
ABSTRACT
Targeting antigens with antibodies exhibiting pH/Ca2+-dependent binding against an antigen is an attractive strategy to mitigate target-mediated disposition and antigen buffering. Studies have reported improved serum exposure of antibodies exhibiting pH/Ca2+-binding against membrane-bound receptors. Asialoglycoprotein receptor 1 (ASGR1) is a membrane-bound receptor primarily localized in hepatocytes. With a high expression level of approximately one million receptors per cell, high turnover, and rapid recycling, targeting this receptor with a conventional antibody is a challenge. In this study, we identified an antibody exhibiting pH/Ca2+-dependent binding to ASGR1 and generated antibody variants with increased binding to neonatal crystallizable fragment receptor (FcRn). Serum exposures of the generated anti-ASGR1 antibodies were analyzed in transgenic mice expressing human FcRn. Contrary to published reports of increased serum exposure of pH/Ca2+-dependent antibodies, the pH/Ca2+-dependent anti-ASGR1 antibody had rapid serum clearance in comparison to a conventional anti-ASGR1 antibody. We conducted sub-cellular trafficking studies of the anti-ASGR1 antibodies along with receptor quantification analysis for mechanistic understanding of the rapid serum clearance of pH/Ca2+-dependent anti-ASGR1 antibody. The findings from our study provide valuable insights in identifying the antigens, especially membrane bound, that may benefit from targeting with pH/Ca2+-dependent antibodies to obtain increased serum exposure.
Introduction
Development of hybridoma technology by Kӧhler and Milstein and subsequent advancements in antibody engineering techniques have led to a rapid advancement of antibodies into the clinic.Citation1,Citation2 More than 100 antibody-based therapies are approved worldwide with a similar or greater number of antibodies, or their derivatives, currently in late-stage clinical trials.Citation3 In addition to providing high affinity binding and specificity to their desired targets resulting in minimal off-target toxicities, antibodies have a long serum persistence. The desirable pharmacokinetic (PK) properties of antibodies and crystallizable fragment (Fc)/albumin-based therapeutics stem from FcRn-mediated salvage from lysosomal degradation.Citation4 Linear clearance of antibodies arises from ‘off-target’ interactions primarily from Fc receptors. Nevertheless, like any other targeted therapies, antibodies experience non-linear clearance from target-mediated drug disposition (TMDD) effecting their serum exposure.Citation5,Citation6 Linear clearance of antibodies is constant and can be estimated with reasonable accuracy, whilst TMDD is dependent on target expression, internalization, and turnover of the antibody:target complex. For reduced cost and increased patient compliance, enhanced serum exposure of therapeutic antibodies is desirable and can be achieved by decreasing linear and non-linear clearances of antibodies.Citation7–9
FcRn protects IgG antibodies and albumin from intracellular catabolism through pH-dependent interactions, thereby maintaining homeostasis of IgG and albumin levels in serum.Citation10 As the key regulator of half-life of IgG and albumin, multiple engineering efforts have been directed toward increasing the affinity of Fc/albumin:FcRn interactions at acidic pH.Citation11 Increased affinity of engineered Fc/albumin variants at acidic pH without impacting binding at neutral pH not only improved PK, but also the pharmacodynamics (PD) of the engineered variants.Citation12,Citation13 However, exceeding the affinity threshold of Fc:FcRn interaction at acidic pH concurrently increased the affinity at neutral pH, thereby impeding efficient release of the antibodies back into circulation.Citation14 This highlights the need to balance affinity enhancements at acidic pH without introducing affinity at neutral pH, resulting in a technical limitation in the improvement in Fc:FcRn affinity in acidic pH to ~ 11 fold. This affinity enhancement results in an approximately 3.5-fold half-life enhancement in humans for a monoclonal antibody against a soluble target, respiratory syncytial virus (RSV).Citation11,Citation15 These and other studies have shown that altering biophysical properties of antibodies can have a favorable impact on improving their non-TMDD linear clearance.Citation16
An attractive strategy to mitigate target-mediated non-linear clearance is to generate “catch-and-release” (CAR) antibodies, which exhibit pH/Ca2+-sensitive binding toward their target.Citation17–19 The pH/Ca2+-dependent binding enables a CAR antibody to bind to the target with high affinity at neutral pH/high calcium concentration (~2 mM) while dissociating from the target at acidic pH/low calcium concentration (~2 μM).Citation18 Therefore, a CAR antibody binds to its target at the cell surface where the pH is neutral and calcium concentrations are high, resulting in endocytosis of the complex into the endosomes, exposing the complex to acidic pH and low calcium concentration enabling dissociation of the complex. Unbound antibody then returns to circulation through binding to endosomal FcRn, recycling, and then releasing at the cell surface, allowing the antibody to interact with additional target molecules.Citation18 Currently, only a few CAR antibodies against membrane-bound targets have been developed that result in enhanced serum persistence in comparison to parental antibody.Citation8,Citation17,Citation20,Citation21 In addition to the technical challenges of developing CAR antibodies, there is limited data on the FcRn affinity requirements within target cells for efficient salvage of CAR antibodies.Citation20 To understand the applicability of this approach for targeting highly expressed membrane-bound receptors, we chose to target the rapidly recycling asialoglycoprotein receptor 1 (ASGR1) with CAR antibodies engineered for modulated binding to FcRn.
ASGPR is a hetero-oligomer transmembrane glycoprotein composed of C-type lectins ASGR1 and ASGR2.Citation22,Citation23 ASGPR is primarily expressed in the liver, but a low level of extra-hepatic tissue expression is also observed. At the cellular level, a majority of the receptor is expressed on hepatocytes while liver resident macrophages also express ASGPR at lower levels.Citation24 ASGPR is a constitutively recycling receptor that binds galactose- and N-acetylgalactosamine (GalNAc)-containing ligands through Ca2+ coordinating loops of ASGR1. The binding of defective glycoproteins by ASGR1 in a calcium-sensitive manner maintains serum glycoprotein homeostasis by directing defective glycoproteins to lysosomal compartments for degradation.Citation25 Additionally, ASGPR is known to clear desialylated platelets, apoptotic cells, lipoproteins, IgA, and hepatic fibronectin from circulation. With a high expression level and rapid endocytosis, ASGPR has been used for hepatocyte-specific delivery of therapeutic moieties, including nucleotides and siRNAs.Citation25 In a recent large-scale genetic study, a loss of function (LOF) variant of ASGR1 was determined to be associated with decreased levels of non-high-density lipoprotein cholesterol, which led to ~ 34% lower risk of coronary heart disease. The cardioprotective effect of ASGR1 LOF has generated interest in developing therapeutics that can antagonize ASGR1 function.Citation26 Approximately one million ASGR1 copies are expressed per human hepatocyte, the cells which contribute to about 80% of liver mass.Citation24,Citation27 The combination of high target expression, rapid turnover and internalization, high and frequent dosing regimen of anti-ASGR1 antibody would be required to neutralize ASGR1, as antibodies targeting ASGR1 are anticipated to undergo significant TMDD.Citation28
One of the primary objective of our study was to understand the target-mediated clearance of an anti-ASGR1 antibody and to assess whether a CAR anti-ASGR1 antibody would exhibit reduced TMDD and enhanced serum exposure. Antibodies binding to ASGR1 were screened and identified for pH/Ca2+ sensitivity in their binding to ASGR1. To further investigate the influence of FcRn affinity on both linear and non-linear clearance of anti-ASGR1 antibodies, variants with increased or ablated binding to FcRn were also generated. Dose-dependent PK studies of CAR and non-CAR anti-ASGR1 antibodies were conducted in transgenic mice expressing human FcRn to characterize their serum exposure. To understand the in vivo serum clearance data, sub-cellular trafficking of the anti-ASGR1 antibodies was analyzed in HepG2 and AML12 cells. The in vivo PK data revealed a stark difference in clearance profiles of CAR and non-CAR antibodies, and a limited benefit of increased serum exposure of the CAR antibodies at low doses in comparison to non-CAR antibodies. Our study highlights the importance of considering administration doses and target cell FcRn expression and IgG salvage capacity when developing catch-and-release antibodies against membrane-bound targets.
Results
Characterization of CAR and non-CAR anti-ASGR1 antibodies and their FcRn binding variants
To generate antibodies binding to ASGR1 in a pH/Ca2+-dependent manner, XenoMouse® animals and Brown Norway rats were immunized with human ASGR1 and mouse ASGR1/ASGR2 recombinant proteins. The antibodies from the immunization campaign were converted to human IgG1 and screened for binding to mouse ASGR1 at neutral pH with 2 mM CaCl2 and at acidic pH with 2 μM CaCl2 using surface plasmon resonance (SPR). A high-affinity antibody exhibiting no pH/Ca2+-dependent binding to ASGR1 is defined as the non-CAR-WT antibody. The non-CAR-WT antibody bound to immobilized ASGR1 with an affinity of 2.2 nM and 4.8 nM at neutral pH/high calcium and acidic pH/low calcium conditions, respectively (). In contrast, a separate antibody, which is defined as the CAR-WT antibody, bound to ASGR1 with an affinity of 21 nM at neutral pH/high calcium condition whilst exhibiting no detectable binding at acidic pH/low calcium condition (). FcRn binding enhanced (YTE) and ablated (IHH) mutations that modulate FcRn-mediated PK were introduced into the Fc of the non-CAR antibody, while only the enhancing YTE mutations were introduced in the CAR antibody Fc.Citation29,Citation30 The FcRn affinity modulating mutations did not have any significant effect on ASGR1 binding characteristics (). As expected, the non-CAR-YTE and CAR-YTE antibodies exhibited enhanced binding to human FcRn at acidic pH, while the non-CAR-IHH antibody exhibited no detectable binding to human FcRn at acidic pH (data not shown). To inhibit any FcγR interactions, mutations to ablate Fcγ receptor effector functions were introduced in all the generated antibodies.Citation31,Citation32 Additionally, the antibody affinities to ASGR1 expressed on mouse hepatocytes were analyzed at neutral pH with 2 mM CaCl2 to assess their cellular binding to membrane-bound ASGR1. The non-CAR antibody variants exhibited on-cell binding affinities ranging from 5.7 to 6.3 nM, whilst the CAR antibodies bound with affinities of 51.2 and 69.5 nM for WT and YTE variants, respectively (Supplementary Figure S1).
