to hE10 with a KD of 4.2 ± 1.5 uM, highlighting the importance of both residues in the interaction with hE10.
Design of a pro-TCE
The tight affinity of the CD3ε peptide for the CDR loops of hE10 suggested that it could be used as an effective masking domain to create a conditionally activatable TCE. In the crystal structure, the N-terminus of the E10 antibody LC is located 14 Å from the end of the CD3ε peptide. To gauge the feasibility of linking the peptide to the antibody, we performed molecular modeling simulations with glycine serine linkers of various lengths, also including an MMP-2 cleavage site in the linker (; Figure S1). The simulations indicated that a 4-residue linker combined with two MMP-2 cleavage sites (sequence PLG/LAGPLG/LAG-GGGS) is sufficient to allow the CD3ε peptide to interact with its binding site.
To mask hE10, we used a masking sequence (PCA-DGQESM) that differs slightly from the wild-type (WT) sequence of CD3ε (PCA-DGNEEM). The serine was incorporated at position 6 (in place of a glutamate) to enable an alternative design goal not relevant in this study and the asparagine at position 4 was mutated to a glutamine to prevent the incorporation of a glycosylation site. Our crystal structure and alanine scan of the peptide indicated that these mutations should not degrade the effectiveness of the mask. Consistent with this analysis, no binding is observed to CD3ε peptide when hE10 is masked with either the WT CD3 sequence (PCA-DGNEEM) or the alternative sequence (PCA-DGQESM) (Figure S2). All further experiments were performed with the PCA-DGQESM mask. We used FP to measure the affinity of the masked hE10 Fab for TAMRA-CD3ε peptide before and after treatment with MMP-2 protease (). The masked Fab exhibited no affinity for the peptide, while the protease-treated Fab bound with a measured KD to TAMRA-CD3ε peptide of 21 ± 16 nM, similar to the binding affinity measured between TAMRA-CD3ε peptide and hE10 with no mask (KD = 45 ± 25 nM).
Pro-TCE production and biophysical characterization
Next, we created an inducible TCE by engineering a bispecific IgG antibody that incorporates the masked hE10 as one arm of the antibody and an anti-human epidermal growth factor 2 (HER2) antibody (pertuzumab) as the other arm of the antibody. To promote efficient assembly of the IgG antibody during expression, we incorporated OrthoMab mutations at the HC/LC interfaces to favor the hE10 LC pairing with the hE10 HC and the pertuzumab LC with the pertuzumab HC ().Citation21 Similarly, mutations were included in the Fc region of the antibody to promote the formation of the hE10/pertuzumab heterodimer over hE10 and pertuzumab homodimers. We selected HER2, a protein expressed in several solid tumors and a well-established clinical target in oncology, to direct the prototype pro-TCE activity to a relevant target tumor antigen.Citation22–24
Figure 2. Recombinant expression, assembly and biophysical characterization of pro-TCE. (a) Interface mutations across the antibody fab and Fc framework regions facilitate proper recombinant assembly of IgG bispecific antibodies. (b) SDS-PAGE gel electrophoresis showing recombinant MMP-2 before and after activation with APMA, and the pro-TCE before and after proteolysis with the active MMP-2. Protein samples treated with βMe. (c) Fluorescence polarization binding data with pro-TCE IgG before and after MMP-2 proteolysis. The reported KD values are averages with errors reflecting the standard deviation of the mean for three replicates. (d) Biolayer interferometry data of the immobilized pro-TCE and parental anti-HER2 pertuzumab binding to HER2 extracellular domain before proteolysis. (e) Biolayer interferometry data of the immobilized pro-TCE and parental anti-CD3 hE10 mAb binding to CD3γε extracellular domain before and after proteolysis. In (d-e) the reported KD values are averages along with the standard deviation of the mean for two replicates.
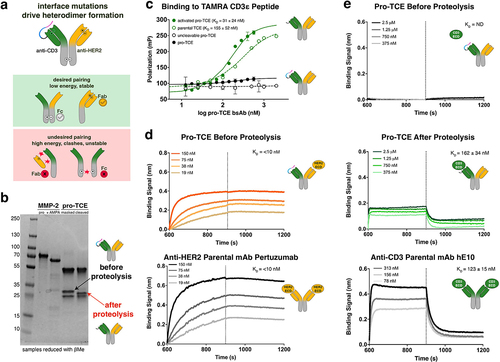
The pro-TCE bispecific antibody was transiently expressed in Expi293F mammalian cells and purified over protein A resin, followed by gel filtration to obtain a final, highly homogenous sample of bispecific antibody as observed by intact high-resolution mass spectrometry analysis (Figures S3, S4). We used a biochemical mobility gel-shift assay to visualize proteolysis through changes in gel mobility/protein molecular weight before and after incubation of the pro-TCE with MMP-2 protease. After proteolysis with purified recombinant MMP-2, we observed a mass shift of the anti-CD3 LC, corresponding to successful cleavage of the prodrug anti-CD3 component on the pro-TCE (). During these experiments, we noticed that the mask was slowly cleaved from the antibody if it was stored for several weeks at 4°C, and a small amount of cleavage product was observed in the mass spectrometry data (Figure S4). Including additional protease inhibitors or ethylenediaminetetraacetic acid (EDTA) in the storage buffer reduced background cleavage, suggesting that protease from the expression system was cleaving the antibody. All further experiments were performed with antibody that was expressed and purified immediately before use. For control experiments, we also expressed and purified the TCE without a mask (“parental TCE”) and a masked form of the TCE with a scrambled MMP-2 cleavage sequence (“uncleavable pro-TCE”).
