
ABSTRACT
Microorganisms are present in the universe and they play role in beneficial and harmful to human life, society, and environments. Plant microbiome is a broad term in which microbes are present in the rhizo, phyllo, or endophytic region and play several beneficial and harmful roles with the plant. To know of these microorganisms, it is essential to be able to isolate purification and identify them quickly under laboratory conditions. So, to improve the microbial study, several tools and techniques such as microscopy, rRNA, or rDNA sequencing, fingerprinting, probing, clone libraries, chips, and metagenomics have been developed. The major benefits of these techniques are the identification of microbial community through direct analysis as well as it can apply in situ. Without tools and techniques, we cannot understand the roles of microbiomes. This review explains the tools and their roles in the understanding of microbiomes and their ecological diversity in environments.
Introduction
Microorganisms have a variety of roles in both human and environmental life. Despite their small size, these organisms have a significant impact on society, either positively or negatively [Citation1]. Small organisms, on the other hand, can be found in almost any environment, including water, soil, and air, making microorganisms pervasive [Citation2]. Microbe research is difficult due to our lack of understanding of them and their environments. Because microorganisms occur in large quantities in the natural environment, conventionally based methods do not provide enough data to fully comprehend microbes and their impact on the ecosystem, including plants [Citation3]. However, due to a lack of relevant and up-to-date instruments and methodologies for understanding these microbes, numerous microbial species have remained unknown till today [Citation4,Citation5].
Different techniques/procedures are available for characterization and identification of microbes, such as isolation, purification of visible colonies, and microbe rearing, however, all methods are applied at the laboratory desk, and these methods are old [Citation6,Citation7]. Some techniques and instruments, such as culture technique and microscopy, have been widely employed in the past, but they provide a limited picture of the microbial world [Citation8,Citation9]. Many bacteria seem the same under a microscope, and many won’t thrive outside of their natural habitats. Because of a lack of tools and methodologies, only around 1% of microbes have been discovered. This is because certain microbes are viable but non-culturable (VBNC) while others are found in extreme environments such as very high or low temperature, pH, pressure, salinity, and so on [Citation10,Citation11]. As a result, early scientists only investigated a small number of bacteria, and a certain region of microbial habitat refers to just those microbes that have been produced in a microbial laboratory. However, increased culture is required for a complete investigation of microorganisms, which can only be done in the lab [Citation12,Citation13]. For the study of microbial diversity, there are commonly three methodologies. 1) methods that are culture-specific 2) Techniques that are not culture-specific 3) methods based on molecular biology. Culture-dependent approaches lose the majority of microbial species, making it impossible to investigate microbial ecology [Citation14,Citation15], but culture-independent and molecular techniques allow us to research microbial ecology more easily [Citation16]. For bacterial identification, PCR amplification of the universal 16S rRNA gene is often utilized. 16 sRNA sequencing offers data at the taxonomic level of bacterial species, and it is a commonly used technology [Citation17,Citation18]. The comparable rRNA gene known as 18 sRNA is used to research higher microorganisms such as fungus [Citation19]. If 18 sRNA does not provide enough information on the fungi, we go on to the internally transcribed spacer (ITS). ITS gives complete data for fungus taxonomy identification [Citation20,Citation21]. Although neither technique can identify a new species, this is a significant drawback of both techniques because they both rely on primers. Although both procedures cannot identify new species, this is a significant limitation of both techniques because both techniques rely on primers that detect just the target species/organisms [Citation22–24].
All realms of life, on the other hand, are populated in a complex environment. Prokaryotic grazers, such as protozoa, fungi, nematodes, and oomycetes, can be important plant symbionts or destructors, while others are prokaryotic grazers found in most soils around the world [Citation25,Citation26]. In agricultural soils, the archaea domain participates in metabolic activities such as ammonia production, oxidation, and methanogenesis. In the environment, bacteria in the last domain serve a variety of activities, including plant growth and development, pathogen defense, bioremediation, biodegradation, and other industrial sectors [Citation27]. They also operate in the manufacturing and service industries, producing ethanol, enzymes, acids, fragrances, and medications, among other things. Bacteria interact with the host plant and other bacteria in the ecosystem, therefore capturing as much variation of a microbiome as possible is critical [Citation28]. To accomplish so, international methodologies such as metagenomics, meta-transcriptomic, meta-proteomic, and metabolomics must be used, which allow for the simultaneous appraisal and judgment of indigenous microflora across all domains of life [Citation29]. Metagenomics is the direct extraction and cloning of genetic material from samples to determine the genomic diversity of microbes (culturable and unculturable), whereas metaproteomics, metatranscriptomics, and metabolomics, respectively, provide a sketch of community-wide protein abundance, gene expression, and metabolic activities [Citation30,Citation31].