Table 1. Equilibrium dissociation constants of the interactions between mouse ASGR1 and anti-ASGR1 antibodies at pH 7.4 with 2 mM CaCl2 or at pH 6.0 with 2 μM CaCl2. KDs were determined by immobilizing receptor on the chip and antibodies as analytes. N.D – no detectable binding.
“Catch-and-release” binding mechanism did not have appreciable enhancement in serum exposure of the anti-ASGR1 antibody
PK parameters of therapeutic antibodies observed in homozygous huFcRn Tg32 mice have high correlation to parameters observed both in non-human primates and in humans.Citation33 For this reason, PK parameters were analyzed for the CAR and non-CAR antibodies in homozygous huFcRn Tg32 mice following intravenous bolus administration. The antibodies were dosed at a range of 0.3–30 mg/kg, anticipating that the antibodies dosed at 0.3 mg/kg would be below the level required to saturate ASGR1, while the 30 mg/kg dose was expected to saturate the target.Citation28 Serum exposure of the non-CAR-WT antibody did not increase in a dose-proportional manner, indicating substantial target-mediated clearance at the lower doses (, Supplementary Table S1). The rapid decline with no detectable antibody concentration post 24 hours after administration at doses of 0.3 mg/kg and 3 mg/kg non-CAR-WT antibody indicates rapid binding to ASGR1 and depletion of antibody circulating in the serum. At 10 mg/kg the clearance profile is biphasic, where the rate of clearance marginally decreased 48 hours after administration (). The typical four phases of target-mediated antibody clearance were observed for the non-CAR-WT antibody dosed at 30 mg/kg ().Citation34 In contrast, the CAR-WT antibody exhibited a different clearance profile (). At all dose levels, the CAR-WT antibody exhibited a rapid decline in serum concentration within 24 hours after IV administration (). We observed almost one-log fold depletion of CAR-WT antibody serum concentrations within 6 hours of administration for doses 3 and 10 mg/Kg, and a two-log fold depletion of the CAR-WT antibody dosed at 30 mg/kg. As soon as 24 hours after administration, the CAR-WT antibody serum concentration reached a range of 36 nM to 0.7 nM for doses 30 mg/kg to 0.3 mg/kg, which is either close to or below the ASGR1 binding affinity at neutral/high Ca2+ (, Supplementary Figure S1). After this point, the clearance rate decreased substantially, suggesting minimal ASGR1-mediated clearance during this period.
Figure 1. Differential clearance of non-CAR and CAR anti-ASGR1 antibodies in human FcRn Tg mice. (a) mice were intravenously administered with antibodies and bled at indicated time points to measure antibody concentrations over time. Individual measurements were marked by (▼) for 0.3 mg/kg, (▲) 3 mg/kg, (■) 10 mg/kg, and (●) 30 mg/kg. (b) the inset presents a zoom in of the non-CAR and CAR antibody data from 0 to 24 hours.
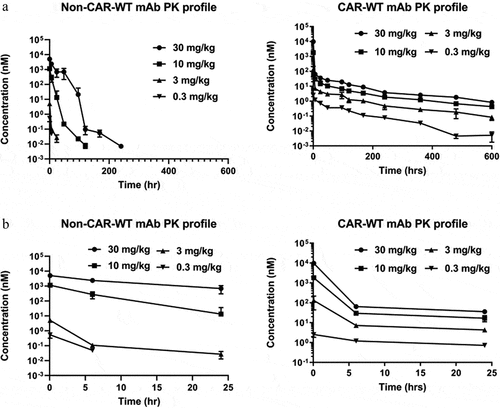
The CAR-WT antibody, which was designed to overcome ASGR1-mediated clearance by disassociating from ASGR1 in the acidic endosome, thus enabling recycling from these compartments by binding to FcRn, has 85- and 58–fold excess of non-CAR antibody serum exposure at 0.3 and 3 mg/kg doses, respectively, as measured by AUC (Supplementary Table S1, ). Higher exposure of the CAR-WT antibody at lower doses could be because of the lower affinity of the CAR-WT antibody to ASGR1, resulting in lower TMDD (, Supplementary Figure S1). At the 10 mg/kg dose, both CAR-WT and non-CAR-WT antibodies have similar serum exposure (Supplementary Table S1, ), while at the 30 mg/kg dose CAR-WT antibody serum exposure is ~ 0.4-fold relative to non-CAR-WT antibody (, Supplementary Table S1).
Figure 2. Moderate increase in serum exposure of anti-ASGR1 antibodies incorporating YTE mutations in comparison to WT variant antibodies in human FcRn Tg mice. (a) Mice were intravenously administered with antibodies and bled at indicated time points to measure antibody concentrations over time. Individual measurements for CAR and non-CAR antibodies were marked by (●) and (■) for WT and YTE, respectively. Non-CAR-IHH antibody concentrations over time are marked by (▲). (b) Serum exposure of antibodies AUClast was plotted against the administered dose. (c) Fold increase in serum exposure [mean AUC(CAR-antibody) /mean AUC(non-CAR antibody)] for WT and YTE antibodies by introducing catch-and-release binding to ASGR1 (left). Fold increase in serum exposure [mean AUC(antibody-YTE) /mean AUC(antibody-WT)] for non-CAR antibody and CAR antibody by introducing YTE mutations (right).
![Figure 2. Moderate increase in serum exposure of anti-ASGR1 antibodies incorporating YTE mutations in comparison to WT variant antibodies in human FcRn Tg mice. (a) Mice were intravenously administered with antibodies and bled at indicated time points to measure antibody concentrations over time. Individual measurements for CAR and non-CAR antibodies were marked by (●) and (■) for WT and YTE, respectively. Non-CAR-IHH antibody concentrations over time are marked by (▲). (b) Serum exposure of antibodies AUClast was plotted against the administered dose. (c) Fold increase in serum exposure [mean AUC(CAR-antibody) /mean AUC(non-CAR antibody)] for WT and YTE antibodies by introducing catch-and-release binding to ASGR1 (left). Fold increase in serum exposure [mean AUC(antibody-YTE) /mean AUC(antibody-WT)] for non-CAR antibody and CAR antibody by introducing YTE mutations (right).](/cms/asset/acd029b2-3485-46ab-b311-92179445c3a1/kmab_a_2383013_f0002_oc.jpg)
Whole-body and serum radioactivity of mice administered with CAR-WT antibody labeled with non-residualizing radioactive dye (I-125) was conducted to analyze the catabolism and excretion of antibody from the body following rapid depletion from the serum. A decrease in whole-body radioactivity counts in mice administered with radiolabeled CAR-WT antibody mirrored the rapid fall of radioactivity from serum, suggesting instantaneous catabolism of antibody following uptake of antibody from serum (Supplementary Figure S2). Importantly, non-CAR and CAR antibody dosed in ASGR1 knock-out mice have dose-dependent exposure with no target-mediated clearance (Supplementary Figure S3), indicating the initial rapid loss of serum concentration observed for CAR-WT antibody in mice expressing ASGR1 is indeed mediated through ASGR1. Glycosylation on the Fc of IgGs leads to interactions with multiple cell membrane-bound receptors like FcγRs, scavenger and ASGPR, which can affect their serum exposure.Citation35,Citation36 The antibodies in our study were generated on the SEFL2.2 IgG1 backbone, which includes N297G mutation resulting in lack of glycosylation in the Fc domain of the antibodies. Analysis of the sequences of the antibodies failed to reveal any additional glycosylation sites, indicating that interaction of antibodies to ASGR1 is indeed through the Fab domain.Citation31,Citation32
Among the natural ligands of ASGPR, sugar-based ligands exhibit maximum affinity with pH/Ca2+ dependence to the receptor. Calcium-dependent binding to the ligands emanates from the two high affinity Ca2+ ions, which are coordinated by the loops of the receptor. As CAR antibodies exhibit analogous pH/Ca2+-dependent binding to ASGR1, competitive binding analyses were performed to study the impact of ligands on antibodies binding to the receptor and potentially affecting serum clearance. For these analyses, CHO-S cells transfected with mouse ASGR1 were used. CHO-S-expressing mouse ASGR1 were incubated with ASGR1 ligands GalNAc and asialofetuin at neutral pH with 1 mM CaCl2. GalNAc and asialofetuin bound with EC50 values of 2.7 nM and 53 nM, respectively, to CHO-S cells expressing mouse ASGR1. There was no detectable binding of these ligands to CHO-S cells not expressing mouse ASGR1, indicating the specificity of the ligands to mouse ASGR1 (Supplementary Figure S4). To analyze the competitive binding of antibodies to ASGR1 in the presence of its natural ligands, CHO-S cells expressing mouse ASGR1 were preincubated with the antibodies followed by incubation of the ligands at their respective EC50 concentrations to determine the concentration of the antibody at which it can inhibit the ligand binding to the receptor. The CAR-WT antibody exhibiting pH/Ca2+-dependent binding to ASGR1 exhibited competitive binding to both the ligands with IC50 values of 44 nM and 3.7 nM to GalNAc and asialofetuin, respectively (Supplementary Figure S4). Surprisingly, the non-CAR-WT antibody with no pH/Ca2+-dependent binding to ASGR1 also exhibited competitive binding to the ligands with IC50 values of 24 nM and 4 nM for GalNAc and asialofetuin, respectively (Supplementary Figure S4). Competitive binding analyses indicate a potential overlap of epitopes for both the antibodies with the ligand binding domain on ASGR1, which mainly encompasses the Ca2+ binding loops of ASGR1.Citation37 As the anti-ASGR1 antibodies were dosed in vivo at much higher concentration than the observed IC50 values (, Supplementary Figure S4), dosed antibodies effectively compete with ligands and undergo ASGR1-mediated clearance. The natural ligands may affect ASGR1-mediated clearance of antibodies when the dosed antibody concentrations are at or below the IC50 values.