We confirmed that the pro-TCE bispecific antibody retains high affinity and specificity for the target antigen CD3 using two spectroscopic techniques: FP measurements with TAMRA-CD3ε peptide and biolayer interferometry (BLI) with recombinant full-length CD3 ectodomain (CD3γε). In the FP experiments, binding to CD3ε peptide is undetectable up to a concentration of 4 uM for the masked antibody, while the protease-treated TCE (“activated pro-TCE”) bound with a KD of 31 ± 24 nM (). To perform BLI, the pro-TCE or the activated pro-TCE antibody was immobilized to a protein A biosensor and binding was measured with full-length CD3 ectodomain (CD3γε). Binding of the masked pro-TCE to CD3 was undetectable, while activated pro-TCE bound to CD3 with similar binding kinetics and affinity as the parental anti-CD3 hE10 monoclonal antibody. BLI was also used to confirm binding of the anti-HER2 arm of the pro-TCE bispecific antibody to recombinant full-length HER2 ectodomain. The pro-TCE antibody bound to HER2 with the same affinity as the parental anti-HER2 antibody pertuzumab, and binding was not perturbed by protease treatment (). Our combined FP and BLI data show functional in vitro binding behavior for our pro-TCE prototype where CD3 binding is masked before proteolysis and fully restored through incubation with recombinant MMP-2.
Engineering the pro-TCE required addition of the CD3 masking domain tethered to the anti-CD3 LC and a total of 17 interface mutations across the antibody framework to facilitate proper bispecific antibody heterodimer formation. To confirm that our modifications did not affect the stability of the bispecific antibody, we compared the thermostability of the pro-TCE to its parental anti-CD3 and anti-HER2 monoclonal antibodies (Figure S5). The bispecific pro-TCE remains well-folded above physiological temperatures and has comparable stability to the parental monoclonal antibodies.
Conditional activation of Jurkat NFAT-Luciferase T cells with pro-TCE
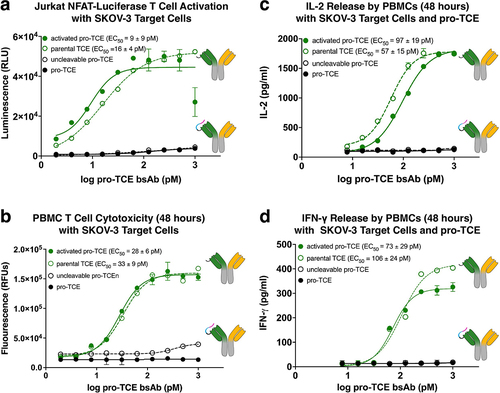
Next, we evaluated the ability of the pro-TCE bispecific antibody to activate Jurkat T cells in co-culture with HER2-expressing SKOV-3 cells in an NFAT luciferase reporter co-culture assay. We measured the luminescence signal after adding pro-TCE or pro-TCE cleaved with recombinant MMP-2 to the cell mixture. The recombinant MMP-2 was removed from the sample before adding to cells. The pro-TCE significantly reduced the luminescence signal, reinforcing our findings that our design is functional and able to mask binding to CD3 on the surface on T cells (, S6). The Jurkat T cells treated with proteolytically activated pro-TCE readily achieved maximal signal and activation (pro-TCE EC50 = 9 ± 9 pM).
Figure 3. Preclinical functional validation of pro-TCE with recombinant MMP-2 protease treatment. All experiments were performed at an effector cell to target cell ratio of 1:1. Each point represents the mean value of triplicates. (a) T cell activation induced by pro-TCE after proteolysis using a Jurkat NFAT-Luciferase T cell model. (b) HER2+ target cell killing by hPBMCs induced by pro-TCE after proteolysis after 48 hours. (c–d) IL-2 and IFN-γ secretion by hPBMCs induced by pro-TCE after 48 hours. The reported errors for the EC50 values are the 95% confidence intervals from fitting each individual experiment.
Pro-TCE cytotoxicity
To show that the pro-TCE not only induces T cell activation but can effectively kill tumor cells that express the target HER2 antigen, we investigated the ability of the pro-TCE and the proteolytically activated pro-TCE to induce human peripheral blood mononuclear cells (PBMCs) to kill HER2-expressing SKOV-3 target cells (, S7). We observed potent cytotoxicity after 48-hour incubation with the proteolytically activated pro-TCE, reaching complete target cell killing with a 50% effective concentration of 28 ± 11 pM. Importantly, cell death was not triggered by the pro-TCE, our control with an uncleavable pro-TCE, or by simply mixing the PBMCs with SKOV-3 cells without any pro-TCE. These results demonstrate the functional protection that our engineered pro-TCE provides against cellular cytotoxicity.
Secretion of PBMC IL-2 and IFN-γ
There are a multitude of elevated pro-inflammatory cytokines released from T cells during CRS, so we evaluated if the pro-TCE reduced the release of interleukin-2 (IL-2) and interferon (IFN)-γ cytokines. Using our co-culture model with PMBCs and target SKOV-3 cells, we incubated the cell mixture with the pro-TCE or proteolytically active pro-TCE and used Promega’s Lumit Immunoassay to quantify the secreted IL-2 and IFN-γ cytokines in the cellular medium after 48 h (, S8 and S9). In agreement with our T cell activation and cytotoxicity studies, the pro-TCE did not trigger robust cytokine secretion, while the proteolytically active pro-TCE induced cytokine secretion with EC50 for IL-2 and IFN-γ of 97 ± 19 and 73 ± 23 pM, respectively.