In this review, we will look at the know-how and methods that have been used in the isolation and characterization of bacteria and microbiomes on a morphological and molecular level. We’ll also learn how to use metagenomics to identify VBNC organisms. These methods can also be used to investigate microbiomes, or microbe populations, in various contexts [Citation32].
Plant microbial community
To address the importance of the soil-associated plant microbiome to plant trait expression and ecosystem functions, the aspects of the plant microbiome covered in this overview are limited to the rhizosphere, rhizoplane (epiphytes), and internal endosymbiosis (endophytes) of the belowground organs of the plant [Citation33–35]. However, this ignores the rhizosphere microbiome’s effects on aboveground interactions, including herbivory, pollination, and seed predation, as well as pathogen attack from aboveground structures [Citation36,Citation37].
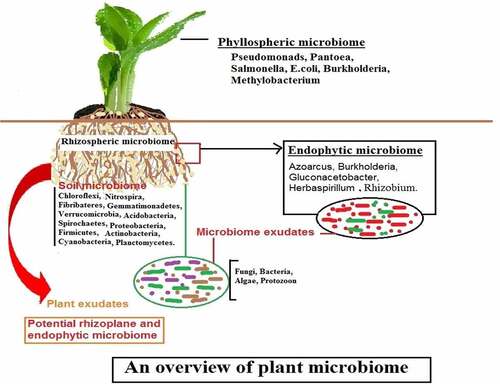
Plant-microbe interactions are important for plant growth and yield, and they have gotten a lot of attention recently [Citation38]. This type of interaction is seen in all regions of plants, according to microbiologists and ecologists, but it is called a plant microbiome when it occurs in a specific portion of the plant [Citation39]. “A plant microbiome is a specific place/region/habitats, such as roots, leaves, stems, and floral sections, where varied bacteria exist and display various interactions with the plants,” according to the definition [Citation40]. These interactions may be mutual/beneficial/harmful given in .
Figure 1. Role of the plant microbiome.
Types of plant microbiome
Plant microbiomes are diverse, containing both beneficial and harmful rhizospheric microorganisms, as well as endophytic and pathogenic pathogens [Citation41]. Plant microbiome is categorized into three kinds based on their occurrence and interactions with the plant: 1) Rhizospheric plant microbiome; 2) Phyllospheric plant microbiome; and 3) Endophytic plant microbiome.
Rhizospheric plant microbiome
Soil has a variety of microorganisms, which divide into two zones based on their availability to the plant [Citation42]. The rhizosphere is a zone characterized by increased microbial mass and soils that directly surround plant roots. The rhizosphere is further separated into two parts: a) edaphosphere (one side bordered by soil region) and b) histosphere (the other side surrounded by plant tissues), both of which play a vital role in rhizosphere development [Citation43,Citation44]. Rhizospheric soils have a high water-holding capacity, indicating that there are many mutual interactions between microorganisms and plant roots in the soil, as well as improved nutrient availability. Rhizoplane refers to soil particles that are closely adhered to the root surface [Citation45,Citation46].