Enhanced FcRn binding affinity resulted in moderate increase in serum exposure of anti-ASGR1 antibodies
Previous studies reported increased serum exposure for antibody therapeutics engineered to bind FcRn with increased affinity at acidic pH. A majority of these studies were geared toward antibodies targeting soluble targets with limited knowledge of the impact of modulation to FcRn binding of antibodies binding to high expression membrane-bound targets. Both CAR-WT and non-CAR-WT antibodies displayed significant target-mediated clearance by binding to membrane-bound ASGR1 (). To assess the impact of varying FcRn affinity (at acidic pH) on clearance of anti-ASGR1 antibodies, mutations (YTE or IHH) were introduced in the Fc domain of CAR and non-CAR antibodies. CAR-YTE and non-CAR-YTE antibodies exhibited increased binding to human FcRn at acidic pH relative to WT antibodies whilst non-CAR-IHH antibody had no detectable binding to FcRn (data not shown). We next compared the serum exposures of non-CAR-IHH, non-CAR-WT and non-CAR-YTE antibodies in vivo to inform the impact of FcRn salvage on clearance of anti-ASGR1 antibodies exhibiting high target-mediated clearance.
The non-CAR-YTE antibody demonstrated increased serum exposure in comparison to the non-CAR-WT antibody at all dose levels (, Supplementary Table S1, Supplementary Figure S5). The increase in exposure for the non-CAR-YTE antibody ranged from 1.2 to 2-fold when compared to the non-CAR-WT antibody, with higher fold increases observed at lower doses (). This increased exposure for the non-CAR-YTE antibody stemmed from reduced clearance 48 hours after administration (). During this phase, the non-CAR-YTE antibody serum concentration is approximately 1 nM, a concentration at which the non-CAR-YTE antibody is anticipated to undergo non-target-mediated clearance where increased FcRn binding can enhance the salvage of the antibody. The non-CAR-IHH antibody exhibited rapid clearance and diminished exposure compared to the non-CAR-WT and non-CAR-YTE antibodies (, Supplementary Table S1). This reduced exposure is more prominent for the higher doses of (10 and 30 mg/kg) at which ASGR1 is saturated and the antibody is expected to undergo substantial non-target-mediated clearance.
For the CAR antibody, enhanced FcRn binding resulted in increased serum exposure in the range of 2.7–3.1-fold for doses between 3 and 30 mg/kg, while at 0.3 mg/kg, the CAR-YTE antibody had reduced exposure by 0.5-fold compared to CAR-WT antibody (, Supplementary Table S1, Supplementary Figure S5). The increased serum exposure for the CAR-YTE antibody between 3 and 30 mg/kg stemmed from the non-linear antibody clearance phase and a higher Cmax for the CAR-YTE antibody. After 48 hours, the clearance rate of the CAR-YTE antibody was comparable to the CAR-WT antibody.
To confirm that the observed slow clearance 48 hours post administration of CAR-WT, CAR-YTE and non-CAR-YTE antibodies was not due to ASGR1 saturation but because of low serum concentration of antibodies leading to reduced interaction with ASGR1, the CAR-YTE and non-CAR-YTE antibodies were dosed every week for 3 weeks (Supplementary Figure S6). A similar rapid clearance was observed after repeat dosing, indicating that ASGR1 was indeed not saturated and that the administered antibodies after 48 hours were primarily undergoing non-target-mediated clearance (Supplementary Figure S6). To analyze the effects of FcRn-mediated salvage on antibodies undergoing substantial target-mediated clearance like anti-ASGR1 antibodies, we generated non-CAR anti-ASGR1 antibody variant (non-CAR-IHH) ablated of FcRn-mediated salvage. Comparing the clearance values of IHH, WT and YTE variants of non-CAR anti-ASGR1 antibodies suggested that a substantial amount of the dosed antibodies was being endocytosed into FcRn-positive cells in a fluid phase manner and subsequently rescued by FcRn. Additionally, the amount of salvaged antibody can be increased by enhancing the binding to FcRn at acidic pH ().
Pulsed anti-ASGR1 antibodies localize to endo-lysosomal compartments and decrease ASGR1 levels
To gain mechanistic insights into the differential in vivo clearance of CAR and non-CAR antibodies, we conducted cellular uptake and localization studies by fluorescence microscopy. Undesirable auto-fluorescence from primary hepatocytes of mouse and human origin prompted us to use HepG2 (expressing human ASGR1) and AML12 (expressing mouse ASGR1) cell lines for fluorescence-based imaging experiments. Multiple reports indicate that ASGR1 is primarily expressed on hepatocytes and undergoes rapid internalization and recycling, but its sub-cellular localization is not well understood. To examine the steady-state localization of human ASGR1, fixed and permeabilized HepG2 cells were stained with an anti-ASGR1 antibody along with staining for Rab11 (early endosomal marker) and LAMP1 (late endosomal marker) (). In HepG2 cells, there is an approximately equal distribution of intracellular and extracellular ASGR1 (, ). Consistent with this, ASGR1 expression was equally distributed intracellularly and on the cell membrane in primary human hepatocytes, despite variable expression across donors (). Intracellular ASGR1 predominantly colocalized with Rab11-positive endosomal compartments with minimal colocalization being observed with LAMP1-positive compartments ().
Figure 3. Sub-cellular localization of human ASGR1 and its colocalization with early and late endosomal markers. HepG2 cells were fixed with paraformaldehyde and permeabilized prior to staining with anti-ASGR1 antibody labeled with alexa-647, anti-Rab11 antibody labeled with alexa-568, and anti-LAMP1 antibody labeled with alexa-488. Stained cells were imaged and pseudo-colored red (alexa-647) or green (alexa-488 or alexa-568). An image of a representative field of view of cells is shown. Fluorescence intensities along the lines in the overlays are shown in the fluorescence intensity plot. The scale bars represent 20 µm.
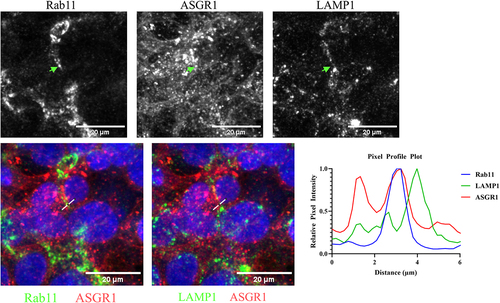
Table 2. ASGR1 expression is several folds higher than FcRn in hepatocytes. Receptor quantification is performed by flow-based assay in fixed or fixed and permeabilized cells to quantify the surface and total receptors per cell, respectively.
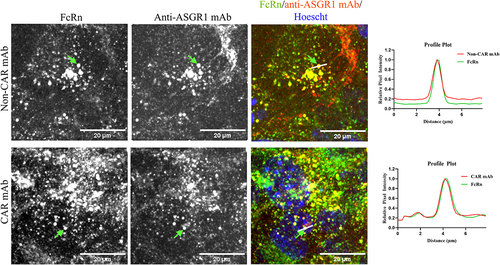
To understand the initial rapid drop in serum concentration of the CAR antibody in vivo (), we conducted an imaging-based study to analyze cellular localization of anti-ASGR1 antibodies and their association with endosomal FcRn. In vivo-dosed antibodies exhibited rapid serum clearance at concentrations ranging from ~ 104 nM to 10 nM, so in vitro cells were pulsed with antibodies at a concentration of 66.6 nM (10 µg/ml) to mimic the rapid uptake of antibodies by ASGR1 into the cells (). AML12 cells were co-pulsed with CAR or non-CAR antibodies along with an FcRn marker (Abdeg, an engineered Fc fragment that binds to FcRn with high affinity at neutral and acidic pH) and cellular localization of pulsed antibodies and FcRn was visualized using confocal microscopy.Citation38 Aligning with the observed sub-cellular distribution of the ASGR1 receptor (), pulsed non-CAR-WT and CAR-WT antibodies localized to endosomal compartments as well as to the cell membrane (). However, in contrast to the non-CAR antibody, the bulk of the pulsed CAR-WT antibody appeared to localize to the endosomal compartments, as indicated by punctate intracellular staining, with limited cell membrane localization (). Endocytosed CAR and non-CAR anti-ASGR1 antibodies co-localize with the fluorescently labeled Abdeg, indicating that they do localize to FcRn+ compartments, and that a CAR antibody upon dissociation from ASGR1 in endosomes can potentially recycle back to cell surface through FcRn-mediated salvage mechanism ().
Figure 4. Internalized anti-ASGR1 antibodies colocalize with intracellular FcRn. AML12 cells were co-pulsed with 10 µg/mL of alexa-568 labeled FcRn marker (Abdeg) and alexa-647 labeled CAR and non-CAR anti-ASGR1 antibodies at 37°C, followed by wash and fix with paraformaldehyde. Fixed cells were imaged, and a representative field of view is presented. Alexa fluorophores 568, 647 and hoescht were pseudo-colored green, red, and blue, respectively. Fluorescence intensity along the dotted line over an endosomal compartment is presented in the fluorescence intensity plot. The scale bar for the panel of cells is 20 µm.
To rule out cross-species binding anomalies between mouse FcRn in AML12 cells and to human Fc, and to avoid potentially confounding competition between anti-ASGR1 antibodies and Abdeg binding to FcRn, colocalization experiment was also conducted in HepG2 cells, which express human FcRn (Supplementary Figure S7). HepG2 cells were co-pulsed with CAR-WT antibody and engineered human serum albumin (HA-HSA, high affinity HSA with a mutation K573P that exhibits enhanced binding to human FcRn at acidic pH).Citation39 FcRn ligands albumin and Fc are capable of binding simultaneously to FcRn, and binding of either of the ligands does not influence binding or recycling of the other ligand.Citation4 As observed in AML12 cells, CAR-WT antibody endocytosed into endosomal compartments of HepG2 cells co-localized to HA-HSA-labeled FcRn+ compartments (Supplementary Figure S7).
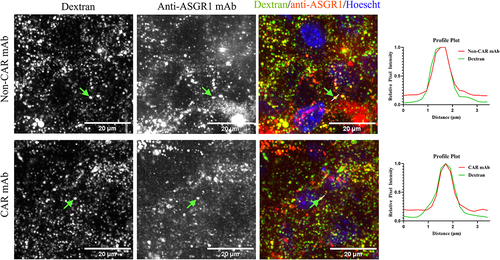
Upon confirmation of localization of intracellular CAR-WT antibody in FcRn+ endosomes, cellular FcRn levels were quantified by a flow-based assay and mass-spectrometry to investigate whether sufficient FcRn expression levels are present to salvage endosomal CAR antibodies ( and Supplementary Table S2). In human and cynomolgus monkey hepatocytes and in HepG2 cells, there is a 5- to 60-fold excess of ASGR1 molecules relative to FcRn ( and Supplementary Table S2). In hepatocytes of human FcRn transgenic mice, ASGR1 levels are ~ 10-fold higher than FcRn, suggesting that there could be an excess pool of the CAR antibody in endosomes that could not be rescued by FcRn when administered at high doses (Supplementary Table S2). To investigate whether the anti-ASGR1 antibodies internalized to the endosomes are recycled back to cell surface or routed to lysosomes for degradation, AML12 cells were pre-pulsed with dextran, a lysosomal marker, followed by a pulse-chase with CAR-WT and non-CAR-WT antibodies and cells were then analyzed for colocalization of antibodies with dextran-positive lysosomes (). After prolonged chase of anti-ASGR1 antibodies, the anti-ASGR1 antibodies not recycled back to cell surface eventually localized to dextran-labeled lysosomal degradative compartments.
