
ABSTRACT
Chemotactic/chemotropic cells follow accurately the direction of gradients of regulatory molecules. Many G-protein-coupled receptors (GPCR) function as chemoattractant receptors to guide polarized responses. In “a” mating type yeast, the GPCR Ste2 senses the α-cell’s pheromone. Previously, phosphorylation and trafficking of this receptor have been implicated in the process of gradient sensing, where cells dynamically correct growth. Correction is often necessary since yeast have intrinsic polarity sites that interfere with a correct initial gradient decoding. We have recently showed that when actively dividing (not in G1) yeast are exposed to a uniform pheromone concentration, they initiate a pheromone-induced polarization next to the mother–daughter cytokinesis site. Then, they reorient their growth to the intrinsic polarity site. Here, to study if Ste2 phosphorylation and internalization are involved in this process, we generated receptor variants combining three types of mutated signals for the first time: phosphorylation, ubiquitylation and the NPFX1,2D Sla1-binding motif. We first characterized their effect on endocytosis and found that these processes regulate internalization in a more complex manner than previously shown. Interestingly, we showed that receptor phosphorylation can drive internalization independently of ubiquitylation and the NPFX1,2D motif. When tested in our assays, cells expressing either phosphorylation or endocytosis-deficient receptors were able to switch away from the cytokinesis site to find the intrinsic polarity site as efficiently as their WT counterparts. Thus, we conclude that these processes are not necessary for the reorientation of polarization.
Introduction
Cells integrate environmental and internal information to produce robust responses. In the case of chemotaxis/chemotropism, cells use external cues to decide where to polarize. However, most cells also present intrinsic polarization cues. These external and internal cues might oppose each other or synergize through mechanisms that are is still under study. We recently showed in the Cdc42-signaling system of the budding yeast S. cerevisiae, that intrinsic polarization cues actually interfere with mating pheromone gradient decoding, as yeast genetically modified to lack all known internal landmarks track external gradients better [1].
Haploid yeast exists in two mating types, MATa and MATα, which communicate with each other to trigger mating behavior through the secretion of short peptide pheromones (a- and α-factor, respectively). These pheromones bind G protein-coupled receptors on the opposite mating type (Ste2 in MATa and Ste3 in MATα) that stimulate the dissociation of a trimeric G-protein into Gα and Gβγ [Citation2]. Free Gβγ (Ste4+Ste18) recruits the scaffold protein Ste5 and the adaptor protein Far1 to the plasma membrane. Ste5 activates a MAPK-dependent signaling that induces mating-related genes, stimulates polarization and arrests the cell cycle in G1 phase [Citation3,Citation4]. Far1 links activated pheromone receptors to the Cdc42-polarization module [Citation5,Citation6] by associating with Cdc24, a Cdc42 guanine nucleotide exchange factor (GEF), which locally increases the concentration of Cdc42-GTP. Cdc42-GTP further activates itself by positive feedback mechanisms, resulting in the formation of a cortical polarity “patch” [Citation7]. At this location, Cdc42-GTP recruits the formins Bni1 and Bnr1, which nucleate linear actin filaments. Bni1 is part of the polarisome, a multiprotein complex organized by the Spa2 and Pea2 proteins, which acts as the focal point for polymerization of actin monomers into actin cables [Citation8,Citation9]. Finally, transport of membrane vesicles along these cables allows polarized cell growth in the direction of the pheromone gradient, causing the formation of a mating projection (MP).
When cells are exposed to a uniform pheromone concentration (i.e., not in a gradient), yeast cells do not polarize their growth randomly but use “default” sites marked by intrinsic cues: specific cortical landmarks, called budding cues, that define the position of bud emergence in mitotically growing cells. These mitotic landmarks remain active during mating and also locally activate Cdc42. In the W303 genetic background, the default site under α-factor stimulation is defined by Rax1/Rax2- and Rsr1-dependent complexes in the “distal” pole, the region of the membrane opposite of the site of the cytokinesis event that gave birth to the cell [Citation1] ().
Figure 1. (a) Model that depicts the distinctive behavior of cycling or G1 daughter cells after uniform pheromone exposure (high or low concentration). Light blue points represent the polarity-patch proteins. In cycling daughters, the cytokinesis-related patch is used for MP formation. Polarization within the birth scar is inhibited. At high α-factor, due to low polar cap mobility cells make proximal MPs. At low α-factor, increased cap mobility permits the patch to detach from the neck and to reach, moving through the membrane, the default sites (distal). G1 daughters make an MP by de novo polarization independently of α-factor concentration. Distal polarization is guided by Rsr1 and Rax1. (b) Diagram of Ste2 protein. Ubiquitylatable lysines (K) are shown in green and phosphorylatable serines and threonines (ST) are in yellow. The NPFX1,2D signal recognized by Sla1 endocytic adaptor correspond to the sequence GPFAD and is depicted in light blue. The S331INDAKKS339 sequence originally proposed as the internalization signal is also shown. In the box below, we detailed the mutations performed in this study starting at position 331 (black arrow and dashed line).