Fungi, protozoa, archaea, nematodes, oomycetes, bacteria, algae, and viruses are all frequent creatures found in the rhizosphere, and these organisms are referred to as rhizo-microbiomes [Citation37,Citation47]. These creatures dwell in the rhizosphere and feed on the plant’s nutrients (organic acids, sugars, amino acids, fatty acids, vitamins, and growth hormones) [Citation48]. Plant growth-promoting rhizobacteria (phosphate solubilizing bacteria, nitrogen-fixing bacteria, biological control bacteria), cyanobacteria, fungi, mycorrhiza, protozoa, and pathogenic bacteria, fungi, virus, and nematodes are among the organisms that have been well studied for their beneficial effects on plant growth and development [Citation27]. Another type of bacteria to consider is Pseudomonas aeruginosa, which improves plant growth and production but can cause sickness in people [Citation38,Citation49]. Plant growth and fitness are influenced by rhizospheric microbiomes, which can be beneficial or harmful. It is positively influenced by phytohormone secretion (indole-3 acetic acid, gibberellin), production of siderophore and ammonia, solubilization of phosphate, zinc, and potassium, and indirectly by decomposition of detritus, nutrient cycling, pathogen inhibition, secretion of stress hormone (ACC deaminase), and stimulation of the plant immune response [Citation50]. However, certain bacteria function as pathogens, reducing crop productivity [Citation51].
Phyllospheric plant microbiome/aerial plant surface microbiome
The phyllosphere is the world’s second-largest microbial habitat. By colonizing severe, stressful, and changing settings, the microbial community in this region performs a dynamic function [Citation52,Citation53]. It is a significant point of entry for phytopathogens into plant tissues, where they cause disease. Furthermore, they offer a unique location for easily comprehending the interaction between microbiota and plants, as well as the methods by which distinct microbial populations sustain their populations in nature [Citation54,Citation55]. In comparison to the rhizosphere or endophytic zone, the phyllosphere has a low nutritional content [Citation56].
On the phyllosphere, microbial population colonization is different, but it is influenced by the leaf’s stomata, hairs, and veins [Citation57]. On the leaf surfaces, 107 microorganisms per cm2 colonize [Citation58]. The phyllosphere is a much more interesting place, where microbes live in the presence of large fluctuations in temperature, radiation, moisture, and light throughout the day and night. Plant metabolism changes as a result of these environmental factors and the phyllosphere microbiome are affected [Citation59].
Endophytic/ root interior plant microbiomes
Endophytes are microorganisms that dwell inside plant tissue, whereas the endosphere is the surrounding environment [Citation60]. Endophytic bacteria are typically thought to be non-disease causing agents because they cause no obvious symptoms on plants [Citation61], but researchers have recently uncovered several pathogens that are dependent on the host genotype and environmental factors.
Endophytes are thought to have started in the rhizosphere, although they exhibit unique characteristics from rhizospheric microbes, implying that not all rhizospheric microbes can penetrate the plant [Citation62]. When bacteria penetrate a plant, they change their physiological/metabolic processes and adapt to the host’s inner environment [Citation3].
Tools and techniques to the understanding of plant microbiome
There are new methodologies and procedures available to assist in the research of microorganisms that reside in a biome, allowing for accurate microbial ecology values [Citation63]. The abundance of species, population size, species consistency, and species distribution are all factors in microbial ecology [Citation64]. Measurement of microbial diversity is routinely done using morphological, biochemical, and molecular approaches. Molecular-based approaches, for example, can provide taxonomy-level information [Citation65].
Some molecular approaches, including G + C percentage, restriction fragment length polymorphism (RFLP), DNA hybridization, and community-level physiological profiles (CLPP), are useful for identifying aquatic populations without providing any data [Citation66]. Many DNA fingerprinting techniques, such as denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis (TGGE), repetitive PCR, arbitrarily PCR, and terminal-restriction length polymorphism (T-RFLP), are now available and widely used in the identification of microbial species by retaining polymerase chain reaction from environmental samples [Citation67,Citation68]. Researchers, on the other hand, can’t utilize diversity metrics since microbiomes don’t have a specific diversity and fluctuate as the environment changes [Citation69]. First and foremost, the origin of the community must be known to comprehend the secret of microbes and plants in a specific habitat [Citation70]. Different tools and strategies are required to do this. lists the most important instruments and strategies for learning about plant microbiomes.
Table 1. Major tools and techniques for studying of plant microbiome and their merits and demerits [Citation96].