Figure 5. Anti-ASGR1 antibodies localize to lysosomal compartments at the end of the chase phase. AML12 cells were pre-pulsed with alexa-568 labeled dextran for four hours and chased overnight at 37°C. Following the dextran pulse-chase, alexa-647 labeled CAR and non-CAR anti-ASGR1 antibodies were pulsed for 30 minutes and chased for four hours at 37°C. Chased cells were washed and fixed with paraformaldehyde and imaged. Alexa-647 labeled antibodies and alexa-568 labeled dextran are presented in pseudo-colored red and green, respectively. Fluorescence intensity along the dotted line is presented in the fluorescence intensity plot. The scale bars for the panel of cells is 20 µm.
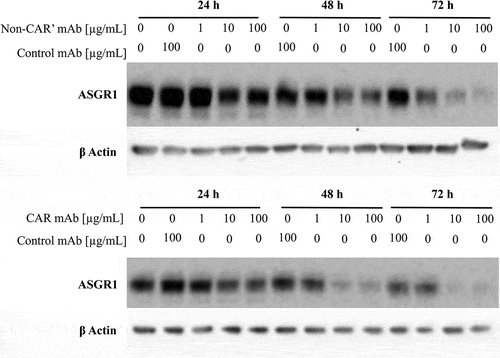
ASGR1 is a recycling receptor, but it is not known whether the internalized receptor bound by either CAR or non-CAR antibody is recycled back to the cell surface or shunted to lysosomal compartments for degradation. Western blot analysis was performed on HepG2 cells treated with anti-ASGR1 antibodies to determine if CAR or non-CAR antibody treatment influences degradation of ASGR1. The non-CAR antibody used in the in vivo PK studies does not exhibit similar binding characteristics to human ASGR1, while the CAR antibody binds to both human and mouse ASGR1 with similar affinity (data not shown). The observed differential binding of the non-CAR antibody to ASGR1 from mouse and human could be due to minor differences in sequence identity between both species, especially in the carbohydrate binding domain (Supplementary Figure S8). Because of the difference in binding to mouse and human ASGR1 by the non-CAR antibody, we used a variant of the non-CAR antibody (non-CAR’ antibody) that binds to human ASGR1 with no pH/Ca-sensitivity in in vitro experiments using HepG2 cells. HepG2 cells were pulsed with concentrations ranging from 1 µg/ml to 100 µg/ml of non-CAR’ and CAR antibodies for 24 to 72 hours. The concentrations of anti-ASGR1 antibodies pulsed in vitro correspond to the in vivo antibody concentrations at which ASGR1 is anticipated to be saturated with dosed anti-ASGR1 antibodies undergoing ASGR1-mediated clearance (). The pulsed CAR and non-CAR anti-ASGR1 antibodies resulted in a dose-dependent decrease in cellular ASGR1 levels (), but there was no appreciable difference in the amount of ASGR1 degraded by either the CAR or non-CAR’ antibody. This data suggests that ASGR1 internalized by a bivalent anti-ASGR1 antibody, irrespective of being bound or free of antibody in the endosome, may not recycle back to the cell surface and is shunted to lysosomes for degradation ().
Figure 6. Treatment of HepG2 cells with CAR and non-CAR’ anti-ASGR1 antibodies decreases ASGR1 levels. HepG2 cells were pulsed with varying concentrations (1, 10 and 100 µg/mL) of CAR antibody and non-CAR’ antibody (an antibody binding to human ASGR1 with no pH/Ca-sensitivity) for 24, 48 and 72 hours. Treated cells were lysed and analyzed for ASGR1 levels through western blot.
Discussion
With long serum persistence, high affinity, and specificity toward their targeted antigens, developing antibodies as drugs to treat diseases is an attractive strategy.Citation3 Targeting antigens with high expression or rapid turnover requires large and/or frequent dosing, leading to inconvenience for patients and increased treatment costs.Citation40 To mitigate target-mediated clearance and improve the PK properties of an antibody, Igawa et al. introduced pH/Ca2+-dependent binding to antigen.Citation17 Several groups followed this approach to generate antibodies having switchable binding properties to the targeted antigens, resulting in reduced target-mediated clearance and improved PK. Antibodies exhibiting this property are termed either pH-dependent, catabolic or catch-and-release (CAR) antibodies. Targeting ASGR1, a highly abundant membrane-bound receptor that exhibits fast internalization and rapid turnover, with a conventional antibody has been challenging.Citation28 We aimed to generate a CAR antibody against ASGR1 to determine if the switchable binding property of a CAR antibody and FcRn-mediated recycling would improve PK relative to a non-CAR anti-ASGR1 antibody. We also evaluated the impact of enhanced FcRn binding on the serum clearance of CAR and non-CAR anti-ASGR1 antibodies by dosing antibodies with modified FcRn binding in transgenic mice expressing human FcRn.
Traditionally, generating CAR antibodies is a laborious task with limited success of identifying an antibody with the desired pH/Ca2+ switchable binding properties. Various display techniques and histidine scanning approaches have been used previously to generate CAR antibodies against antigens from their parent non-CAR antibodies.Citation17,Citation18,Citation41 Even with extensive histidine scanning of parental antibodies, candidate CAR antibodies often retained substantial binding to antigens at acidic pH or had reduced binding affinities to antigen at neutral pH.Citation17,Citation18,Citation41 Alternative strategies to isolate CAR antibodies using de novo phage display enriched in histidine content, or directly from immunization campaigns were more successful.Citation20,Citation42 Literature reports indicate that the success rate of identifying of CAR antibodies is about 3%, but Yang et al. reported greater success with an immunization campaign that yielded over a hundred clones exhibiting reduced binding affinities at acid pH against connective tissue growth factor and protein convertase subtilisin/kexin type 9.Citation20
To identify a CAR anti-ASGR1 antibody, we screened clones from an immunization campaign using SPR assays at neutral pH with high calcium concentration and at acidic pH with low calcium concentration. Our screening approach yielded two antibodies out of approximately 100 candidates that exhibited reduced to no detectable binding to ASGR1 at acidic pH and low calcium concentration while maintaining desirable binding affinity at neutral pH and high calcium concentration. ASGR1 binds its ligands in a pH/Ca2+ dependent manner through a carbohydrate binding domain consisting of flexible loops coordinated by calcium ions.Citation37,Citation43,Citation44 These flexible loops might undergo structural conformational changes in the endosomes where the calcium concentration drops by a thousand-fold in comparison to extracellular fluid.Citation37 For efficient generation of pH/Ca2+-dependent antibodies, selecting antibodies against domains consisting of histidine and Ca2+ ion coordinating residues could yield desirable CAR antibodies. The obtained CAR anti-ASGR1 antibodies epitopes could be fortuitously directed against the calcium binding loops of ASGR1, leading to reduced binding to ASGR1 in the environment mimicking endosomal compartments, thus reducing the need for further engineering of the clones. Among the two CAR anti-ASGR1 clones we identified, the clone with the tightest binding at neutral pH with high calcium concentration was chosen for further analysis in in vivo and in vitro assays.
After CAR antibodies dissociate from bound antigen in the endosome, the free antibodies recycle back to the extracellular space via association with FcRn through tubulovesicular transport carriers.Citation18 The efficiency of dissociation of antigen by CAR antibody is governed by the magnitude of the change in antigen:CAR-antibody affinity at the acidic endosomal conditions relative to affinity at neutral pH. For example, an anti-IL-6 antibody exhibiting an approximately 2-fold reduction in affinity at acidic pH compared to neutral pH resulted in only about 25% of the bound IL-6 being dissociated in the endosomes, while an antibody with no detectable binding to IL-6 at acidic pH led to about 81% IL-6 dissociation from the antibody-cytokine complexes in the endosomes.Citation18 Previous studies have demonstrated that, rather than the degree of binding affinity at endosomal conditions, it is instead the rate of dissociation, in conjunction with tight binding to antigen at the extracellular milieu, that leads to efficient antigen clearance and mitigation of antigen-mediated clearance of CAR antibodies.Citation17,Citation41 Additionally, the clearance of antigens can be enhanced by introducing receptor-mediated uptake termed as “sweeping mechanism” even without the complete loss of binding to antigens in endosomal conditions.Citation20,Citation45 The selected CAR anti-ASGR1 antibody used in this study exhibited no detectable binding at endosomal conditions, but had reduced affinity to ASGR1 at extracellular conditions in comparison to non-CAR anti-ASGR1 antibody (, Supplementary Table S1). Nevertheless, with the high expression load of ASGR1 both CAR and non-CAR antibodies are expected to have high receptor-mediated uptake at the in vivo dosed concentrations.
The anti-ASGR1 antibodies generated in this study contain the human IgG backbone, and some also contain mutations in the Fc domain that affect affinity to human FcRn. We used transgenic mice expressing human FcRn to characterize the in vivo PK of antibodies bearing human Fc because a good correlation has been observed in relation to PK obtained in higher order species.Citation33 We administered mice with CAR and non-CAR antibodies at doses ranging from 0.3 mg/kg to 30 mg/kg in an effort to investigate antibody PK at both target saturating and sub-target saturating concentrations.Citation28 Others have reported that a non-CAR anti-ASGR1 antibody saturated the receptor with doses ≥5 mg/kg and the percentage of the available free receptor decreased as the dose increased. Nevertheless, even with a 30 mg/kg dose level of a non-CAR antibody the majority of ASGR1 was available for binding one-week post-dosing, suggesting a higher dose levels and frequencies would be required in the clinical setting.Citation28 Similarly, our non-CAR anti-ASGR1 antibody exhibited a clearance profile consistent with high levels of ASGR1-mediated TMDD ().