Secreted cancer proteases trigger conditional activation of Jurkat NFAT-Luciferase T cells with pro-TCE
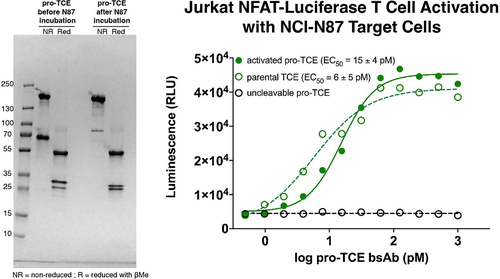
To demonstrate that the presence of protease does not affect the activity of the activated pro-TCE and that secreted levels of MMP-2 are sufficient for triggering proteolysis of the pro-TCE and subsequent T cell activation, we co-cultured the pro-TCE with a HER2-expressing gastric cancer cell line that secretes MMP-2 protease, NCI-N87, and monitored pro-TCE proteolysis and Jurkat NFAT-luciferase T cell activation.Citation25 First, we confirmed the presence of secreted MMP-2 in the tumor cell culture after 1 h using a fluorescent MMP-2 substrate test kit (Figure S10). Next, we incubated pro-TCE and an uncleavable control pro-TCE in NCI-N87 tumor cell culture medium for 1 h, repurified the pro-TCE and visualized the tumor cell-mediated proteolysis using gel electrophoresis. Our results show a single shifted band for the activated pro-TCE LC, suggesting complete proteolysis of the pro-TCE in 1 h while the uncleavable control pro-TCE remained intact for the duration of the experiment (, left panel). Furthermore, the pro-TCE activated Jurkat NFAT-luciferase T cells in co-culture with NCI-N87 cells while our uncleavable pro-TCE control remained inert, demonstrating that pro-TCE proteolysis induced by tumor-secreted MMP-2 can trigger T cell activation in cell culture (, right panel).
Figure 4. NCI-N87 tumor cell secreted proteases trigger pro-TCE activity. SDS-PAGE gel electrophoresis of pro-TCE before and after incubation with NCI-N87 cellular supernatant for 1 h (left). Jurkat NFAT-Luciferase T cell activation assay with NCI-N87 target cells (right). The reported EC50 value is an average of three replicates with errors reflecting the 95% confidence intervals of the average measurements.
Expanding the pro-TCE platform
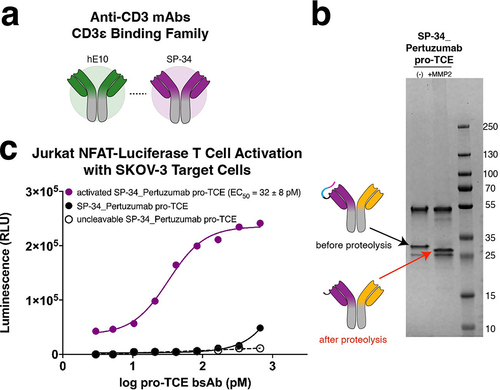
Clone hE10 recognizes the same epitope on CD3ε as the antibody SP34, from which many TCEs in the clinic are derived.Citation26 Based on this similarity, we reasoned that SP34 could be masked using the same strategy and exact sequence that we used to mask hE10 (). To test this hypothesis, we made a pro-TCE that recognizes CD3ε using the SP34 antibody sequence instead of hE10, including the alternative CD3ε peptide (sequence PCA-DGQESM) tethered to the antibody with our optimized flexible linker containing an MMP-2 protease cleavage site. The other binding arm recognizes HER2 with the same sequence as the original pro-TCE derived from pertuzumab. We readily expressed the SP34 pro-TCE and observed a change in molecular weight after MMP-2 cleavage in our SDS-PAGE gel shift assay (). To confirm the SP34 pro-TCE was functional, we used the Jurkat NFAT luciferase reporter co-culture assay to show that before cleavage there was no activation of Jurkat T cells in co-culture with HER2-expressing SKOV-3 cells (). T cell activation is restored after MMP-2 proteolysis, suggesting that both anti-CD3 clones hE10 and SP34 can be used in the pro-TCE format because they bind the same CD3ε epitope.
Figure 5. Expanding the pro-TCE conditionally active T cell engager platform to clone SP34. (a) Anti-CD3 mAbs hE10 and SP34 both bind the N-terminus of CD3ε. (b) SDS-PAGE gel electrophoresis of SP34_Pertuzumab pro-TCE before and after proteolysis. (c) Designed pro-TCE using anti-CD3 mAb with SP34 CD3-binding arm activates Jurkat NFAT-Luciferase T cells when proteolyzed but not when masked. The reported EC50 value is an average of three replicates with errors reflecting the 95% confidence intervals of the average measurements.
Discussion
In this study, we developed modular pro-TCEs that only bind to CD3 and activate T cells after unmasking with the protease MMP-2. Our design strategy for masking the anti-CD3 antibody hE10 was informed by our crystal structure of the parental antibody, E10, bound to CD3εγ. In the structure, the antibody engages CD3 by forming interactions with the flexible N-terminal tail of CD3εγ. This result suggested that sequences derived from the N-terminal region of CD3ε could serve as a potent mask if fused to the antibody. This hypothesis was confirmed using CD3 binding experiments, T cell activation assays, and cell killing assays.
Our pro-TCE prototype joins a growing set of recombinant proteolytically activated TCE prodrugs for preferential activation of T cells in the tumor microenvironment. Proteolytically activated TCEs have demonstrated enhanced tolerability in preclinical studies while retaining antitumoral efficacy toward different solid tumor targets.Citation12,Citation12,Citation14,Citation15,Citation27,Citation28 Our approach is distinct from published methods due to our use of different tumor-associated protease cleavage sequences, including sequences for multiple serine and cysteine proteases in addition to matrix metalloproteases. Unlike some existing proteolytically activated TCE prodrugs, HER2 binding affinity is not masked in our pro-TCE design, potentially having the advantage of facilitating tumor accumulation. Four proteolytically active prodrug TCEs, CytomX’s CX-904 (NCT05387265), Janux’s JANX008 (NCT05783622), and Takeda’s TAK-186 and TAK-280 (NCT04844073 and NCT05220098, respectively) are undergoing Phase 1 trials.Citation29 Detailed reviews of these protease-sensitive prodrug TCEs can be found elsewhere.Citation30,Citation31 Notably, these prodrugs do not use masks derived from CD3, but rather the masks were identified using selection techniques such as phage display.Citation32 Using a mask derived from CD3 has two advantages: 1) we avoided the need for high throughput screening, and 2) sequences derived from human proteins may be less immunogenic as a drug.