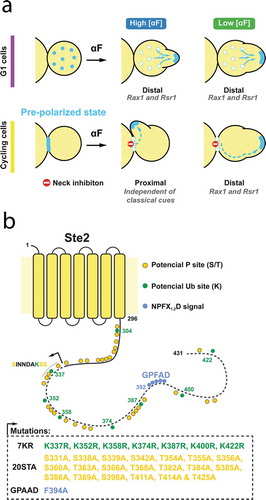
Our recent observations revealed that the mechanism of polarization in response to pheromone depends on cell-cycle position. In response to uniform external pheromone, cells in the G1 phase assemble de novo polarization patches and form their MPs directly at the site of the dominant distal internal landmark [Citation1] (). In contrast, if cells are in the middle of a cell division (i.e., out of G1) at the time of stimulation, they first finish mitosis and then utilize the already assembled cytokinesis-related polarity patch at the bud-neck. The use of this patch depends on the recruitment of Far1-Cdc24 complex by Gβγ, which is normally concentrated around the neck during cytokinesis. The future behavior of this initial patch depends on α-factor concentration. If the concentration of α-factor is high, the MP emerges next to the cytokinesis site, forming a proximal projection (i.e., proximal to the division site). However, if the concentration of α-factor is low (at or below the Kd between α-factor and its receptor), this initial patch can move away from the bud-neck scar to form an MP at the location of the dominant internal landmark.
Previously, we presented evidence supporting two means that help yeast “escape” the initial cytokinesis-related patch in low α-factor conditions [Citation1]: (a) the patch is highly mobile at low α-factor [Citation10] and (b) tethering of the pheromone response machinery to the cytokinesis-related patch is weaker, because at low α-factor the fraction of dissociated Gβγ dimers is low, and thus fewer Gβ-Far1-Cdc24 tethering complexes can form. Artificially increasing patch mobility by destabilizing the polarisome (using a Δspa2 strain) enables escape at high α-factor, while artificially strengthening Cdc42 activity at the neck (using a strain with a deletion in Rga1, a GTPase activating protein for Cdc42-GTP located at the cytokinesis site) blocks escape at low α-factor. As in the case of uniform low pheromone concentrations, cycling cells in a mating situation are able to move their polarization patch away from the initial cytokinesis-related location to track the direction of the α-factor gradient [Citation1]. Thus, the mechanisms that allow the polarity patch to escape the cytokinesis-related patch are also important for gradient sensing.
Previous evidence suggested that phosphorylation and endocytosis of the pheromone receptor Ste2 are needed to switch from an initial polarization at the neck to the chemotropic site in cells exposed to natural α-factor gradients [Citation11,Citation12]. Based on this observation, here we tested if Ste2 phosphorylation and/or endocytosis are important for the proximal to distal re-localization of the polarity patch under uniform low α-factor concentrations, as a proxy for studying this underlying mechanism in gradient sensing.
Both the activity and trafficking of the GPCR Ste2 are regulated by post-translational modifications (PTMs) at its intracellular C-terminal domain (CTD). Serine/threonine hyperphosphorylation by Yck1/2 kinases is proposed to be a prerequisite for mono-ubiquitylation [Citation13] and to regulate binding of regulatory proteins, such as Sst2, the Gα GTPase Activating Protein [Citation14]. CTD ubiquitylation, performed by the Rsp5 E3 ligase with the help from α-arrestins, participates in the recognition of Ste2 by the cargo receptors that mediate endocytosis through clathrin-coated pits, and in the subsequent trafficking to the lumen of the vacuole, where Ste2 is degraded [Citation15–Citation20]. A redundant mechanism for Ste2 endocytosis relies on the Sla1 adaptor protein that recognizes the NPFX1,2D signal in membrane proteins, which in Ste2 is GPFAD [Citation21,Citation22].
Ste2 CTD has 33 phosphorylatable S/Ts which may have other roles besides being a hallmark for ubiquitylation. By mutating these residues to alanine in six groups (CT-1 to CT-6), Kim et al. uncovered different roles for CTD phosphorylation [Citation23]. S/Ts in the first two groups (CT-1, −2, covering the 14 amino-terminal residues) are critical for signaling but dispensable for internalization. Residues from each of the central groups CT-3, −4 and −5 have impact in constitutive internalization only. However, when mutated together (CT-345), pheromone-induced internalization is greatly impaired. The distal group, CT-6, is not clearly involved in endocytosis but may regulate signaling through interactions with residues in the proximal groups (CT-12). Interestingly, mutation of all 33 phosphorylatable residues increases the sensitivity to pheromone greatly.
In this work, we showed that Ste2 phosphorylation (of central and distal sites, CT-3456) or endocytosis is not required for the polarity patch to move away from the cytokinesis-related polarization in cycling cells at low α-factor concentrations, suggesting that receptor migration through the plasma membrane is sufficient.
Materials and methods
Strains
Yeast strains used in this study are listed in . Standard yeast molecular and genetic procedures were used to generate the strains. Strains were generated from ACL379 which is a derivative of W303-1A [Citation24].
Table 1. Strains used in this study
STE2 alleles were introduced at the endogenous locus by integrating STE2WT-CFP-pRS406, STE27KR-CFP-pRS406, STE220STA-CFP-pRS406, STE220STA−7KR-CFP-pRS406, STE27KR&GPAAD-CFP-pRS406 or STE220STA−7KR-GPAAD-CFP-pRS406 plasmids. In all cases, plasmids were linearized by ClaI digestion. STE2 endogenous promoter was replaced by GAL1 promoter by one-step PCR-mediated method using plasmid pFA6a-kanMX6-PGAL1 as template [Citation25].
Plasmids
Plasmid pBB13 was constructed by cloning a DNA fragment amplified by PCR from yeast genomic DNA containing the 403–916 nt region of the STE2 ORF and flanked with KpnI and RsrII sites, respectively, into a pRS406 plasmid containing CFP and the ADH1 terminator (tADH1). This resulted in the construct KpnI-STE2403 − 916-RsrII-CFP-tADH1.