Contrary to the published reports of increased serum exposure of CAR antibodies against membrane-bound antigens, the CAR anti-ASGR1 antibody exhibited rapid serum depletion (), despite sufficient switchable binding properties and the presence of FcRn in the target-expressing hepatocytes (, Supplementary Table S2). The CAR antibody did exhibit slightly increased serum exposure at lower doses in comparison to the non-CAR antibody (), but this increase in serum exposure could be attributed to lower affinity of the CAR antibody to ASGR1, resulting in reduced ASGR1 engagement (, Supplementary Figure S1).Citation17 A TMDD model developed by Bon et al. suggested that, as the dose of non-CAR antibody increased, a greater amount of antibody undergoes non-target clearance.Citation28 Validating the published model, the non-CAR-YTE antibody exhibited increased serum exposure compared to the WT variant. Despite rapid binding and catabolism of anti-ASGR1 antibody by ASGR1, a substantial amount of the dosed antibody undergoes FcRn-mediated salvage, as demonstrated by poor serum exposure of the anti-ASGR1 variant (non-CAR-IHH antibody) with ablated binding to FcRn ().
To further understand the potential reasons for why the CAR antibody targeting ASGR1 did not exhibit improved PK, we quantified expression levels and analyzed sub-cellular localization of ASGR1 and FcRn in the hepatocytes. The availability of sufficient endosomal FcRn levels is not only necessary but a prerequisite for efficient recycling of the CAR antibody following dissociation from ASGR1. The observed cellular ASGR1 levels corroborated with published literature of about million receptors per human hepatocyte with variability being observed among individuals.Citation46,Citation47 Analysis of the ASGR1/FcRn ratio in hepatocytes indicates a massive overexpression (~5–60-fold) of ASGR1 in comparison to FcRn in analyzed HepG2 cells and hepatocytes from humans, cynomolgus monkeys, and transgenic human FcRn mice (, Supplementary Table S2). Additionally, the intracellular levels of FcRn were far less compared to ASGR1 in our analysis (). Apart from cellular receptor levels, endosomal co-location of receptors is crucial for co-trafficking of a CAR antibody with FcRn.Citation18 Although we could not achieve the microscopic resolution to differentiate between the membrane and lumen of endosomes, our immunofluorescence analysis indicated that ASGR1 localized in recycling and endolysosomal compartments, analogous to the sub-cellular localization of FcRn ().Citation48 We also determined that the intracellular ASGR1 CAR antibody indeed colocalized to the FcRn+ subcellular compartments ( and Supplementary Figure S7), suggesting that the CAR antibody can potentially recycle with FcRn through tubulovesicular transport carriers. These findings indicate that the low levels of FcRn in hepatocytes could be the limiting factor preventing salvage of the CAR anti-ASGR1 antibody, resulting in its rapid clearance. Other studies emphasized the importance of affinity of CAR antibodies toward FcRn at acidic pH and the expression of FcRn for CAR antibodies to efficiently recycle from endosomal compartments to the cell surface.Citation20,Citation21 A CAR antibody targeting carcinoembryonic antigen exhibited target-mediated clearance in tumor-bearing mice lacking mouse FcRn in tumors, suggesting FcRn presence at the antigen expression site is essential for the functionality of a CAR antibody targeting a membrane-bound antigen.Citation21 This study analyzed the absence of FcRn at the antigen expression site, but further studies are required to analyze the relative level of FcRn required in comparison to the antigen expression level for effective CAR antibody salvage.Citation21 Another potential factor not analyzed in our study is the IgG recycling capacity of hepatic FcRn. In one study, conditional deletion of hepatic FcRn led to hypoalbuminemia and albumin loss to bile without affecting total IgG levels in mice.Citation49 Additional studies are warranted to fully understand the recycling efficiency of IgG through hepatic FcRn.
We have shown that CAR antibodies with a complete loss of binding to antigen in endosomal environments can be obtained directly from an immunization campaign, especially for an antigen containing calcium binding loops. The serum exposure of CAR antibodies targeting a membrane antigen greatly depends on the FcRn expression and sub-cellular localization in the antigen-expressing cell. Due to low expression of hepatic FcRn in comparison to high expression of ASGR1, anti-ASGR1 CAR antibody does not exhibit the expected increase in serum exposure. On the contrary, rapid elimination of anti-ASGR1 CAR antibody from circulation is observed with localization of CAR antibody to endo-lysosomal compartments of hepatocytes. Recent studies had demonstrated increased intracellular accumulation of payloads to cells expressing membrane-bound antigens when targeted with payloads conjugated to CAR antibodies for therapeutic purposes.Citation50,Citation51 Given the importance of targeting ASGR1 for multiple diseases, additional studies are warranted to determine if rapid accumulation and catabolism of the CAR anti-ASGR1 antibody in hepatocytes can be exploited for enhancing the delivery of payloads to hepatocytes or depleting pathogenic antigens for therapeutic applications.Citation25,Citation50,Citation52
Materials and methods
Cloning
The antibodies against human and mouse ASGR1 were identified in an antibody discovery campaign using the Brown Norway rats and XenoMouse® platform (patent publication number – WO2017058944A1). Gene fragments for antibody heavy chains (HC) or light chains (LC) were synthesized by Twist Bioscience or Integrated Technologies Inc. and cloned via Golden Gate assembly into mammalian stable expression vectors.Citation53,Citation54 Antibody HC was cloned into a pTT5-derived vector carrying a puromycin selection marker, while antibody LC was cloned into a pTT5-derived vector with a hygromycin selection marker. Transfection-grade plasmids containing the LC or HC were miniprepped using QIAprep 96 Plus Miniprep Kits and sequence confirmed by Sanger sequencing. Sequence verified clones were used for stable mammalian expression. The mutations M252Y/S254T/T256E and I253A/H310A/H435 were introduced in the Fc of antibodies to either enhance (at acidic pH) or ablate binding to human FcRn.Citation29,Citation55 The mutations to make antibodies effector-functionless are SEFL2.2 (R292C, N297G, and V302C) introduced in the Fc of the antibodies.Citation31,Citation32
Transfection and expression
For anti-ASGR1 antibodies production, CHO-K1 cells transfection was done by Lipofectamine LTX reagent from ThermoFisher Scientific (cat # 15338500) in a 24-well format. Non-linearized DNA and Lipofectamine LTX were diluted in 0.5 ml Opti-MEM media, respectively. Then the diluted DNA and Lipofectamine LTX were mixed thoroughly and incubated at room temperature for 15–20 minutes. Two million viable CHO-K1 cells were washed with phosphate-buffered saline (PBS) and resuspended in 1 mL of Opti-MEM. The resuspended CHO-K1 cells were then mixed with the DNA/Lipofectamine complex and incubated for 5–6 hours at 37°C with 5% CO2 with shaking. After the incubation, an additional 2 mL of CHO-K1 media (50% CS9 media with 50% ExCell302, SAFC Biosciences, cat# 14324C) with 2 mM L-glutamine (Gibco, cat# 25030–081)) was added. After 2 days, media was changed to selection media (CHO-K1 media with 10 µg/mL puromycin, Gibco, cat #A11138–03) and 600 µg/mL hygromycin (Invitrogen, cat# 10687–010)). Cell viability was measured, and selection media was changed every 2 days until viability reached 90%. The recovered cells are expanded and seeded at 1.5 × 106/mL density in 6D CD media (6D CD media: ABM025, SAFC Biosciences, cat# 66674) with 5% AFM028 (SAFC Biosciences, cat# 66683) and 0.5 mg/mL Tyrosine (SAFC Biosciences, cat# 91135) for protein expression at 31°C with 5% CO2. CHO-S cells were transfected with the vector encoding mouse ASGR1 FLAG tagged sequence to be used for ligand:antibody competition assays.
Protein purification
Recombinant antibodies were purified using standard methods on an Akta Explorer or Akta Pure chromatography instrument (GE Healthcare LifeSciences/Cytiva). Briefly, proteins were affinity purified from conditioned media (cell supernatant) using either MabSelect SuRe or KappaSelect resin (Cytiva) with acidic pH elution buffer (0.5% acetic acid, 150 mM NaCl, pH 3.5 for MSS or 0.1 M Glycine, pH 2.7 for KS). Pooled eluate was neutralized and further purified by size exclusion chromatography (320 mL HiLoad 26/600 Superdex 200 pg; Cytiva) and formulated in 30 mM HEPES, 150 mM NaCl, pH 7.6.
Affinity determination by surface plasmon resonance and on cell binding
Equilibrium binding affinities of the interaction between mouse/human ASGR1 and anti-ASGR1 antibodies were determined using BIAcore 3000. Mouse and human ASGR1 proteins were obtained from R&D-systems (cat # 2755-AS/CF and 4394-AS/CF, respectively) and immobilized on CM5 chips using amine coupling chemistry to a density of ~ 1500 RU. On each CM5 chip a reference flow cell is used which was coupled with coupling buffer only. Antibodies were injected over immobilized ASGR1 at a concentration range of 1000 nM − 0.2 nM, with a two-fold serial dilution. To determine pH/Ca-sensitive binding runs were performed using PBS with 0.01% (v/v) Tween20 and 0.05% azide with either pH 7.4/2 mM calcium chloride or pH 6.0/2 μM calcium chloride. The chip was regenerated using 0.15 M NaCl, 0.1 M glycine pH 1.5 buffer between each injection cycle. Equilibrium binding affinities between human FcRn (R&D-systems, cat# 8639-FC) and antibody variants were determined by immobilizing FcRn on CM5 chip using amine chemistry and injecting antibodies as described above. PBS at pH 6.0 with 0.01% (v/v) Tween 20 and 0.05% azide was used as a running buffer. Equilibrium dissociation constants were determined using 1:1 interaction model using BIAevaluation.