At the beginning our study, we were concerned that using a mask derived from CD3ε would result in a mask that does not effectively dissociate from the antibody following protease cleavage, but this does not appear to be a limitation of the system. In the T cell activation studies with the NCI-N87 target cells, cleavage of the mask relied on proteases secreted from the cells and there was no step in the protocol aimed at removing cleaved mask from the solution. Even with cleaved mask still present in the system, the T cells were activated at similar concentrations of TCE as a control experiment performed with a TCE that did not include a mask (). This result is consistent with our biophysical measurements. First, BLI experiments show that hE10 dissociates rapidly from CD3ε, with >90% dissociation within 60 s and therefore the mask derived from CD3ε is likely to dissociate with similar off rates. Second, the equilibrium dissociation constant for the interaction between the CD3εγpeptide and hE10 was measured to be 45 nM, which is higher than the concentration of TCE needed to activate T cells. In the T cell activation studies with the NCI-N87 target cells, full activity was achieved with TCE concentrations of 100 pM. During this experiment, the released mask should be present at the same concentration as unmasked TCE, but, since the KD of the mask for the TCE is greater than 1 nM, only a small fraction of the TCE was likely bound to the mask at concentrations needed to activate the T cells. One amazing property of TCEs highlighted by our study is that T cell activation can occur at TCE concentrations that are significantly lower than the KD between the TCE and CD3. Similar results have been observed for other TCEs, including the clinically approved TCE blinatumomab.Citation33 These results suggest that only a fraction of CD3 molecules need to be engaged on T cells to promote activation.
The N-terminus of CD3ε is an immunodominant cross-reactive epitope recognized by other anti-CD3 antibodies including the commonly employed SP34.Citation34 While no structure of SP34 bound to CD3ε is publicly available, our masking data with SP34, using residues from the N terminus of CD3ε as the mask, provides biophysical experimental evidence that the SP34 CDR loops engage the N-terminus of CD3ε. Of the TCEs in the clinic, many have sequences that are derived from SP34Citation26 and it is likely that these drugs can be converted to protease-sensitive prodrugs by masking the CD3 arm with sequences derived from the N terminus of CD3ε.
Recently, another crystal structure was solved of an anti-CD3 antibody, ADI-26906 (PDB: 8F0L), bound to a peptide derived from the N-terminus of CD3ε.Citation26 In this study, a Fab derived from ADI-26906 was crystallized with the first 13 residues of CD3ε. Like our structure of E10 complexed with CD3εγ, only the first seven residues of CD3εγwere resolved in the structure and the N-terminal pyroglutamate of CD3ε was buried in a cleft formed between the VH and VL domains. In a cryo-EM structure of the T cell receptor-CD3 complex (PDB: 6JXR),Citation35 the first 11 residues of CD3ε are not resolved and are presumed to be disordered. This suggests that there is unlikely to be a fixed orientation between antibodies that bind this region of CD3ε and the rest of the TCR-CD3 complex. This conclusion is supported by the lack of density observed for most of CD3εγ in our crystal structure of E10 bound to CD3εγ (PDB code: 8VY4). These results suggest that activation of T cells by TCEs does not require a rigid connection between the antibody and the TCR-CD3 complex. This mode of action is consistent with a model in which cancer cells coated with a TCE promote multivalent engagement of the TCR/CD3 complex and activate the T cell through TCR clusterization.Citation36
Materials and methods
Crystal structure of anti-CD3 rabbit E10 antibody
The rabbit E10 antibody Fab was recombinantly produced for crystallization. Crystals were obtained by screening with a sparse matrix grid screen, Index® from Hampton Research. Crystallization experiments were performed in a sitting drop plate at 21°C, mixing 0.3 µL of protein solution at 10.8 mg/ml with an equal volume of reservoir solution, and equilibrating against a 70 µL reservoir. The crystallization trays were setup using a Mosquito® liquid handler from SPT Labtech. Plate shaped crystals grew in a drop with a reservoir solution containing 100 mM Bis-Tris pH 6.5 and 20% Polyethylene glycol monomethyl ether 5000. Crystals were harvested directly from the sparse matrix screening tray, transferred to a cryoprotectant solution supplemented with 20% ethylene glycol, and flash frozen in liquid nitrogen for data collection.
Data were collected on the Lilly Research Laboratories Collaborative Access Team beamline 31-ID at the Advanced Photon Source at Argonne National Laboratory, Argonne, IL. Crystals belonged to the P212121 space group with unit-cell parameters a = 48.7, b = 72.7, c = 126.7 Å, and diffracted to 1.7 Å.
The structure was determined by Molecule Replacement using Phaser,Citation37 and a search model generated from an internal Fab structure. The structure was refined using Refmac5Citation38 and Buster.Citation39 The model was built using COOT.Citation40
The structure was refined to R-factors of Rworking = 17.2% and Rfree = 21.4%. The final structure had good geometry with 97% of the residues in the most favored regions of the Ramachandran map and no residues in disallowed regions (PDB code: 8VY4). Additional crystallography details are included in Table S1.