STE27KR-CFP-pRS406, STE220STA-CFP-pRS406 plasmid we constructed by Gibson assembly as previously reported for pRS406-STE2WT and pRS406-STE220STA−7KR [Citation26]. Briefly, synthetic DNA fragments were recombined into plasmid pBB13 (STE2403−916-RsrII-CFP-tADH1) linearized by RsrII digestion. These fragments contain the sequence coding for different variants of the C-terminal domain of Ste2p, flanked by 40 bp regions homologous to the plasmid DNA surrounding the RsrII site. 7KR refers to mutations K337R, K352R, K358R, K374R, K387R, K400R and K422R. 20STA refers to mutations S331A, S338A, S339A, S342A, T354A, T355A, S356A, S360A, T363A, S366A, T368A, T382A, T384A, S385A, S386A, T389A, S398A, T411A, T414A and T425A. A 19STA mutant with all but S342A was described in Ballon et al. [Citation14]. All mutations were confirmed by DNA sequencing.
STE27KR-GPAAD-CFP-pRS406 or STE220STA−7KR-GPAAD-CFP-pRS406 plasmids were constructed based on STE27KR-CFP-pRS406 and STE220STA−7KR-CFP-pRS406 plasmids by site-directed mutagenesis. In both cases, the GPFAD endocytosis signal was mutated to GPAAD to block the Sla1-dependent endocytosis.
Microscopy methods
Cultures in exponential growth were sonicated and diluted to a concentration of 2.5 × 105 cells/mL. Polyethylene glycol (avg MW = 3550, Sigma cat #P4338) was added to all media at 0.1% to avoid nonspecific binding of α-factor [Citation27]. Then, 20 μL of cell suspension was applied to individual wells of 384-well glass-bottom plates pre-coated with 1 mg/mL concanavalin A (Con A, Sigma cat #C7275). Plates were centrifuged to assist the attachment of cells. In the microscope, two to three image fields per well were selected, and the time-lapse imaging was started. After the first time point, 20 μL of α-factor (Anaspec, custom made) was added to final concentrations of 0–1000 nM, depending on the experiment.
For imaging, a fully motorized Olympus IX-81 microscope with an Olympus UplanSapo objective (63×; N.A. = 1.35), coupled with an HQ2 (Roper Scientific) cooled CCD camera was used, with filter sets for YFP and CFP (41,028, 31044v2 and 41,004, Chroma Technologies).
CFP fluorescence in individual cells was analyzed by two approaches. First, fluorescence profiles were obtained for individual cells by measuring CFP signal in 4-pixel-wide line-scans over the vacuole. After background subtraction, fluorescence was normalized relative to the total fluorescence from each cell. Second, we manually segmented the cells and measured both the total and internal fluorescence (internal corresponds to total fluorescence excluding the two most external pixels). Then, we defined the ‘internalization index’ as the ratio between internal and total CFP signal. Due to cellular autofluorescence, out-of-focus light and the different internal localization of CFP in the strains analyzed (vacuolar lumen vs internal membranes), these measures are only semi-quantitative. As an internal control, we performed the same procedure on two strains: one expressing cytosolic CFP (TCY3154) and one bearing a membrane-targeted version of CFP (a fusion with the carboxy-terminal transmembrane domain (CTM) of yeast Snc2 v-SNARE [Citation4]).
Angle determination
Angles were manually measured using custom-written macros for ImageJ on brightfield time-lapse image stacks. Daughter cells were classified G1 or cycling cells relative to the α-factor stimulation. We only measure cycling cells which comprised daughter cells that were buds at the time of pheromone exposure. Cells that rotated or moved led to inexact determination of the proximal pole or the MP, and thus were excluded from the analysis.
Dose-response curves
Cells expressing the PPRM1-YFP reporter were grown in synthetic media until mid-exponential phase. Then, the cultures were mildly sonicated, diluted to 6 × 105 cells/mL and treated with different α-factor concentrations in 384-well glass-bottom plates (Brooks) for 2 hours at 30°C. All media was supplemented with 0.1% w/v PEG (MW3550, Sigma) to block pheromone unspecific binding to plastic material. Finally, cycloheximide was added to a final concentration of 100 μg/ml to inhibit translation and incubated at 30°C for at least 2 more hours to allow complete maturation of YFP. Fluorescence microscopy-based cytometry was done as described elsewhere [Citation28].
Fluorescent α-factor uptake
Cells were grown on synthetic media supplemented with 2% glucose or 2% galactose/1% raffinose and prepared for imaging in 384-well glass-bottom plates as described. Once in the microscope, cells were stimulated with 50 nM fluorescently labeled α-factor [Citation29] and followed over time. Two hours later, since fluorescent α-factor has low biological activity, unlabeled pheromone was added at a final concentration of 50 nM to further stimulate Ste2 internalization.
Data analysis
All data analysis was performed in R [Citation28].