Anti-ASGR1 antibody binding affinities to mouse hepatocytes at neutral pH and 2 mM CaCl2 were determined using a flow-based assay. Anti-ASGR1 antibodies and isotype control were fluorescently labeled with Alexa 647 dye (Invitrogen, cat#A30009), according to the protocol. Mouse cryosuspension hepatocytes from BIOIVT (cat # M005052) were thawed according to the protocol and were washed with ice cold DPBS supplemented with 2 mM CaCl2 followed by centrifugation at 50 g for five minutes. The hepatocytes pellet was suspended with DPBS containing 2 mM CaCl2 at a density of one million cells per mL. The experiment was conducted in 96-well plates, with 50,000 cells per well. The cells were blocked with CD16/CD32 (BD Biosciences, cat# 553141), mouse serum, and human serum and stained with hepatocyte marker CK18 AF 350 (Bioss, cat#BSM33103MA350, 1:200 dilution) and live/dead cell marker APC-Cy7 (Invitrogen, cat #L10119, 0.2 µl/well). Anti-ASGR1and isotype control antibodies were prepared at a concentration range from 0.05 nM − 100 nM and 0.05 nM − 1600 nM (four-fold dilution series) for non-CAR and CAR antibodies, respectively, in ice-cold DPBS buffer supplemented with 2 mM CaCl2. The hepatocytes were incubated in the antibody solutions on ice for 30 minutes. Post incubation the cells were washed with an excess of ice-cold DPBS containing 2 mM CaCl2 and spun at 100 g for five minutes. The supernatant was aspirated and cells were fixed using IC fixative buffer (eBioscience, cat#00-8222-49), which was diluted 1:1 with DPBS containing 2 mM CaCl2. The fluorescence signal was measured by analyzing the cells through a cytometer and background fluorescence intensity (cells stained with isotype control at identical concentration to antibody) was subtracted. Mean fluorescence intensity data was plotted against the incubated antibody concentration and analyzed in GraphPad Prism (version 10.2.2, 397). Binding dissociation constants (KD) were determined by fitting the curves to a nonlinear regression one site – total fit model.
Receptor quantification by flow cytometry, mass spectrometry and western blot
The number of FcRn and ASGR1 receptors expressed by the cell were quantified using a bead-based assay QIFIKIT (Agilent, cat# K007811–8). Cryopreserved cynomolgus monkey hepatocytes (Triangle Research Labs, cat#TRL-MICC-316) and human hepatocytes (Lonza, cat#HUCPG) were thawed and added to 50 mL of thawing media and centrifuged at 100 g for 5 minutes. Cells were washed with room temperature DPBS and centrifuged at 100 g for 5 minutes. Washed cells were fixed with 4% paraformaldehyde on ice for 15 minutes. Half of the fixed cells were permeabilized by incubating the fixed cells in BD perm wash buffer (BD biosciences, cat# 554723) for 30 minutes at room temperature. HepG2 cells (ATCC, cat# HB-8065) were treated in similar fashion after harvesting following treatment with TrypLE express enzyme (Gibco, cat# 12-604-013). Treated cells were stained with either anti-FcRn antibody, anti-ASGR1 antibody or respective isotype controls. Staining antibodies are anti-human ASGR1 antibody (R&D – Systems, cat#MAB4394), anti-human FcRn antibody (R&D – Systems, cat# MAB8639), IgG1 isotype control (Biolegend, cat# 401402) and IgG2b isotype control (Biolegend, cat# 401201). F(ab’)2 goat anti-mouse IgG (H+L) labeled with alexa 647 (Thermo Fisher, cat# A-21237). Stained cells were analyzed for fluorescence intensity on flow cytometer. Receptors were quantified as per the protocol described in QIFIKIT.
Cellular ASGR1 and FcRn levels were also quantified through mass spectrometry using stable isotope-labeled (SIL) peptides (BIOSYNTH, KY, USA).Citation56 SIL peptides used for the receptor quantification are – WVDGTDYETGFK (human/cynomolgus monkey ASGR1), DLTEDHSSLLLHVK (mouse ASGR1), and QGTWGGDWPEALAISQR (human/cynomolgus monkey FcRn). For mass spectrometry-based analysis, human hepatocytes (ThermoFisher Scientific, cat#HMMCPIS, and Corning, cat454551), cynomolgus monkey hepatocytes (BioIVT, cat#M003055-P), HepG2 (ATCC, cat# HB-8065) and hepatocytes from livers harvested from human FcRn transgenic mice were used. Cells were counted and incubated with NP-40 cell lysis buffer (ThermoFisher Scientific, cat#J60766.AP) for 30 minutes on ice. To precipitate the protein fractions from lysed cells, 100 µl of the cell lysate was incubated with ice-cold acetone (Sigma Aldrich, cat#179124) at −20°C overnight. Cellular proteins were pelleted by centrifugation at 4500 g for 15 minutes at 4°C, followed by reconstitution with 50 µl of 8 M urea (Sigma Aldrich, cat#51457) containing SIL peptides. Cellular proteins were further reduced by incubating the precipitated proteins with 10 mM TCEP (ThermoFisher Scientific, cat#77720) at room temperature for 30 minutes. Cysteine residues in the proteins were then alkylated by incubating with 55 mM 2-iodoacetamide (ThermoFisher Scientific, cat#A39271) for 30 minutes at room temperatures while protected from light. Digestion of the cellular proteins were performed by incubation with 1 µg of Lys C proteases (ThermoFisher Scientific, cat#90307) for 2 hours at room temperature followed by dilution of urea to 1 M by addition of 50 mM ammonia bicarbonate. Additionally, 2.5 µg of trypsin (ThermoFisher Scientific, cat#90059) was added, and digestion was allowed to proceed overnight at 37°C. Following overnight incubation, protein digests were acidified with 1% formic acid (ThermoFisher Scientific, cat#28905) and desalted on a 10 mg Waters HLB cartridge. Samples were then loaded on prepared 96-well SPE plate (Waters, cat#186000128). SPE plates were initially hydrated with 0.5 mL of 10% ethanol followed by equilibration twice with 0.5 mL of 0.1% formic acid solution. Samples were loaded onto the equilibrated SPE plates followed by wash with 0.5 mL of 0.1% formic acid. Bound samples were eluted into a collection plate by adding 0.1 mL of 0.1% formic acid with 70% acetonitrile solution. Samples were dried on collection plates followed by reconstitution in 0.1 mL of 0.1% formic acid. Reconstituted samples were analyzed using a nano-flow UPLC (ThermoFisher Scientific, Ultimate 3000) and a high resolution orbitrap mass spectrometer (ThermoFisher Scientific, Orbitrap Lumos). Samples were initially loaded onto a trapping column (ThermoFisher Scientific, cat# 164750) with a flowrate of 8 µl/minute of 3% mobile phase A for 4 minutes and then separated on a EasySpray C18 column (ThermoFisher Scientific, cat#ES900) with a flow rate of 350 nL/min, the gradient was 3% to 36% mobile phase B over 12 minutes. Easy-spray interface was used to couple the liquid chromatography with a mass spectrometer. The ion transfer tube temperature was set at 275°C and positive mode (+1.9kV) was applied to the EasySpray emitter. Parallel reaction monitoring (PRM) mode of the mass spectrometer was used to target both heavy and light peptides. Targeted ions were isolated with the quadruple with an isolation window of 1.6 m/z and Higher-energy collisional dissociation with a fixed collision energy of 28% to fragment the ions. Orbitrap with a resolution of 60K was used to detect the fragmented ions. Obtained data was analyzed in skyline software.Citation57
To quantify the cellular levels of ASGR1 following treatment with CAR and non-CAR antibodies, western blot methodology was employed. HepG2 cells plated in 6-well plates were treated with CAR and non-CAR antibodies at a concentration of 1, 10 and 100 µg/mL for either 24, 48 or 72 hours. Incubated cells were washed with PBS followed by treatment with RIPA lysis buffer (Millipore, cat# 20–188). Cell lysates were incubated on ice for 20 minutes followed by a 15 minute spin at 13,000 rpm at 4°C. Supernatants were separated from pellet and protein concentration was analyzed by BioRad protein assay reagents (Cat# 500–0113, 500–0114 and 500–0115). To detect the blotted ASGR1, anti-ASGR1 antibody (Sino Biologics Inc, Cat# 10773-R023) was used at a dilution of 1:2000 with an overnight incubation at 4°C followed by detection using secondary anti-rabbit IgG HRP with 1:2000 dilution at room temperature for one hour.
Ligand:antibody competitive binding assays
GalNAc was purchased from Fisher Scientific (cat #NC9024754), while asialofetuin was produced in-house at Amgen (PL-43379) and has been desialylated with neuraminidase (data not shown). Both ASGR1 ligands GalNAc and asialofetuin were biotinylated for the competitive binding assays. To determine the binding EC50 values of the ASGR1 ligands, CHO-S cells were incubated with the biotinylated ligands with a starting concentration of 300 nM and followed by a 12-point dilution series with 1:2 dilution of starting ligand concentrations with the buffer (10 mM Tris, pH 7.4, 137 nM NaCl, 1 mM CaCl2, 2% fetal bovine serum (FBS) and 0.01% NaN3). The assay was conducted in 384-well plates, with 30,000 cells per well. The cells were incubated with the ligands for 30 minutes on ice, followed by washing with excess of the buffer and centrifuging the plate at 1500 rpm for 5 minutes at 4°C. Ligands bound to ASGR1 were detected by staining with 30 µl of 1:10000 dilution of Alexa 647 labeled streptavidin (Jackson ImmunoResearch, cat# 016-600-084) on ice for 30 minutes. Following the wash step, cells were stained with 60 µl of a 1:1000 dilution of propidium iodide (Life Technologies, cat#P3566). Cells were analyzed on a flow cytometer and the observed median fluorescent intensity was plotted against incubated ligand concentration in Graphpad prism. To determine the binding EC50 values, the plot was fitted with a four-parameter curve. To analyze the ligand binding inhibition by anti-ASGR1 antibodies, cells were first incubated with anti-ASGR1 antibodies with a starting concentration of 300 nM followed by a 12-point dilution series with 1:2 dilution with the buffer on ice for 30 minutes. This was followed by incubation of the biotinylated ligands at their EC50 concentrations (2.7 nM for GalNAc and 53 nM for asialofetuin) on ice for 30 minutes. As described above, ligand bound to the receptor was detected by streptavidin-A647. Bound ligand fluorescent intensity was plotted against the competing antibody concentration and the plot was fitted with a four-parameter curve to determine the IC50 values for the anti-ASGR1 antibodies.
In vivo PK studies and isolation of hepatocytes from mice
Mice were housed in groups at an AAALAC, International accredited facility. Animals were cared for in accordance with the Guide for the care and Use of Laboratory Animals, 8th Edition. All research protocols (permit number − 2008–00092) were reviewed and approved by the Amgen Institutional Animal Care and Use Committee (Thousand Oaks, CA). Animals were housed in individual ventilated caging on irradiated corncob bedding (Envigo Taklad 7097). Animals had ad libitum access to irradiated pelleted feed (Enigo Teklad 2029X) and reverse-osmosis chlorinated (2 to 3 ppm) water via an automatic watering system. Lighting in animals holding rooms was maintained on 12:12 hour light:dark cycle, and the ambient temperature and humidity range was 68 to 79 ºF and 30 to 70%, respectively.