Cell lines and recombinant proteins
Human tumor cell lines SKOV-3, NCI-N87, NCI-H929, and Jurkat E6.1 were purchased from American Type Culture Collection (ATCC, #HTB-77, #CRL-5822, #CRL-3580) and cultured in RPMI-1640 (Corning, #10–041-CV) growth media containing 10% fetal bovine serum (Corning, #35–015-CV) and 1% pen/strep (Life Technologies, #15140122). Jurkat NFAT-Luciferase reporter cells (Promega, J1601) and primary human T cells (ALLCELLS frozen normal human peripheral blood CD3+ Pan T cells, include cat#260100.19) were grown in RPMI growth media containing 10% fetal bovine serum and hygromycin B (Gibco, #10687–010). All cell lines were cultured in a humidified incubator at 37°C, 5% CO2. Recombinant human MMP-2 and HER-2 were purchased from Sino Biological (#10082-HNAH, #10004-H08H). Recombinant CD3γε extracellular domain with C-terminal his tag was prepared in-house.
Design of bispecific TCE antibody and monoclonal antibody constructs for recombinant expression
All bispecific antibodies and monoclonal antibodies expressed recombinantly for this study were produced as full-length immunoglobulin IgG1 proteins. The sequences for pertuzumab were obtained from the National Center for Biotechnology Information (NCBI) and the SP34 sequences have been previously published.Citation41 The sequences for the in-house anti-human CD3 clone hE10 are published here in the supplemental materials.
Briefly, we incorporated amino acid point mutations into the conserved framework variable domain and constant domain antibody HC-LC chain interfaces using previously published mutation sets that facilitate proper recombinant assembly of bispecific IgG antibody heavy and light chains (full sequences including point mutations can be found in Table S2).Citation21,Citation42,Citation43 The masked pro-TCE bispecific antibodies contain an additional N-terminal domain that genetically encodes the CD3 masking peptide and MMP-2 cleavage site as part of the CD3 LC. All monoclonal antibody sequences remained unmodified wild type without any interface mutations. A detailed data for all antibody design sequences can be found in the supplemental materials.
Recombinant antibody expression in Expi293F cells
Antibody HC and LC sequences were cloned into the pAH vector by Twist Biosciences. Plasmids were sequenced and scaled-up for protein production using the Expi293F Expression System (Thermo Fisher, #14635). Each antibody chain was cloned into pAH as a single cistronic domain and transiently co-expressed and secreted by Expi293F cells for 4 days at 37°C, 8% CO2. Cells were pelleted, and supernatant was filtered prior to purification. The secreted recombinant antibodies were purified with protein A resin (GenScript, #L00210) followed by gel filtration on a Superdex 200 increase 10/300 GL column and concentrated to 1 mg/ml and stored in phosphate-buffered saline (PBS) at 4°C.
In vitro proteolysis of pro-TCE antibody with recombinant MMP-2
Recombinant MMP-2 (Sino Biological, #10082-HNAH) was first activated with 4 mM 4-aminophenyl mercuric acetate (Sigma, #A-9563) in TNBC buffer (50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05% (w/v) Brij 35), pH 7.5 at 37°C for 2 h.
Pro-TCE antibody used for subsequent binding and cellular assays was cleaved with MMP-2 by incubating 100 ug pro-TCE with activated MMP-2 for 16 h at 37°C in TNBC buffer. MMP-2 was then removed from the cleaved pro-TCE by buffer-exchange back into PBS with a 100 kDa MWCO Amicon Ultra-0.5 Centrifugal Filter Unit (Sigma, #UFC5100). Cleaved pro-TCE is stored in PBS at 4°C.
Reduced and non-reduced intact analysis of pro-TCE
The pro-TCE antibody sample was diluted to 0.25 mg/ml in 50 mM ammonium bicarbonate for non-reduced intact analysis, with the addition of 10 mM dithiothreitol for the reduced sample. The sample was injected onto an HR Chip (908Devices Inc.) using a ZipChip system (908Devices Inc.) interfaced to a Thermo QExactive HF Biopharma mass spectrometer. The MS data were acquired through Tune (v. 2.9) and settings included: scan range 1000–4000 m/z, in-source CID 50 eV, mass resolution 15,000, 5 microscans, AGC target 1e6, 20 ms maximum injection time. The CE settings included: BGE type = peptides, field strength 500 V/cm, injection volume 1 nL, pressure assist start time 0.5 min, analysis run time 5 min. Intact mass spectra were deconvoluted using PMI Intact Mass software (Protein Metrics Inc.).
Fluorescence polarization binding assays
Masking efficiency and binding after MMP-2 cleavage of the pro-TCE to the CD3ε N-terminus (sequence: Pyr-DGNEEM) was measured with fluorescence polarization.
A TAMRA dye-labeled CD3 peptide (TAMRA-CD3) containing the first seven N-terminal amino acids of CD3ε (QDGNEEM-K(5-TAMRA)-NH2) was prepared by solid-phase synthesis and purified with a Chromolith RP18e column. The purified TAMRA-CD3 peptide was lyophilized for long-term storage at −20°C. Lyophilized TAMRA-CD3 peptide was resuspended in FP buffer (Ca/Mg2+ free PBS, 0.1% BSA, 0.5 mM EDTA, 0.1% NaN2) on day of binding experiment. All purified bispecific and monoclonal antibodies were diluted in FP buffer for assay. TAMRA-CD3 peptide was diluted to 200 nM for all binding titrations. Each bispecific and monoclonal antibody titration concentration value was measured in triplicate with 200 nM TAMRA-CD3 peptide in a 384-well low volume black plate (Corning, #3820). Polarization values were measured on a SpectraMax M5 plate reader at Ex/Em 550 nm/580 nm.