Microscopy
Researchers employ a variety of microscopes to study the plant microbiome, including light, compound, dark field, bright field, confocal, and fluorescence microscopes [Citation71]. However, because these microscopes have a smaller focusing point, they are not suitable for in-depth research. Electron microscopes, such as the scanning electron microscopy (SEM) and transmission electron microscope (TEM), can overcome these restrictions since they have a high-resolution power attached to the usage of an electron beam, as opposed to light or compound microscopes [Citation72]. Microorganisms and plants on the surface and inside the cell can be spotted using an electron microscope [Citation73]. Furthermore, the microbiome’s habitat, niche, host, and behavior can all be explored [Citation74]. The microscope aids in the colonization of microorganisms on or inside the plant surface, as well as the understanding of their role in the plants [Citation75]. Because it is a basic requirement of microorganisms for sustenance, and because of the nature of the habitat, effective colonization is a vital point in plant-microbe interactions [Citation3]. Thus, plant colonization has long been an important issue in studies on the routes and roles of these organisms with plants [Citation76], and microscopy aids in the viewing of microorganisms in their natural habitat and relationship with plants [Citation77]. Furthermore, this method enables the tracking of microorganisms [Citation78].
Nucleic acid extraction
Understanding the plant microbiome requires the use of nucleic acid extraction, which is one of the most significant technologies in biology. A nucleic acid extraction is an old approach that has been well refined in the twenty-first century [Citation79]. Nucleic acids are extracted in three processes, depending on the sample and downstream application: a) breaking the samples (tissue or cells); b) removing lipids, proteins, and other contaminants from the nucleic acid; and c) transferring the nucleic acid to a buffer solution for storage [Citation80]. However, numerous commercial molecular kits for nucleic acid extraction are currently available on the market. However, the identification of DNA and RNA can be done using both traditional and kit approaches, and both methods are used to extract quantitative and qualitative nucleic acid analyses [Citation81]. Furthermore, it is expected that the same standardized procedure is employed, as each method will have its preferences in terms of nucleic acid quality and amount [Citation82,Citation83].
Nucleic acid hybridization
In the study of the plant microbiome, nucleic acid hybridization (DNA-DNA and RNA-DNA) is a useful technique [Citation84]. Hybridization reaction occurs when two harmonizing single-stranded nucleic acids form a partial or entire double-stranded nucleic acid by a specific-sequence interface [Citation85]. Nucleic acid is analyzed quantitatively and quantitatively utilizing specific probes in this approach [Citation86]. The probe, which is recognized sequences spanning in specificity from domain to species and is identified with markers at the fifty end position, is often taken [Citation87]. There are a variety of nucleic acid hybridization procedures available, with FISH (fluorescent in situ hybridization) being one of the most popular [Citation88].
This method can be used to investigate the spatial dispersion of a microbial population in a different place [Citation89]. However, due to a lack of sensitivity and the presence of high copy numbers in comparison to dominant species in a sample, we cannot directly extract nucleic acid from environmental materials using this method [Citation90]. As a result, analytical tools can be used to overcome these constraints, and PCR is a good option. Membrane hybridization is another prominent technique utilized by researchers in addition to FISH [Citation91]. Denatured RNA or DNA is immobilized on inner support in such a way that self-annealing is prevented while the residual sequence (bound) is present for hybridization with tagged probes in the membrane method (single or double-stranded). Furthermore, the membrane is extensively washed to remove the unattached probe, and a low case of matched hybrids follows the nucleic acid hybridization reaction [Citation92].
Polymerase chain reaction
One of the most essential techniques for increasing or amplifying a certain target sequence of DNA is the polymerase chain reaction (PCR) [Citation93]. In PCR, the targeted sequences are chosen from the nucleic acid sequence, such as repeated sequences or specific genes [Citation94]. The PCR process takes at least 35–40 cycles to complete and is divided into three parts based on temperature: One cycle consists of 1) denaturation, 2) annealing, and 3) elongation. 16S rRNA gene primers are known as universal or species-strain primers [Citation95].
It’s a promising PCR amplification for bacterial species identification and phylogenetic reasons [Citation96]. The PCR employs nonspecific dyes (SYBER Green I and SYBER Gold) that bind to double-stranded DNA [Citation97]. PCR develops numerous variants based on the type of sample to justify the isolation and quantification of live bacterial numbers at the same time. Multiplex PCR is a fantastic example for separating mixed bacterial pathogens from a sample as well as differentiating multiple species belonging to the same genus [Citation65,Citation98].