Results
To determine if Ste2 phosphorylation and/or endocytosis are required for polarity-patch migration away from the initial cytokinesis-related polarization site, we created strains expressing combinations of three Ste2 CTD mutants ( and ) as CFP fusions. We used CFP due to its relatively slow maturation kinetics (approximately 50 minutes [Citation30]) to minimize the probability that intracellular CFP fluorescence instead of being a result of internalization is due to new receptors in transit to the plasma membrane. The first is the phosphorylation mutant Ste220STA. Of the 33 serines/threonines in the CTD, the 13 amino-terminal ones are required for GPCR signaling [Citation23]. Thus, we constructed a Ste2 version in which alanines replace the remaining 20 serine or threonine residues (from S331 to T425), which are involved in Ste2 trafficking [Citation14,Citation26] (corresponding to CT-3456 in Kim et al. nomenclature [Citation23]). The second is the ubiquitylation mutant Ste27KR, which has arginines replacing all potentially ubiquitylated lysine residues [Citation21,Citation26,Citation31]. The third is Ste2GPAAD, which has the GPFAD Sla1-dependent endocytosis signal mutated and it, therefore, cannot use this ubiquitin-independent pathway for internalization [Citation16,Citation21]. In what follows, we tested Ste2WT, Ste27KR, Ste220STA, Ste220STA−7KR, Ste27KR-GPAAD and Ste220STA−7KR-GPAAD.
Figure 2. (a) Fluorescence and transmission microscopy images of yeast expressing STE2 alleles C-terminally tagged with CFP exposed to 0, 5 or 1000 nM α-factor for two hours: Ste2WT-CFP (YGV5560), Ste27KR-CFP (YGV5562), Ste220STA-CFP (YGV5561), Ste220STA−7KR-CFP (YGV5563), Ste27KR-GPAAD-CFP (YGV5842), Ste220STA−7KR-GPAAD-CFP (YGV5843). Arrowheads indicate fluorescence at the rim of the vacuole. The scale bar represents 5 μm. (b–c) Semi-quantification of CFP fluorescence in cells in (a) exposed to 0 or 1000 nM α-factor. (b) For each cell, a 4-pixel-wide line-scan was measured over the vacuole (see yellow-bar example on Ste220STA cells in (a). Normalized fluorescence is plotted as a function of distance (n = 30 cell per strain and condition). In yeast with WT Ste2 the majority of the signal is intracellular. In the mutants with reduced endocytosis two peaks of fluorescence appear coinciding with the location of the plasma membrane (shaded regions). (c) The ‘internalization index’ estimated by manual segmentation (see yellow contours example on Ste220STA−7KR cells in (a), and Materials and Methods for details). To calibrate this measure, we included yeast expressing either cytosolic (cyt) CFP (TCY3154) and membrane (mem) CFP (fused to the transmembrane domain of Scn2 v-SNARE, TCY3050). The differences between Ste27KR-GPAAD-CFP and Ste220STA−7KR-GPAAD-CFP localization under 1000 nM pheromone stimulation observed in (a) is not captured by this measure but it is better detected in (b).
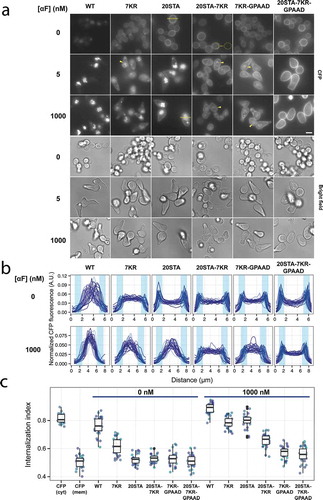
To determine which mutations were more suitable for the patch migration experiments, we assessed Ste2 localization by fluorescence microscopy in unstimulated cells or after 2 hours of a treatment with a low (5 nM) or high (1000 nM) α-factor (). To help us compare Ste2 localization in the different strains, we performed two semi-quantitative analyses of CFP fluorescence at the single-cell level: a spatial signal profile (in a cross section of the cells, ) and an internalization index (). The first approach was more sensitive, allowing us to detect subtler changes in endocytosis between strains, while the second permitted an easier comparison between strains and conditions. Together, these two measures showed that within each strain, there was a little cell to cell variability in Ste2 localization.
Under unstimulated, basal conditions ( and , top rows, and , left), the CFP signal from WT Ste2 was mostly located in the vacuolar lumen. Blocking ubiquitylation (Ste27KR) diminished basal endocytosis, as the CFP signal increased at the plasma membrane. Also, the internalized fluorescence appeared membrane-associated, not luminal (see below). In contrast, when both the ubiquitin and the Sla1 internalization mechanisms were blocked (Ste27KR-GPAAD), the receptor completely failed to be endocytosed, indicating that there is no other internalization pathway during mitotic growth. As previously noted, basal internalization was also completely blocked in the phosphorylation mutant Ste220STA, suggesting that, in the absence of α-factor, this modification is an essential pre-requisite to recruit both endocytosis mechanisms to the receptor [Citation23].
In the presence of α-factor, neither phosphorylation nor ubiquitylation was essential for endocytosis, since even the double mutant Ste220STA−7KR showed intracellular CFP fluorescence (). This suggested that the remaining endocytosis capacity was provided by the Sla1 pathway. In support of this idea, the triple mutant Ste220STA−7KR-GPAAD showed minimal signs of endocytosis. In addition, these results showed that when Ste2 is bound to α-factor, phosphorylation is not indispensable to become a substrate of the Sla1-dependent machinery.
Interestingly, the intracellular CFP fluorescence in all 7KR containing Ste2 mutants we tested was not located in the vacuolar lumen but exhibited a signal that appeared membrane bound. This showed that ubiquitylation was essential for Ste2 entry into the vacuolar lumen. Taking this into account, the fact that the CFP from the internalized Ste220STA did appear in the vacuolar lumen indicated that this receptor was endocytosed via the ubiquitin pathway. Consistent with this hypothesis, Ste20STA−7KR showed less internalization than Ste220STA. This led us to conclude that phosphorylation of these 20 sites is not essential for the ubiquitylation of the α-factor-bound Ste2.