Binding analysis with BLI
Masking efficiency and binding affinities of the masked and cleaved pro-TCEs for the soluble ectodomains of CD3γε and HER2 were measured with BLI on a ForteBio Octet Red384. All BLI measurements were recorded at 25°C with an orbital shaking speed of 1000 rpm. To determine binding of antibodies to soluble antigens, purified masked and cleaved pro-TCEs and monoclonal parental antibodies were diluted to 15 nM in ForteBio Kinetics Buffer (Fisher Sci., #NC1824136) and immobilized on protein A biosensors (Sartorius, #18–5010). A two-fold dilution series of His-tagged HER2 extracellular domain (Sino Biological, #10004-H08H) and His-tagged CD3γε extracellular domain (in-house) ranging from 2500 nM to 156 nM was made in ForteBio Kinetics buffer (Fisher Sci., #NC1824136). Affinity constants were measured using the Octet Analysis Studio software with a 1:1 global fit binding model.
In vitro T cell activation bioassay with NFAT-luciferase Jurkat reporter cells
Jurkat NFAT-luciferase reporter cells (Promega, #J1601) were used to determine the ability of pro-TCE masking to prevent T cell activation and to further restore T cell activation after MMP-2 cleavage. Jurkat NFAT-luciferase reporter cells were cultured following the standard protocols outlined in Promega’s technical manual TM489.
Briefly, target tumor cells (SKOV-3 or NCI-N87) and Jurkat NFAT-luciferase reporter cells were plated in a white, flat-bottom 96-well assay plate at a target: effector cell ratio of 1:5. Intact or pre-cleaved activated pro-TCE is added to the cell mixture (final well volume 50 L) and kept for 6 h in a humidified incubator at 37°C, 5% CO2. After 6 h incubation, luminescence was measured on a ClarioStar plate reader after adding equal volume BioGlo (Promega, #G7941) to sample wells. Data collected in triplicate were analyzed in GraphPad Prism 9.0.
In vitro T cell cytotoxicity assays
Human T cells (ALLCELLS frozen normal human peripheral blood CD3+ Pan T cells, cat #260100.19) were used as effector T cells to measure target tumor cell death (SKOV-3 or NCI-N87) in the presence of our designed pro-TCE before and after MMP-2 proteolysis.
To acquire endpoint cytotoxicity data at 48 h, we used the fluorescent CellTox Green Cytotoxicity Assay (Promega, #G8741) based on a nontoxic cyanine dye that is not permeable to live cells but preferentially stains the DNA of dead cells. Human PMBCs were thawed and rested in RPMI-1640 media Target tumor cells (SKOV-3 or NCI-N87) and human T cells were plated in a white, flat-bottom 96-well assay plate at a target: effector cell ratio of 1:1 with 5,000 of target and effector cells each. Intact or pre-cleaved activated pro-TCE was added to the cell mixture (final well volume 50 L) for 48 h in a humidified incubator at 37°C, 5% CO2.
After 48 h incubation, CellTox Green Reagent was added to the assay plate and the fluorescence intensity values were measured on a ClarioStar plate reader at Ex/Em 480 nm/520 nm. Data collected in triplicate were analyzed in GraphPad Prism 9.0.
IL-2 and IFN-γ cytokine release immunoassays
We used Promega’s Lumit IL-2 and IFN-γ immunoassay kits to detect secreted cytokines from primary human T cells in co-culture with tumor cells and our designed pro-TCE before and after MMP-2 proteolysis.
Human T cells (ALLCELLS frozen normal human peripheral blood CD3+ Pan T cells, cat #260100.19) were used as effector T cells to measure pro-inflammatory cytokine release after co-culture with target tumor cells (SKOV-3 or NCI-N87) in the presence of the masked and cleaved pro-TCE constructs. Target tumor cells (SKOV-3 or NCI-N87) and human T cells were plated in a white, flat-bottom 96-well assay plate at a target: effector cell ratio of 1:5. Intact or pre-cleaved activated pro-TCE was added to the cell mixture (final well volume 50 L) for 48 h in a humidified incubator at 37°C, 5% CO2.
Stability measurements with NanoDSF
Thermal unfolding measurements of the pro-TCE and the monoclonal parental antibodies were performed using a Prometheus NT.48 System. All proteins were diluted to 6 M in 1X PBS pH 7.4 and loaded into capillary tubes in triplicate. Proteins were heated at 1°C/min from 25°C to 95°C and the intrinsic fluorescence signal was measured at 330 nm and 350 nm for each sample. The Tm values obtained were calculated automatically using the first derivative of the ratio of fluorescence signals in the PR.ThermControl software (NanoTemper).
Abbreviations
| ALL | = | acute lymphoblastic leukemia |
| BLI | = | biolayer interferometry |
| CD3 | = | cluster of differentiation 3 |
| CDRs | = | complementarity determining regions |
| CRS | = | cytokine release syndrome |
| Cyno | = | cynomolgus |
| DLBCL | = | diffuse large B cell lymphoma |
| EDTA | = | ethylenediaminetetraacetic acid |
| EGFR | = | epidermal growth factor receptor |
| Fab | = | antigen-binding fragment |
| FP | = | fluorescence polarization |
| HER2 | = | human epidermal growth factor receptor 2 |
| ICI | = | immune checkpoint inhibitor |
| IFN-γ | = | interferon gamma |
| IL-2 | = | interleukin 2 |
| mAb | = | monoclonal antibody |
| MM | = | multiple myeloma |
| MMP-2 | = | matrix metalloprotease 2 |
| NFAT | = | nuclear factor of activated T cells |
| PBMCs | = | (human) peripheral blood mononuclear cells |
| Pro-TCE | = | prodrug T-cell engager |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TCE | = | T-cell engager |
| VH | = | variable heavy chain |
| VL | = | variable light chain |
| WT | = | wild-type |
Author contributions
Conceptualization, B.K., S.D., K.F., and A.C.M.; Methodology, B.K., S.D., and A.C.M.; Data Collection, A.C.M.; S.A., and M.H.; Data Analysis, B.K. and A.C.M.; Writing – draft preparation, B.K. and A.C.M.; Writing – review and editing, B.K., A.C.M., K.F., S.A., and M.H.