The earliest PCR, on the other hand, is unable to detect live or dead bacterial cells. However, this problem can now be rectified using a cutting-edge technology known as reverse transcriptase PCR (RT-PCR) [Citation99]. The reverse transcriptase enzyme powers RT-PCR. This technology, on the other hand, is sensitive yet does not require pre-enrichment operations, and it also takes less time than traditional PCR. Furthermore, VNC cells cannot be cultivated using standard PCR or simple laboratory methods, but we can detect them using RT-PCR [Citation100,Citation101].
Exonuclease activity generates a luminous and detectable signal in RT-PCR. A signal is created with each PCR cycle. The generated signal enables real-time detection through real-time PCR. When a signal permits a specific threshold level, the signal is transformed into forecast target gene numbers based on a pre-established calibration line with ordinary target DNA [Citation102]. RT-PCR also assesses the magnitude of gene effects in local settings as well as the degree of gene expression in microhabitats. As a result, it may be possible to more precisely map microflora and their utility to the soil, plants, and other places using this technique [Citation103].
DNA fingerprinting
Because every organism has unique DNA, DNA fingerprinting is a unique approach for identifying an organism based on DNA properties. It is primarily utilized in forensic sciences, although it is currently used in a variety of fields, including the study of plant-microbe interactions [Citation104]. In the fingerprinting techniques, restriction fragment length polymorphism (RFLP), polymerase chain reaction-restriction fragment length (PCR-RFLP) [Citation105], denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (T-RFLP), temperature gradient gel electrophoresis (TGGE), single-strand conformational polymorphisms (SSCP), ribosomal internal spacer analysis (RISA), length heterogeneity-PCR (LH-PCR) are involved [Citation106,Citation107].
These strategies aid in the study and comparison of data from microbial communities in a sample. However, some PCR-based approaches for microbial community characterization were outmoded, but many new post-PCR analytical techniques have emerged in recent years [Citation108]. The main advantages of this method are that it allows for a comparison of the microbiome’s morphology, composition, and diversity in samples such as soils. Another advantage of this technique is that it can distinguish between viable and nonviable cells in the microbial population, which is something that no other fingerprint technology can do [Citation109].
Denaturing gradient gel electrophoresis
DGGE is a very useful technology for detecting the microbial population directly from environmental materials, which means it does not require microbe rearing. PCR-DGGE, phylogenetic DGGE, and functional gene DGGE are the three most common forms. Only a few samples are required for microbial community characterization in PCR-DGGE [Citation110]. This method detects specific clusters of microorganisms from various plant zones, such as roots and leaves.
On polyacrylamide gels with denaturing gradients, the principle of PCR-DGGE is carried out. However, while this technique was originally developed for mutation analysis, it is currently employed in a wide range of applications, such as detecting microbial communities in environmental samples [Citation111]. This technique has the advantage of simultaneously observing many samples and assessing microbial communities based on ecological and historical differences [Citation112,Citation113].
Phylogenetic study of bacteria using 16 sRNA genes is commonly employed in PCR-DGGE currently. This is not a new notion, as similar techniques have been in use since 1990 [Citation114]. The single species of the community can be identified using phylogenetic DGGE [Citation115,Citation116] by removing DGGE bands from the gel and sequencing them, or by creating clone libraries of 16S rRNA that are separated using DGGE [Citation115]. Muyzer et al. [Citation117] reported that the 16S rRNA gene is employed as a molecular biomarker for microbial species in a population. The most important thing to remember when using partial 16S rRNA gene sequencing is to be cautious when interpreting the results. V4-V5 are appropriate sections for phylogenetic analysis when compared to full-length sequence data [Citation118]. However, the DGGE technique has some limitations, such as microbial DNA extraction and community analysis from environmental samples. Another limitation is that while one strain produces only one band, in some species two or more bands have been observed, and thus we cannot estimate the true microbial diversity data [Citation110].
The data of soil health, quality, and function is provided by the functional gene-based DGGE. We may simply link reduced soil microbial diversity to poor soil functioning this way [Citation119]. The study of coding proteins genes entangled in critical biome practices has gotten a lot of interest in the past few years [Citation120]. Furthermore, functions in which the genes are accommodated by one or more species of bacteria were postulated. In comparison to extremely unnecessary bacteria species/groups, interruption inducing bacteria species/groups affect the functioning of soil [Citation121].