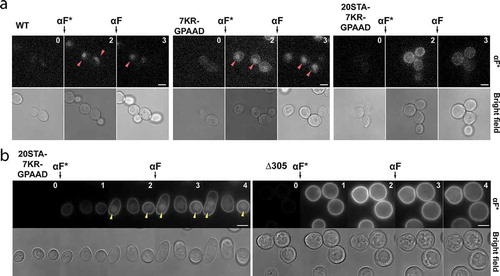
To verify that the low CFP intracellular signal observed in Ste27KR-GPAAD and Ste220STA−7KR-GPAAD was in fact due to endocytosis, we observed the internalization of fluorescent α-factor (). Indeed, Ste27KR-GPAAD mutant cells displayed accumulation of intracellular labeled pheromone released into the vacuole. This activity was further reduced by blocking receptor phosphorylation, since triple mutant Ste220STA−7KR-GPAAD did not show evident fluorescent α-factor internalization. To fully test the endocytic capacity of this Ste2 triple mutant, we changed its endogenous promoter to the galactose-inducible GAL1 promoter and overexpressed it before challenging the cells to labeled α-factor. In this condition, we observed some vacuolar accumulation of the fluorescent ligand ( left). Since no accumulation took place in cells expressing a control Ste2 mutant that lacks the CTD altogether (Ste2-Δ305) ( right), we conclude that the CTD from Ste220STA−7KR-GPAAD still serves as a weak signal for internalization.
Figure 3. (a) Internalization of fluorescently labeled α-factor (αF*) was analyzed by fluorescence microscopy. Ste2 WT cells (ACL379), Ste27KR-GPAAD-CFP (YGV5842) and Ste220STA−7KR-GPAAD-CFP (YGV5843) cells were challenged with 50 nM αF*. After two hours, unlabeled αF was added to further stimulate receptor internalization. Arrowheads indicate accumulation of αF* in the vacuole. Numbers correspond to time in hours. (b) Cells expressing STE2 alleles under the GAL1 promoter (PGAL1-STE220STA−7KR-GPAAD-CFP and PGAL1-STE2 Δ305 -CFP) were grown in galactose/raffinose-supplemented media and treated as in (a).
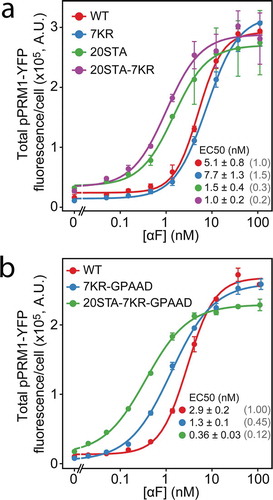
Since Ste2 phosphorylation is known to affect α-factor sensitivity [Citation23,Citation32], we then measured the ability of our mutant receptors to activate an α-factor inducible reporter, PPRM1-YFP [Citation24] (). Strains with the Ste220STA mutation showed half-maximal responses (EC50) 3 to 8 times lower than their phosphorylation-competent counterparts, confirming that receptor phosphorylation was an important determinant of the α-factor response sensitivity. In contrast, cells with Ste27KR had a sensitivity comparable to that of Ste2WT cells. Finally, the GPAAD mutation also caused a small, two-fold increase in α-factor sensitivity, as revealed by comparing the effect of Ste2 mutants with or without the GPAAD mutation relative to Ste2WT (Ste27KR vs. Ste27KR-GPAAD and Ste220STA−7KR vs. Ste220STA−7KR-GPAAD).
Figure 4. Sensitivity to α-factor of strains measured using the transcriptional reporter PPRM1-YFP. Curve corresponds to a fitted Hill-function model to the data (3 independent experiments), from where EC50 ± SE were estimated. EC50 values normalized relative to the STEWT are shown between parenthesis. (a) Ste2WT-CFP (YGV5560), Ste27KR-CFP (YGV5562), Ste220STA-CFP (YGV5561), Ste220STA−7KR-CFP (YGV5563). (b) Ste2WT-CFP (YGV5560), Ste27KR-GPAAD-CFP (YPD6223), Ste220STA−7KR-GPAAD-CFP (YPD6224).
Based on the above results, to test the effect of phosphorylation and endocytosis on the mobility of the polarization patch, we selected two mutants: Ste27KR-GPAAD-CFP and Ste220STA−7KR-GPAAD-CFP, both with minimal endocytic capacity, but the first phosphorylation-competent and the second phosphorylation-defective. We stimulated with high (1000 nM) or low (between 1 and 5 nM) α-factor and monitored MP formation over time. We then selected cycling cells (budding at the time of stimulation) and measured the angles formed between the bud-neck and the MPs (see and Methods).
Figure 5. Distribution of MP angles relative to the proximal pole in cycling cells of strains Ste2WT-CFP (YGV5561), Ste27KR-GPAAD-CFP (YGV5842) and Ste220STA−7KR-GPAAD-CFP (YGV5843) stimulated with the indicated αF concentrations. In all cases, the angle distribution was divided in 30 degrees bins and the fraction of cells in each category was calculated. Data from at least 3 independent experiments (points) was pooled to calculate the mean ± SEM (bars and whiskers). At 1 and 2 nM αF, Ste2WT -CFP Ste27KR-GPAAD-CFP cells fail to form MPs.