All PK experiments were conducted in 8–12-week-old male or female mice. Experiments were conducted in C57BL/6 mice harboring the human FcRn gene (The Jackson Laboratory, strain# 014565) and in-house generated ASGR1 knock-out C57BL/6 mice. There are n = 9 animals per group and antibodies were administered to mice intravenously via the lateral tail vein in their respective antibody buffer at a volume of 5 mL/kg (150 µL in each animal, respectively). Whole blood was collected at desired time points using SARSTEDT Microvette® serum separator tubes via submandibular vein puncture using a sparse serial sampling scheme (n = 3 mice/group/time point). Whole blood was allowed to clot at room temperature for approximately 20 minutes or until fully clotted prior to centrifugation at 11,500 rpm for 15 minutes and resulting serum was stored at −70°C until further analysis. Hepatocytes from livers of transgenic mice expressing human FcRn were isolated as described in published protocol.Citation58 Animals were sacrificed using CO2 asphyxiation followed by cervical dislocation.
Mouse PK analysis
Anti-ASGR1 antibodies serum concentrations were determined by ELISA using Meso Scale discovery platform. MSD plates (MSD, cat# L15SA–1) were coated with in-house generated biotinylated mouse anti-human Fc (clone Ab-35) for 1 hour at room temperature at a concentration of 1 μg/mL with Blocker Blotto in TBS (ThermoFisher, cat# 37530). Coated plates were washed with KPL wash solution containing 0.002 M imidazole buffered saline with 0.02% Tween-20 and blocked with Blocker Blotto in TBS. Standards and quality controls were prepared in naïve mouse serum. Serum samples, standards and quality controls were further diluted at 50× in Blocker Blotto in TBS, plated in duplicates onto the coated plates, then incubated for two hours at room temperature. Bound antibody was detected using sulfo-tagged Ab-35 at 1 μg/mL with Blocker Blotto in TBS. Plates were then washed with KPL wash solution and 150 µL of 1× MSD Read Buffer (MSD, cat# R92TC–1) added to each well. Plates were read using MSD Sector S 600 plate reader. The dose-response curve was constructed using standards ranging from 10,000–0.61 ng/mL, was fitted to a four-parameter logistic regression model. Sample concentrations were interpolated from the fitted curve while taking dilution factor into consideration in Watson Bioanalytical LIMS v.7.6.1 (ThermoFisher). The assay quantitation limit was 0.61 ng/mL. PK analysis of obtained antibody concentration was performed using Phoenix WinNonlin, v6.4.0.768 (Certara). Area under the serum concentration-time curve (AUC) from time zero to the time of last quantifiable concentration, was determined by noncompartmental analysis (NCA) by linear trapezoidal interpolation. For the radioactive-based PK study, antibodies were injected by tail vein. Immediately, the radioactivity of animals was measured in dose calibrator (Capintec, cat# CRC-15 R) for initial (T = 0) whole body activity. At designated times, the animal was read for whole body activity and then a blood sample was taken by a prick to the tail vein and collected in a capillary tube. All capillary tubes were weighed before collection and then after to determine exact blood weight/volume collected. Radioactivity of the serum samples were analyzed by gamma counter.
Immunofluorescence-based imaging studies
HepG2 cells (ATCC, cat# HB-8065) were cultured in 96-well plates in phenol red-free MEM (Gibco, cat# 51200038) supplemented with 2 mM L-glutamine and 10% FBS. AML12 cells (ATCC, cat# CRL-2254) were cultured in 96-well plates in DMEM:F12 medium (Gibco, cat# 11320033) supplemented with 10% FBS, 10 µg/mL insulin, 5.5 µg/mL transferrin, 5 ng/mL selenium, and 40 ng/mL dexamethasone.
For ASGR1 monoclonal antibody uptake studies, media was exchanged with serum-free MEM to avoid saturation of FcRn receptors with albumin. Cells were incubated with 10 µg/mL of the indicated ASGR1 monoclonal antibodies labeled with Alexa Fluor 568 or FcRn binding HSA labeled with Alexa Fluor 647 variants for 1 hour, then washed in PBS and fixed in 4% paraformaldehyde. Fixed cells were counterstained with 1 µM Hoescht 33,342 and 2 µg/mL CellMask Blue (Invitrogen, cat# H32720) for 30 minutes at room temperature. Cells were imaged using an Opera Phenix Plus High Content Screening System confocal microscope (PerkinElmer) using a 64× water emersion objective. Max projections and pixel intensity profile plots were generated from image Z-stacks using ImageJ.
For imaging endogenous ASGR1 subcellular localization, HepG2 cells were fixed with paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with 2% BSA, then incubated with an anti-ASGR1 antibody labeled with Alexa-647, anti-Rab11 (Rabbit polyclonal Ab; US Biological, cat# R0009), and anti-LAMP1 (mouse Ab; eBioscience; cat# eBioH4A3). After incubation with primary antibody, cells were washed and stained with secondary antibody for one hour at room temperature (goat anti-rabbit-Alexa-568; goat-anti-mouse-Alexa-488). Cells were counterstained with 1 µM Hoescht 33342, washed and imaged.
For dextran pulse chase experiments, AML12 cells were pulsed for 4 hours at 37°C with Dextran-Alexa Fluor 647 (Invitrogen, cat # D22914), washed twice in PBS and incubated overnight at 37°C. The next day, cells were pulsed with 10 µg/mL anti-ASGR1 antibody-Alexa Fluor 568 conjugate for 1 hour at 37°C, washed twice then chased for 4 hours at 37°C prior to fixation in paraformaldehyde. Fixed cells were counterstained with Hoescht 33,342 and CellMask Blue as described above. For ASGR1 antibody/FcRn colocalization studies in AML12 cells, cells were pulsed with 10 µg/mL Alexa Fluor 568-labeled engineered Fc bearing mutations (M252Y, S254T, T256E, H433K, N434F) and 10 µg/mL anti-ASGR1 antibodies for 30 minutes at 37°C. After pulse period, cells were washed, fixed in 4% paraformaldehyde, and counterstained with 1 µM Hoescht 33342.
Labelling of proteins with fluorescent and radioactive dyes
Fluorescently labeled proteins were generated using Alexa Fluor-NHS (Invitrogen, Alexa Fluor 647 cat# A37573; Alexa Fluor 568 cat # A20003) as follows: Proteins to be labeled were diluted in 0.5 M sodium borate buffer, pH 8.5 to a concentration of 1 mg/mL. Alexa Fluor-NHS dye was then added at a 3:1 molar ratio of dye to protein and incubated for 1 hour at room temperature. Unreacted dye was then removed from the solution by applying the solution to two subsequent Zeba desalting columns (Fisher cat # PI89882). Concentrations of labeled proteins were then determined using a NanoDrop spectrophotometer (Thermo Fisher). Antibodies were labeled with I-125 using Iodogen (Na125I, Perkin Elmer, cat #NEZ033L) in Iodogen tubes (Pierce, cat#28601) as per the described protocol. The labeling efficiency, as analyzed by thin layer chromatography, was greater than 85%. Radiolabeled antibody was purified using spin column (BioRad, cat#7326221).
Abbreviations
| ASGPR | = | Asialoglycoprotein receptor |
| AUC | = | Area under the curve |
| CAR | = | catch and release |
| FcRn | = | neonatal Fc receptor |
| IV | = | intravenous |
| IgG | = | immunoglobulin G |
| KD | = | dissociated rate constant |
| mAb | = | monoclonal antibody |
| PK | = | pharmacokinetics |
| TMDD | = | target-mediated drugs disposition |
Supplemental Material
Download Zip (12.8 MB)Acknowledgments
The authors thank Simon Jackson, Brooke Rock, and Isabel Figueroa for their scientific inputs during discussions. The authors acknowledge Sirisha Potala from Syngene Amgen Research and Development Center and Anielka Montalvan for their support during antibody production and registering protein lots. The authors recognize the support staff of Flow cytometry core and Imaging core facilities for their technical assistance.
Disclosure statement
The authors declare the following competing financial interest: All authors, except M.D are full time employees and shareholders of Amgen Inc. M.D is a shareholder of Amgen Inc.
Data availability statement
All relevant data and sequences of constructs used in this study are available on request to Siva Charan Devanaboyina.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2024.2383013
Additional information
Funding
References
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–16. doi:10.1038/256495a0.
- Carter PJ, Rajpal A. Designing antibodies as therapeutics. Cell. 2022;185(15):2789–805. doi:10.1016/j.cell.2022.05.029.
- Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. MAbs. 2023;15(1):2153410. doi:10.1080/19420862.2022.2153410.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–25. doi:10.1038/nri2155.
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacom & Syst Pharma. 2017;6(9):576–88. doi:10.1002/psp4.12224.
- Liu L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell. 2018;9(1):15–32. doi:10.1007/s13238-017-0408-4.
- Wu H, Pfarr DS, Losonsky GA, Kiener PA. Immunoprophylaxis of RSV infection: advancing from RSV-IGIV to palivizumab and motavizumab. Curr Top Microbiol Immunol. 2008;317:103–23.
- Heo YA. Satralizumab: first approval. Drugs. 2020;80(14):1477–82. doi:10.1007/s40265-020-01380-2.
- Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1–2):81–88. doi:10.1016/S1359-6446(05)03638-X.
- Ward ES, Ober RJ. Targeting FcRn to generate antibody-based therapeutics. Trends Pharmacol Sci. 2018;39(10):892–904. doi:10.1016/j.tips.2018.07.007.
- Ward ES, Devanaboyina SC, Ober RJ. Targeting FcRn for the modulation of antibody dynamics. Mol Immunol. 2015;67(2):131–41. doi:10.1016/j.molimm.2015.02.007.
- Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. 1997;15(7):637–40. doi:10.1038/nbt0797-637.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28(2):157–59. doi:10.1038/nbt.1601.
- Borrok MJ, Wu Y, Beyaz N, Yu X-Q, Oganesyan V, Dall’Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem. 2015;290(7):4282–90. doi:10.1074/jbc.M114.603712.