Furthermore, functional genes for nitrogen fixation, denitrification, and other processes are abundant in bacterial species. Since the previous few decades, there has been a surge in interest in gene databases, including primer design, gene function, and gene identification [Citation122]. The gene producing nitrate reductase narG, nitrite reductases nirS and nirK, encoding dinitrogenase reductases nifH, and amoA encoding the ammonia monooxygenase have all been used as substitutions to track changes in soil functional gene diversity [Citation123,Citation124]. The nifH has recently been utilized to investigate the influence of GM white spruce on soil N2 fixation communities. However, the authors failed to mention the GM plant’s significant impact on N2 fixation ecosystems [Citation125]. Another study used the PCR-DGGE approach to positively trace the phlD gene, which codes for diacetyl phloroglucinol (DAPG), an antagonistic chemical generated by pseudomonads [Citation126].
Clone libraries
A clone library is a collection of DNA fragments that have been cloned into vectors and have been used by researchers to identify and extract those DNA fragments that they are interested in studying further [Citation127]. cDNA libraries and genomic DNA libraries are the two types of libraries that exist. The cDNA library is made up of clones and contains reverse-transcribed mRNA, but it lacks DNA sequences corresponding to genomic areas that aren’t expressed, such as 5’ and 3’ noncoding regions, and introns. Genomic libraries, on the other hand, contain huge amounts of DNA in the form of bacterial, bacteriophage, or other synthetic chromosomes [Citation128].
Clone libraries provide immediate access to information on the microbiome’s targeted gene sequences. This method involves joining PCR-generated replicons to a suitable vector plasmid [Citation129]. In addition, using the transformation procedure, the synthesized DNA is cloned into an appropriate host such as Escherichia coli. Following transformation, plasmid extraction can be used to remove cloned replicons from the inserted vector, which can then be sequenced and studied using databases [Citation130]. Chimeras (a single bacterial cell with two different genotypes) are generally eliminated during this procedure.
Because the strain’s sequences are evaluated separately, this technique is far superior to phylogenetic analysis or fingerprinting techniques [Citation131]. The capacity to directly obtain and evaluate novel strains is a major benefit of this technology, which improves our understanding of the soil microbial population [Citation132]. However, other researchers claim that clone libraries are a time-consuming method because many techniques have progressed in recent years, allowing for a more comprehensive understanding of bacteria and functional genes in a microbiome [Citation109].
DNA microarrays
For studying the soil microbiome, DNA microarray is an excellent technique. The term “DNA chip” or “biochip” is also used to describe it. This technique is used to examine the genetic constitutions of several sections of an organism’s genome or to evaluate gene expression levels at a large number of genes at the same time [Citation133,Citation134]. The sample’s DNA is extracted and amplified using PCR in this procedure. A microarray is used to analyze additional DNA samples that have been tagged. A thick range of oligonucleotide probes is inserted on the microarray, ranging from 10 to 1000 [Citation135]. Probes could be 16S rRNA gene fragments or functional gene fragments. The DNA samples must be homogeneous to the probes on the chip for them to bind or hybridize. The chip’s signals are digitally analyzed after binding [Citation136]. Chip contributes to our understanding of phylogenetic diversity, functional genes, and community composition in this way. When the material is extremely complex, such as soil, however, analysis can be difficult [Citation137].
Cross-hybridization, on the other hand, is a serious concern with microarrays. At least 11 or more short oligonucleotides have been designed to overcome the challenge, allowing dissimilar matches to be distinguished from similar perfect matches [Citation138,Citation139]. According to the number of probes and design [Citation140], there are two types of microarrays: a) geochips and b) phyloarrays. Over 24, 000 probes cover over 10,000 genes scattered among more than 150 functional categories enmeshed in carbon, sulfur, phosphorous, and nitrogen cycling on the geochips [Citation141,Citation142]. They’re utilized to collect soil samples. Geochips are utilized on Antarctic soils to analyze various cycles such as carbon, nitrogen, and other elements. Microarray binding holds a lot of promise, including the possibility of creating a “universal microarray.” It explains the state and condition of the soil [Citation143].