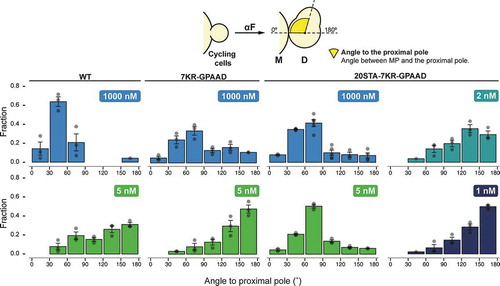
At high pheromone, strains with Ste2WT formed proximal MPs (i.e., low-value angles, around 30°). (Note that there is a no-polarization zone directly at the bud-neck [Citation1], preventing the formation of MPs with lower angles.) Strains with either of the two mutant Ste2 versions also made proximal MPs, though with somewhat higher angles, probably due to the failure of these receptors to polarize properly [Citation33].
At 5 nM α-factor, strains with Ste2WT or Ste27KR-GPAAD tended to make MPs close to the distal pole (angles close to 180°). In contrast, Ste220STA−7KR-GPAAD expressing cells were unable to polarize away from the neck at 5 nM pheromone, suggesting that phosphorylation could be necessary to switch from proximal to distal polarized growth. However, as we showed above, cells expressing Ste220STA−7KR-GPAAD have a significantly higher sensitivity to α-factor (). Therefore, 5 nM might not be a low enough concentration for this mutant strain to move the patch to the distal pole. Thus, we tested lower pheromone concentrations. Indeed, at 2 and 1 nM pheromone, Ste220STA−7KR-GPAAD mutant cells could migrate the patch from the neck and were able to reach the distal pole to form their MPs. Thus, these results indicate that neither receptor endocytosis nor phosphorylation is required for the relocation of polarized growth from the proximal to the distal pole in cycling cells exposed to low concentrations of α-factor.
Discussion
Here we studied the requirement of Ste2 endocytosis and phosphorylation in the proximal to distal relocation of the polarity patch in yeast exposed to uniform pheromone, a process that likely shares underlying mechanisms with gradient sensing. Previous work suggested that endocytosis is necessary for the re-orientation of the patch after an initial misaligned gradient detection [Citation34]. Similarly, others postulated that phosphorylation of Ste2 (in an endocytosis-deficient receptor) was necessary for the re-orientation from the proximal site to a chemotropic growth site in mating mixtures [Citation11,Citation12]. In contrast, in our experimental setup, we found no evidence supporting the role of receptor trafficking or phosphorylation of the C-terminal 20 S/Ts in the process that allows the polarity patch to escape the initial cytokinesis-related polarization site and form MPs in the distal region of the cell. A potential explanation to reconcile these two results is that the version of the receptor they used, Ste2 6SA −7KR-GPAAD (with 6 phosphorylatable serines replaced with alanine) [Citation35], would also confer super-sensitivity to pheromone, just as our Ste220STA−7KR-GPAAD does. In that case, the concentration of pheromone achieved in the crowded mating mixes they used could well be too high for the patches containing Ste2 6SA −7KR-GPAAD to escape the cytokinesis-relate polarization to track the gradient. In this view, their data cannot answer the question of whether receptor trafficking or phosphorylation is necessary to track gradients. A second explanation for the differing results is that our experimental setup (uniform pheromone) would be unable to capture a critical aspect of gradient tracking that makes receptor phosphorylation and trafficking essential. In either case, more experiments would be needed to conclusively attribute the roles of receptor trafficking and phosphorylation in this process.
Other work also studied the re-localization of the polarity patch from the neck to the chemotropic site in cells that were still dividing at the time of exposure to pheromone (which we called cycling cells) [Citation36]. They proposed that signaling controls polarity-patch mobility: lower pheromone concentrations would limit Cdc42 signaling, resulting in an unstable polar cap that permits dynamic axis adjustment toward the gradient. Similarly, another group proposed that low α-factor levels allow polarity patch wandering [Citation10,Citation37]. In this scenario, gradient decoding results from a stochastic search by the polarity patch whose mobility would decrease when the pheromone concentration rises as the polarity-patch approaches the source of α-factor, i.e., the mating partner. Our results certainly support the hypothesis that the strength of signaling positively correlates with lower patch mobility, since the supersensitive Ste2 triple mutant could only wander away from the cytokinesis site at a lower pheromone concentration than WT. Thus, relocation of polarized growth to the distal pole may rely on a high mobility of the polarity patch. If that is the case, receptor polarization does not seem to play a role in guiding this process, since Ste2 crescents cannot form with endocytosis-deficient receptors but cells expressing these receptors still switch to the distal pole. This agrees with the finding that G-proteins can polarize independently of receptor polarization to restrain polarity patch wandering [Citation10].
The role of Ste2 internalization in the pheromone response has been extensively studied in the past by different authors, who used Ste2 point mutants and truncations. However, no single study combined all known modifications that affect Ste2 endocytosis in the context of a full-length protein, and therefore, no side by side evaluation in uniform conditions has been done. Here, we showed that ubiquitylation contributes to but is not essential for basal internalization. This was surprising, since a previous study showed that a truncated version of Ste2 (a STOP codon after amino acid 345) that could not be ubiquitylated, Ste2-K337R-345Stop, could not be endocytosed [Citation13]. However, results obtained with that mutant might be misleading, since it lacks the last 87 residues, where 15 serines/threonines available for phosphorylation, as well as the Sla1-binding signal (GPFAD), are located. In that sense, it is more similar to our triple mutant Ste220STA−7KR-GPAAD than to Ste27KR.