- Robbie GJ, Criste R, Dall’Acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57(12):6147–53. doi:10.1128/AAC.01285-13.
- Conner KP, Devanaboyina SC, Thomas VA, Rock DA. The biodistribution of therapeutic proteins: mechanism, implications for pharmacokinetics, and methods of evaluation. Pharmacol Ther. 2020;212:107574. doi:10.1016/j.pharmthera.2020.107574.
- Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol. 2010;28(11):1203–07. doi:10.1038/nbt.1691.
- Devanaboyina SC, Lynch SM, Ober RJ, Ram S, Kim D, Puig-Canto A, Breen S, Kasturirangan S, Fowler S, Peng L, et al. The effect of pH dependence of antibody-antigen interactions on subcellular trafficking dynamics. MAbs. 2013;5(6):851–59. doi:10.4161/mabs.26389.
- Hironiwa N, Ishii S, Kadono S, Iwayanagi Y, Mimoto F, Habu K, Igawa T, Hattori K. Calcium-dependent antigen binding as a novel modality for antibody recycling by endosomal antigen dissociation. MAbs. 2016;8(1):65–73. doi:10.1080/19420862.2015.1110660.
- Yang D, Giragossian C, Castellano S, Lasaro M, Xiao H, Saraf H, Hess Kenny C, Rybina I, Huang Z-F, Ahlberg J, et al. Maximizing in vivo target clearance by design of pH-dependent target binding antibodies with altered affinity to FcRn. MAbs. 2017;9(7):1105–17. doi:10.1080/19420862.2017.1359455.
- Engler FA, Polli JR, Li T, An B, Otteneder M, Qu J, Balthasar JP. “Catch-and-release” anti-carcinoembryonic antigen monoclonal antibody leads to greater plasma and tumor exposure in a mouse model of colorectal cancer. J Pharmacol Exp Ther. 2018;366(1):205–19. doi:10.1124/jpet.117.246900.
- Braun JR, Willnow TE, Ishibashi S, Ashwell G, Herz J. The major subunit of the asialoglycoprotein receptor is expressed on the hepatocellular surface in mice lacking the minor receptor subunit. J Biol Chem. 1996;271(35):21160–66. doi:10.1074/jbc.271.35.21160.
- Ashwell G, Morell AG. The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv Enzymol Relat Areas Mol Biol. 1974;41:99–128.
- Li Y, Huang G, Diakur J, Wiebe LI. Targeted delivery of macromolecular drugs: asialoglycoprotein receptor (ASGPR) expression by selected hepatoma cell lines used in antiviral drug development. Curr Drug Deliv. 2008;5(4):299–302. doi:10.2174/156720108785915069.
- D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting - strategies and applications. J Control Release. 2015;203:126–39. doi:10.1016/j.jconrel.2015.02.022.
- Nioi P, Sigurdsson A, Thorleifsson G, Helgason H, Agustsdottir AB, Norddahl GL, Helgadottir A, Magnusdottir A, Jonasdottir A, Gretarsdottir S, et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N Engl J Med. 2016;374(22):2131–41. doi:10.1056/NEJMoa1508419.
- Schwartz AL, Fridovich SE, Knowles BB, Lodish HF. Characterization of the asialoglycoprotein receptor in a continuous hepatoma line. J Biol Chem. 1981;256(17):8878–81. doi:10.1016/S0021-9258(19)52477-2.
- Bon C, Hofer T, Bousquet-Melou A, Davies MR, Krippendorff BF. Capacity limits of asialoglycoprotein receptor-mediated liver targeting. MAbs. 2017;9(8):1360–69. doi:10.1080/19420862.2017.1373924.
- Dall’acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal fc receptor: biological consequences. J Immunol. 2002;169(9):5171–80. doi:10.4049/jimmunol.169.9.5171.
- Kim JK, Firan M, Radu CG, Kim C-H, Ghetie V, Ward ES. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol. 1999;29(9):2819–25. doi:10.1002/(SICI)1521-4141(199909)29:09<2819:AID-IMMU2819>3.0.CO;2-6.
- Jacobsen FW, Stevenson R, Li C, Salimi-Moosavi H, Liu L, Wen J, Luo Q, Daris K, Buck L, Miller S, et al. Engineering an IgG scaffold lacking effector function with optimized developability. J Biol Chem. 2017;292(5):1865–75. doi:10.1074/jbc.M116.748525.
- Liu L, Jacobsen FW, Everds N, Zhuang Y, Yu YB, Li N, Clark D, Nguyen MP, Fort M, Narayanan P, et al. Biological characterization of a stable effector functionless (SEFL) monoclonal antibody scaffold in vitro. J Biol Chem. 2017;292(5):1876–83. doi:10.1074/jbc.M116.748707.
- Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan Y-Y, Dowty ME, Zhang M, Zhang Y, Cheng A, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs. 2016;8(6):1064–78. doi:10.1080/19420862.2016.1193660.
- Peletier LA, Gabrielsson J. Dynamics of target-mediated drug disposition: characteristic profiles and parameter identification. J Pharmacokinet Pharmacodyn. 2012;39(5):429–51. doi:10.1007/s10928-012-9260-6.
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog. 2008;21(1):11–16. doi:10.1021/bp040016j.
- Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci. 2015;104(6):1866–84. doi:10.1002/jps.24444.
- Onizuka T, Shimizu H, Moriwaki Y, Nakano T, Kanai S, Shimada I, Takahashi H. NMR study of ligand release from asialoglycoprotein receptor under solution conditions in early endosomes. The FEBS J. 2012;279(15):2645–56. doi:10.1111/j.1742-4658.2012.08643.x.
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23(10):1283–88. doi:10.1038/nbt1143.
- Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A, Bunting K, Antunes F, Williamson R, Athwal S, et al. Extending serum half-life of albumin by engineering neonatal fc receptor (FcRn) binding. J Biol Chem. 2014;289(19):13492–502. doi:10.1074/jbc.M114.549832.
- Lowe PJ, Tannenbaum S, Gautier A, Jimenez P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br J Clin Pharmacol. 2009;68(1):61–76. doi:10.1111/j.1365-2125.2009.03401.x.
- Chaparro-Riggers J, Liang H, DeVay RM, Bai L, Sutton JE, Chen W, Geng T, Lindquist K, Casas MG, Boustany LM, et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem. 2012;287(14):11090–97. doi:10.1074/jbc.M111.319764.
- Bonvin P, Venet S, Fontaine G, Ravn U, Gueneau F, Kosco-Vilbois M, Proudfoot AE, Fischer N. De novo isolation of antibodies with pH-dependent binding properties. MAbs. 2015;7(2):294–302. doi:10.1080/19420862.2015.1006993.
- Meier M, Bider MD, Malashkevich VN, Spiess M, Burkhard P. Crystal structure of the carbohydrate recognition domain of the H1 subunit of the asialoglycoprotein receptor. J Mol Biol. 2000;300(4):857–65. doi:10.1006/jmbi.2000.3853.
- Hoober JK. ASGR1 and Its enigmatic relative, CLEC10A. Int J Mol Sci. 2020;21(14):4818. doi:10.3390/ijms21144818.
- Igawa T, Maeda A, Haraya K, Tachibana T, Iwayanagi Y, Mimoto F, Higuchi Y, Ishii S, Tamba S, Hironiwa N, et al. Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PLOS ONE. 2013;8(5):e63236. doi:10.1371/journal.pone.0063236.
- Miki K, Kubota K, Inoue Y, Vera DR, Makuuchi M. Receptor measurements via Tc-GSA kinetic modeling are proportional to functional hepatocellular mass. Gastroenterol. 2001;120(5):733–37. doi:10.1016/S0016-5085(08)82723-0.
- Shi B, Abrams M, Sepp-Lorenzino L. Expression of asialoglycoprotein receptor 1 in human hepatocellular carcinoma. J Histochem Cytochem. 2013;61(12):901–09. doi:10.1369/0022155413503662.
- Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004;172(4):2021–29. doi:10.4049/jimmunol.172.4.2021.
- Pyzik M, Rath T, Kuo TT, Win S, Baker K, Hubbard JJ, Grenha R, Gandhi A, Krämer TD, Mezo AR, et al. Hepatic FcRn regulates albumin homeostasis and susceptibility to liver injury. Proc Natl Acad Sci USA. 2017;114(14):E2862–71. doi:10.1073/pnas.1618291114.
- Kang JC, Sun W, Khare P, Karimi M, Wang X, Shen Y, Ober RJ, Ward ES. Engineering a HER2-specific antibody–drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat Biotechnol. 2019;37(5):523–26. doi:10.1038/s41587-019-0073-7.
- Polli JR, Balthasar JP. Cell penetrating peptides conjugated to Anti-Carcinoembryonic Antigen “catch-and-release” monoclonal antibodies alter plasma and tissue pharmacokinetics in colorectal cancer xenograft mice. Bioconjug Chem. 2022;33(8):1456–66. doi:10.1021/acs.bioconjchem.2c00152.
- Ahn G, Banik SM, Bertozzi CR. Degradation from the outside in: targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem Biol. 2021;28(7):1072–80. doi:10.1016/j.chembiol.2021.02.024.
- Engler C, Kandzia R, Marillonnet S, El-Shemy HA. A one pot, one step, precision cloning method with high throughput capability. PLOS ONE. 2008;3(11):e3647. doi:10.1371/journal.pone.0003647.
- Li D, Partin AC, Zhao L, Chen I, Michaels ML, Wang Z, Garces F, Gong D, Riley TP. Protocol for high-throughput cloning, expression, purification, and evaluation of bispecific antibodies. Star Protoc. 2022;3(2):101428. doi:10.1016/j.xpro.2022.101428.
- Qiao SW, Kobayashi K, Johansen F-E, Sollid LM, Andersen JT, Milford E, Roopenian DC, Lencer WI, Blumberg RS. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci USA. 2008;105(27):9337–42. doi:10.1073/pnas.0801717105.
- Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochem. 2013;52(22):3797–806. doi:10.1021/bi400110b.
- Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, MacCoss MJ. The skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev. 2020;39(3):229–44. doi:10.1002/mas.21540.
- Charni-Natan M, Goldstein I. Protocol for primary mouse hepatocyte isolation. Star Protoc. 2020;1(2):100086. doi:10.1016/j.xpro.2020.100086.