Next generation OR omics tools and techniques
Metagenomics is the most recent technology to be developed. It’s a cutting-edge technique. Metagenomics is the study of bacteria’ whole genomes retrieved from environmental materials. Metagenome gives a demonstrated understanding of species composition, genetic diversity, inter-species interactions, and species evolution in the context of typical civilizations’ environments [Citation30]. Several sophisticated omics sequencing techniques, like pyrosequencing and Illumina sequencing, were established before the last several decades. These methods are useful in metagenomics and metatranscriptomics research.
For example, soil samples were identified using 454-based pyrosequencing and DNA or RNA extraction [Citation144]. Multi parallel sequencing by synthesis is used in this technique, with luciferase enzyme detection and pyrophosphate release detection of the produced light [Citation145]. Both Illumina and pyrosequencing, on the other hand, go through three steps of traditional sequencing: template preparation, library preparation, and actual capillary sequencing [Citation146]. Due to multi parallelity, the sequencer can generate anything from 100 to millions of 450 base pair (bp) readings in a single run [Citation147].
The sensitivity of next-generation sequencing, when collected straight from a soil sample, is investigated for its competence and impartiality [Citation148]. As a result, the depiction and conclusion bias of the results are determined by the extraction of DNA from the soil sample [Citation149]. Although the 454 sequencers are used directly for the development of base reads because it produces longer reads, the Illumina sequencer may be utilized to fill in gaps in the 454 generated sequence data because of its high throughput [Citation150,Citation151].
Metabolomics
Metabolomics research aims to learn more about a biological system’s small molecule metabolites under specific conditions. Primary and secondary metabolites make up the metabolome in general. Plant defence mechanisms have a large diversity of secondary metabolites compared to other complex biological systems [Citation152–154]. Herbivores and microorganisms find the majority of them poisonous or repulsive. Metabolomic compound analysis yields metabolic profiles and fingerprints, as well as the identification of novel biomarkers, which can be combined into microbiome research for a more holistic understanding of the plant microbiome [Citation155].
Analytical technologies used in metabolomics
Nuclear magnetic resonance (NMR), gas chromatography-mass spectrometry (GC-MS), and liquid chromatography-mass spectrometry (LC-MS) are the most common technologies utilised in metabolomics (LC-MS). MS-based approaches are substantially more sensitive than NMR at detecting metabolites [Citation156]. MS samples, on the other hand, necessitate extensive preparation, and detection is limited to metabolites that can ionise into the detectable mass range. For chemicals that are difficult to ionise or dissolve, or that require derivatization for MS, NMR has certain advantages [Citation157].
Targeted and untargeted techniques to metabolomics have hitherto been separated, however this may change in the future [Citation158]. Pre-processing, annotation, post-processing, and statistical analysis are all steps in the analysis of data generated using these technologies (NMR and MS). These procedures are generally customised to the analytical technology [Citation159]. To adjust discrepancies in peak shape width and location caused by noise, sample differences, or instrument parameters, pre-processing procedures are used. There is currently no gold standard pipeline for data pre-processing [Citation160].
A metabolite must be compared to at least two orthogonal properties of an authentic chemical standard evaluated in the same laboratory using the same analytical methodologies as experimental data, according to the Metabolites Standard Initiative (MSI) [Citation161]. Because most metabolites do not have chemical criteria, they cannot be completely identified. As a result, MS annotation tools are classified into several levels of annotation. Metabolites can be detected using NMR by simply comparing data from internet databases [Citation162]. This restricts the results to the content of the relevant databases [Citation163].
Metatranscriptomics
Metatranscriptomics is a method for identifying active genes or pathways in a microbial community by sequencing the total message RNA (mRNA). This procedure entails extracting total RNA from microbial communities, eliminating ribosomal RNA (rRNA) to obtain high amounts of mRNA transcripts, reverse transcribing mRNA into cDNAs, ligating adapters, and sequencing with NGS [Citation164]. Metagenomics and metatranscriptomics are frequently used jointly to give assembled genomes as templates for transcript mapping. Top Hat and HISAT, as well as Cufflinks and HTSeq, have been developed for this purpose. Metatranscriptomics has been successfully applied to a wide range of settings, including soil, sediment, gut microbiomes, and activated sludge. It’s a useful method for deducing community function and activity, as well as identifying novel pathways in uncultured microorganisms [Citation165].