2024_mAbs_supplement_Revision_ACM_V1.docx
Download MS Word (2.4 MB)Acknowledgments
This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly and Company, which operates the facility.
This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by NCI Center Core Support Grant (2P30CA016086-45) to the UNC Lineberger Comprehensive Cancer Center.
Disclosure statement
S.D., K.F., S.A., and M.H. are or were employees of Eli Lilly. S.D. is an employee of Tentarix Biotherapeutics.
Data availability statement
Coordinates and structure factors for E10 fab complexed to the CD3ε-N7 peptide at pH 6.5 have been deposited in the Protein Data Bank under accession code 8VY4. All other data are available in the manuscript or supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2024.2373325
Additional information
Funding
References
- Goebeler M-E, Bargou RC. T cell-engaging therapies — BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–12. doi:10.1038/s41571-020-0347-5.
- Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–75. doi:10.1016/j.pharmthera.2017.08.005.
- Vafa O, Trinklein ND. Perspective: designing T-Cell engagers with better therapeutic windows. Front Oncol. 2020;10:446. doi:10.3389/fonc.2020.00446.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. doi:10.1038/s41577-020-0306-5.
- Full FDA Approval for Blinatumomab for ALL - NCI [Internet]. [cited 2023 Oct 2]. https://www.cancer.gov/news-events/cancer-currents-blog/2017/blinatumomab-all-fda-full-approval.
- Dickinson MJ, Carlo-Stella C, Morschhauser F, Bachy E, Corradini P, Iacoboni G, Khan C, Wróbel T, Offner F, Trněný M, et al. Glofitamab for relapsed or refractory diffuse large B-Cell Lymphoma. N Engl J Med. 2022;387(24):2220–31. doi:10.1056/NEJMoa2206913.
- Nathan P, Hassel JC, Rutkowski P, Baurain J-F, Butler MO, Schlaak M, Sullivan RJ, Ochsenreither S, Dummer R, Kirkwood JM, et al. Overall survival benefit with tebentafusp in Metastatic Uveal Melanoma. N Engl J Med. 2021;385(13):1196–206. doi:10.1056/NEJMoa2103485.
- Moreau P, Garfall AL, van de Donk NWCJ, Nahi H, San-Miguel JF, Oriol A, Nooka AK, Martin T, Rosinol L, Chari A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495–505. doi:10.1056/NEJMoa2203478.
- Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154:102–17. doi:10.1016/j.ymeth.2018.10.026.
- Leclercq G, Steinhoff N, Haegel H, De Marco D, Bacac M, Klein C. Novel strategies for the mitigation of cytokine release syndrome induced by T cell engaging therapies with a focus on the use of kinase inhibitors. Oncoimmunology. 2022;11(1):2083479. doi:10.1080/2162402X.2022.2083479.
- Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, Przepiorka D, Farrell AT, Pazdur R. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. The Oncologist. 2018;23(8):943–47. doi:10.1634/theoncologist.2018-0028.
- Cattaruzza F, Nazeer A, To M, Hammond M, Koski C, Liu LY, Pete Yeung V, Rennerfeldt DA, Henkensiefken A, Fox M, et al. Precision-activated T-cell engagers targeting HER2 or EGFR and CD3 mitigate on-target, off-tumor toxicity for immunotherapy in solid tumors. Nat Cancer. 2023;4(4):485–501. doi:10.1038/s43018-023-00536-9.
- Skrombolas D, Sullivan M, Frelinger JG. Development of an Interleukin-12 fusion protein that is activated by cleavage with matrix metalloproteinase 9. J Interferon Cytokine Res. 2019;39(4):233–45. doi:10.1089/jir.2018.0129.
- Boustany LM, Wong L, White CW, Diep L, Huang Y, Liu S, Richardson JH, Kavanaugh WM, Irving BA. Abstract A164: EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose> 60-fold in preclinical studies. Mol Cancer Ther. 2018;17(1_Supplement):A164–A164.
- Panchal A, Seto P, Wall R, Hillier BJ, Zhu Y, Krakow J, Datt A, Pongo E, Bagheri A, Chen T-H, et al. COBRA™: a highly potent conditionally active T cell engager engineered for the treaTMent of solid tumors. In MAbs. 2020;12(1):1792130. doi:10.1080/19420862.2020.1792130.
- Autio KA, Boni V, Humphrey RW, Naing A. Probody therapeutics: an emerging class of therapies designed to enhance on-target effects with reduced off-tumor toxicity for use in immuno-oncology. Clin Cancer Res. 2020;26(5):984–89. doi:10.1158/1078-0432.CCR-19-1457.
- Pejchal R, Cooper AB, Brown ME, Vásquez M, Krauland EM. Profiling the biophysical developability properties of common igg1 fc effector silencing variants. Antibodies (Basel). 2023;12(3):12. doi:10.3390/antib12030054.
- https://patentimages.storage.googleapis.com/8e/09/8c/72cfaa03fdd3c6/WO2018213113A1.pdf.
- Bortoletto N, Scotet E, Myamoto Y, D’Oro U, Lanzavecchia A. Optimizing anti-CD3 affinity for effective T cell targeting against tumor cells. Eur J Immunol. 2002;32(11):3102–07. doi:10.1002/1521-4141(200211)32:11<3102:AID-IMMU3102>3.0.CO;2-C.
- Dushek O, Aleksic M, Wheeler RJ, Zhang H, Cordoba S-P, Peng Y-C, Chen J-L, Cerundolo V, Dong T, Coombs D, et al. Antigen potency and maximal efficacy reveal a mechanism of efficient T cell activation. Sci Signal. 2011;4(176):ra39. doi:10.1126/scisignal.2001430.