We also confirmed the participation of ubiquitylation in α-factor-induced endocytosis [Citation31] and we showed that it is required for receptor translocation from the vacuolar membrane into the vacuolar lumen for degradation, consistent with the proposed role of ubiquitin in sorting cargos in the endocytic transport [Citation20]. However, we demonstrate that the ubiquitin-dependent pathway works redundantly with the NPFX1,2D-dependent pathway in basal endocytosis, similar to what is known to happen in pheromone-stimulated endocytosis [Citation21]. In addition, our results indicate that phosphorylation is a pre-requisite for both ubiquitylation- and NPFX1,2D-dependent basal endocytosis, consistent with previous evidence [Citation23]. Given the great number of potential phospho-sites in the Ste2 CTD, the participation of this modification in ligand-induced internalization remained elusive. Original data suggested that three serines from the S331INDAKSS339 sequence were required for internalization of the 345 truncated Ste2 [Citation17]. However, mutation of SINDAKSS’ serines together with three extra neighboring serines in the full-length receptor slowed internalization dynamics but did not block it completely [Citation13,Citation35]. Further studies [Citation23] showed that elimination of all phospho-sites has a similar effect to mutating 15 sites (from S338 through T389) of our 20STA mutant, blocking up to 70% of Ste2 internalization. Even with our 20STA mutant, there is still ligand-induced internalization. We showed that the remaining endocytosis relies both on ubiquitylation- and Sla1-dependent mechanisms, since minimal intracellular receptor was seen in Ste220STA−7KR-GPAAD. This also implies that phosphorylation also explains part of the residual internalization observed in Ste27KR-GPAAD by us and others [Citation21], suggesting that Ste2 hyper-phosphorylation can also be recognized as a weak endocytosis signal by an as-yet uncharacterized mechanism.
Comparing the absolute requirement of phosphorylation for basal versus its lesser role played during ligand-induced endocytosis, we conclude that the α-factor-bound Ste2 must be a significantly better substrate than unliganded receptor for both the ubiquitin and the Sla1 mechanisms. Furthermore, these results support the notion [Citation15] that phosphorylation of the CTD’s function is not to create a docking surface but rather to produce a conformational change in the CTD akin to the one generated by α-factor binding, a conformation that makes the CTD more available to be acted upon by the two internalization machinery.
In agreement with previous results, our Ste2 non-phosphorylatable mutants showed increased sensitivity to α-factor [Citation23]. This is most likely due to their inability to be phosphorylated and not a side consequence of their increased plasma membrane abundance, since we have previously shown that Ste2 regulates signaling by a ratiometric control mechanism that makes the response robust to changes in receptor abundance [Citation26]. This robustness was further confirmed here by the behavior of cells bearing Ste27KR, which accumulated prominently at the plasma membrane, yet these cells showed WT sensitivity. How phosphorylation regulates sensitivity to α-factor is not clear. In its unphosphorylated state, Ste2 might have a higher intrinsic GEF activity or it may have altered interactions with G-proteins or other regulatory proteins. Note that Sst2, a strong negative regulator of the G-protein seems to bind preferentially to non-phosphorylated Ste2 [Citation14], and thus cannot explain the increased α-factor sensitivity of cells bearing the Ste220STA receptor versions. In any case, we suggest that WT Ste2 normally signals downstream to the G-protein in a phosphorylated state (at least in some of the sites we mutated). Indeed, many of the S/T sites appear to be constitutively phosphorylated [Citation38,Citation39]. In agreement with this idea, as mentioned above, cells with Ste27KR, which is expected to be fully phosphorylated in the presence of α-factor, displayed WT sensitivity.
In conclusion, we found that Ste2 endocytosis and phosphorylation impact GPCR-dependent cellular responses in a complex manner. Perhaps most surprising is that receptor trafficking seemed to be unnecessary for the movement of the patch from its origin at the bud-neck to the opposite side of the cell. Uncovering the interaction of different modifications of GPCRs and its functions is key for an integral understanding of GPCR-related cellular responses. We expect these findings to be applicable to other polarity processes in eukaryotes that depend on GPCRs.
Author contributions
G.V. and A.C.-L. designed research; G.V., P.D. and A.C. performed research; G.V. and A.C.-L. analyzed data; and G.V. and A.C.-L. wrote the paper.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. D.G. Drubin for kindly providing fluorescently labeled α-factor. We thank Dr. Alan Bush and Pablo S. Aguilar for helpful discussions and comments on the manuscript.
Additional information
Funding
References
- Vasen G , Dunayevich P , Colman-Lerner A. Mitotic and pheromone-specific intrinsic polarization cues interfere with gradient sensing in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2020;117:6580–6589.
- Bardwell L . A walk-through of the yeast mating pheromone response pathway. Peptides. 2004;25:1465–1476.
- Dohlman HG , Thorner JW . Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. AnnuRevBiochem. 2001;70:703–754.
- Pryciak PM , Huntress FA . Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gβγ complex underlies activation of the yeast pheromone response pathway. Genes Dev. 1998;12:2684–2697.
- Butty AC , Pryciak PM , Huang LS , et al. The role of far1p in linking the heterotrimeric G protein to polarity establishment proteins during yeast mating. Science. 1998;282:1511–1516.
- Nern A , Arkowitz RA . A Cdc24p-Far1p-Gβγ protein complex required for yeast orientation during mating. J Cell Biol. 1999;144:1187–1202.