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. 2014;32(2):191–98. doi:10.1038/nbt.2797.
- Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. 2014;2014:852748. doi:10.1155/2014/852748.
- Galogre M, Rodin D, Pyatnitskiy M, Mackelprang M, Koman I. A review of HER2 overexpression and somatic mutations in cancers. Crit Rev Oncol Hematol. 2023;186:103997. doi:10.1016/j.critrevonc.2023.103997.
- Copeland-Halperin RS, Liu JE, Yu AF. Cardiotoxicity of HER2-targeted therapies. Curr Opin Cardiol. 2019;34(4):451–58. doi:10.1097/HCO.0000000000000637.
- Yoshioka T, Shien K, Namba K, Torigoe H, Sato H, Tomida S, Yamamoto H, Asano H, Soh J, Tsukuda K, et al. Antitumor activity of pan-HER inhibitors in HER2-positive gastric cancer. Cancer Sci. 2018;109(4):1166–76. doi:10.1111/cas.13546.
- Liu CY, Ahonen CL, Brown ME, Zhou L, Welin M, Krauland EM, Pejchal R, Widboom PF, Battles MB. Structure-based engineering of a novel CD3ε-targeting antibody for reduced polyreactivity. MAbs. 2023;15(1):2189974. doi:10.1080/19420862.2023.2189974.
- Geiger M, Stubenrauch K-G, Sam J, Richter WF, Jordan G, Eckmann J, Hage C, Nicolini V, Freimoser-Grundschober A, Ritter M, et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat Commun. 2020;11(1):3196. doi:10.1038/s41467-020-16838-w.
- Rocha SS, Lin R, Dayao MR, Banzon RR, Thothathri S, Wright KJ, Aaron W, Xiao Y, Bergo N, To L, et al. Abstract 2928: TROP2 ProTriTAC™, a protease-activated T cell engager prodrug targeting TROP2 for the treaTMent of solid tumors. Cancer Res. 2023;83(7_Supplement):2928–2928. doi:10.1158/1538-7445.AM2023-2928.
- https://www.cancer.gov/research/participate/clinical-trials-search/v?id=NCI-2023-02213&r=1. [cited 2023 Dec 18].
- Chen TT. Conditionally active T cell engagers for the treatment of solid tumors: rationale and clinical development. Expert Opin Biol Ther. 2022;22(8):955–63. doi:10.1080/14712598.2022.2098674.
- Liu Y, Nguyen AW, Maynard JA. Engineering antibodies for conditional activity in the solid tumor microenvironment. Curr Opin Biotechnol. 2022;78:102809. doi:10.1016/j.copbio.2022.102809.
- US Patent Application for BISPECIFIC. ANTI-CD3 ANTIBODIES, BISPECIFIC ACTIVATABLE ANTI-CD3 ANTIBODIES, and METHODS of USING the SAME Patent Application (Application #20210047406 issued February 18, 2021) - Justia Patents Search [Internet]. [cited 2024 May 27]. https://patents.justia.com/patent/20210047406.
- Mocquot P, Mossazadeh Y, Lapierre L, Pineau F, Despas F. The pharmacology of blinatumomab: state of the art on pharmacodynamics, pharmacokinetics, adverse drug reactions and evaluation in clinical trials. J Clin Pharm Ther. 2022;47(9):1337–51. doi:10.1111/jcpt.13741.
- Zorn JA, Wheeler ML, Barnes RM, Kaberna J, Morishige W, Harris M, Huang RYC, Lohre J, Chang YC, Chau B, et al. Humanization of a strategic CD3 epitope enables evaluation of clinical T-cell engagers in a fully immunocompetent in vivo model. Sci Rep. 2022;12(1):3530. doi:10.1038/s41598-022-06953-7.
- Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, Xie S, Wang Y, Gao N, Huang Z. Structural basis of assembly of the human T cell receptor–CD3 complex. Nature. 2019;573(7775):546–52. doi:10.1038/s41586-019-1537-0.
- Menon AP, Moreno B, Meraviglia-Crivelli D, Nonatelli F, Villanueva H, Barainka M, Zheleva A, van Santen HM, Pastor F. Modulating T cell responses by targeting CD3. Cancers (Basel). 2023;15(4):15. doi:10.3390/cancers15041189.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(4):658–74. doi:10.1107/S0021889807021206.
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(5):760–63. doi:10.1107/S0907444994003112.
- BUSTER version 2.10.0 – ScienceOpen [Internet]. [cited 2024 Feb 2]. https://www.scienceopen.com/document?vid=34a668bc-6e6f-4572-a548-6d19e78e1e30.
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(4):486–501. doi:10.1107/S0907444910007493.
- Wu X, Sereno AJ, Huang F, Lewis SM, Lieu RL, Weldon C, Torres C, Fine C, Batt MA, Fitchett JR, et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. Mabs-austin. 2015;7(3):470–82. doi:10.1080/19420862.2015.1022694.
- Froning KJ, Leaver-Fay A, Wu X, Phan S, Gao L, Huang F, Pustilnik A, Bacica M, Houlihan K, Chai Q, et al. Computational design of a specific heavy chain/κ light chain interface for expressing fully IgG bispecific antibodies. Protein Sci. 2017;26(10):2021–38. doi:10.1002/pro.3240.
- Leaver-Fay A, Froning KJ, Atwell S, Aldaz H, Pustilnik A, Lu F, Huang F, Yuan R, Hassanali S, Chamberlain AK, et al. Computationally designed bispecific antibodies using negative state repertoires. Structure. 2016;24(4):641–51. doi:10.1016/j.str.2016.02.013.