- Irazoqui J , Gladfelter A , Lew D . Scaffold-mediated symmetry breaking by Cdc42p. Nat Cell Biol. 2003;5:1062–1070.
- Chenevert J , Valtz N , Herskowitz I . Identification of genes required for normal pheromone-induced cell polarization in Saccharomyces cerevisiae . Genetics. 1994;136:1287–1296.
- Sheu YJ , Santos B , Fortin N , et al. Spa2p interacts with cell polarity proteins and signaling components involved in yeast cell morphogenesis. Mol Cell Biol. 1998;18:4053–4069.
- McClure AW , Minakova M , Dyer JM , et al. Role of polarized G protein signaling in tracking pheromone gradients. Dev Cell. 2015;35:471–482.
- Ismael A , Tian W , Waszczak N , et al. Gbeta promotes pheromone receptor polarization and yeast chemotropism by inhibiting receptor phosphorylation. Sci Signal. 2016;9:ra38.
- Wang X , Tian W , Banh BT , et al. Mating yeast cells use an intrinsic polarity site to assemble a pheromone-gradient tracking machine. J Cell Biol. 2019;218:3730–3752.
- Hicke L , Zanolari B , Riezman H . Cytoplasmic tail phosphorylation of the alpha-factor receptor is required for its ubiquitination and internalization. J Cell Biol. 1998;141:349–358.
- Ballon DR , Flanary PL , Gladue DP , et al. DEP-domain-mediated regulation of GPCR signaling responses. Cell. 2006;126:1079–1093.
- Alvaro CG , O’Donnell AF , Prosser DC , et al. Specific α-arrestins negatively regulate saccharomyces cerevisiae pheromone response by down-modulating the G-protein coupled receptor Ste2. Mol Cell Biol. 2014;34:2660–2681.
- Dunn R , Hicke L . Domains of the Rsp5 ubiquitin-protein ligase required for receptor-mediated and fluid-phase endocytosis. Mol Biol Cell. 2001;12:421–435.
- Hicke L , Riezman H . Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287.
- Odorizzi G , Babst M , Emr SD . Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998;95:847–858.
- Schandel KA , Jenness DD . Direct evidence for ligand-induced internalization of the yeast alpha-factor pheromone receptor. Mol Cell Biol. 1994;14:7245–7255.
- Shih SC , Katzmann DJ , Schnell JD , et al. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat Cell Biol. 2002;4:389–393.
- Howard JP , Hutton JL , Olson JM , et al. Sla1p serves as the targeting signal recognition factor for NPFX(1,2)D-mediated endocytosis. J Cell Biol. 2002;157:315–326.
- Mahadev RK , Pietro SMD , Olson JM , et al. Structure of Sla1p homology domain 1 and interaction with the NPFxD endocytic internalization motif. Embo J. 2007;26:1963–1971.
- Kim K-M , Lee Y-H , Akal-Strader A , et al. Multiple regulatory roles of the carboxy terminus of Ste2p a yeast GPCR. Pharmacol Res. 2012;65:31–40.
- Colman-Lerner A , Gordon A , Serra E , et al. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437:699–706.
- Longtine MS , McKenzie A III , Demarini DJ , et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961.
- Bush A , Vasen G , Constantinou A , et al. Yeast GPCR signaling reflects the fraction of occupied receptors, not the number. Mol Syst Biol. 2016;12:898.
- Liu B , Huang P-J-J , Zhang X , et al. Parts-per-million of polyethylene glycol as a non-interfering blocking agent for homogeneous biosensor development. Anal Chem. 2013;85:10045–10050.
- Bush A , Chernomoretz A , Yu R , et al. Using cell-ID 1.4 with R for microscope-based cytometry. Curr Protoc Mol Biol. 2012; 100:14.18.1-14.18.26.
- Toshima JY , Toshima J , Kaksonen M , et al. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc Natl Acad Sci U S A. 2006;103:5793–5798.
- Gordon A , Colman-Lerner A , Chin TE , et al. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat Methods. 2007;4:175–181.
- Terrell J , Shih S , Dunn R , et al. A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol Cell. 1998;1:193–202.
- Chen Q , Konopka JB . Regulation of the G-protein-coupled alpha-factor pheromone receptor by phosphorylation. Mol Cell Biol. 1996;16:247–257.
- Suchkov DV , DeFlorio R , Draper E , et al. Polarization of the yeast pheromone receptor requires its internalization but not actin-dependent secretion. Mol Biol Cell. 2010;21:1737–1752.
- Vallier LG , Segall JE , Snyder M . The alpha-factor receptor C-terminus is important for mating projection formation and orientation in Saccharomyces cerevisiae. Cell Motil Cytoskeleton. 2002;53:251–266.
- Toshima JY , Nakanishi J-I , Mizuno K , et al. Requirements for recruitment of a G protein-coupled receptor to clathrin-coated pits in budding yeast. Mol Biol Cell. 2009;20:5039–5050.
- Hegemann B , Unger M , Lee S , et al. A cellular system for spatial signal decoding in chemical gradients. Dev Cell. 2015;35:458–470.
- Dyer JM , Savage NS , Jin M , et al. Tracking shallow chemical gradients by actin-driven wandering of the polarization site. Curr Biology Cb. 2013;23: 32–41.
- Ficarro SB , McCleland ML , Stukenberg PT , et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. NatBiotechnol. 2002;20:301–305.
- Holt LJ , Tuch BB , Villén J , et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686.