Proteomics
The study of proteins in a microbial community extracted from an environmental sample is known as metaproteomics. Metaproteomics, unlike other -omics techniques, gives direct evidence for proteins, post-translational modifications, protein-protein interactions, and protein turnover, all of which represent the structure, dynamics, and metabolic activities of microbial communities [Citation166]. Metaproteomics mostly use mass spectrometry-based proteomics technologies [Citation167,Citation168]. Metaproteomics has been used in plant microbiome studies to assess bacterial communities in the phyllospheres of tree species in a pristine Atlantic Forest [Citation169], to investigate the response of the plant PGPB Bacillus amyloliquefaciens FZB42 to the presence of plant root exudates [Citation170], and to determine the differences in soil protein abundance in plant sugarcane and rat Despite its success, metaproteomics in the plant microbiome is limited due to reduced protein expression in plant microbial samples and limited database information [Citation171].
Metaproteomics is a technique for assessing microbial activity in an ecosystem at a certain moment using protein expression [Citation172]. Metaproteomics, unlike metagenomics and metatranscriptomics, uses liquid chromatography tandem mass spectrometry (LC-MS/MS). The procedure begins with protein extraction, followed by LC-MS/MS 1generation of MS spectra, and finally, comparison of spectra with peptides from thousands of proteins from various taxonomic groups [Citation173]. These comparisons can be made in two ways: by exploring current protein/peptide databases or by comparing to theoretical peptide spectra created in silico from metagenomes from the same sample or from similar environments [Citation174,Citation175]. Metaproteomics is a potent technique for deciphering active metabolic processes in various contexts in a more direct manner than metagenomics or metatranscriptomics. This method has been used to study soils [Citation176], sediments, marine habitats, freshwater systems [Citation177].
Biosensor
Environmental samples such as air and seawater can be monitored for hazardous compounds using biosensors [Citation178]. Nucleic acid amplification techniques, mass spectrometry methods, and receptor-ligand binding assays are the three most used approaches for multiplex detection from complicated matrices. By improving signal transduction, nanotechnology has been used in biosensors to improve sensitivity and performance of assays [Citation179]. Nanotechnology is the process of creating structures, gadgets, and systems that have unique properties that can be controlled by changing the size and form of materials at the nanoscale scale [Citation180].
Advantages
To utilize these instruments to arrest and study the accumulative multifaceted data and information, new ways and approaches for evidence progress are required to build new methods and approaches [Citation181].
Tools and procedures improve the research of microbial communities and evidence of these communities [Citation113].
It aids in time management and data organization.
Some approaches, such as metagenomics, allow us to glimpse into the hidden microbial world, which aids in the study of viable but nonculturable (VBNC) bacteria [Citation30].
Tools and techniques can be used to understand the connection between microbiomes and their hosts, as well as the link between symbiotic, mutualistic, and commensalism variety and functions [Citation182].
Modern omics approaches to aid in the development of novel tactics and the conduct of research to obtain a detailed report on the microbiome and its expression, as well as the level of plant genome expression monitoring [Citation183].
The finding of numerous new sequences, the ultra-high throughput of sequences, and the lack of preferences are all advantages of metagenomics [Citation184].
Although the huge sequence of data obtained will necessitate the use of sophisticated bioinformatics software for processing, metagenomics does not [Citation185].
Tools and procedures are also useful in the study of microbial metabolites and their interactions with plants [Citation186].
The use of approaches can be seen in changes in diversity as a result of specific treatments, as well as in management strategies for soil diversity and production [Citation187].
Disadvantages
When working with a complex sample, such as soil, tools can cause issues.
One of the biggest drawbacks of this method is the high cost of the instrument used in microbiome research [Citation32].
Until now, important tools that will be employed by the majority of researchers are unknown, implying that a researcher’s lack of skills is a big difficulty in comprehending the plant microbiome [Citation188].
For a microbiome researcher, data interpretation, such as metagenomics, is a big difficulty [Citation189].
An omics technology like pyrosequencing or Illumina is a lengthy procedure [Citation190].
Concluding remarks
We provide an overview of almost traditional molecular methods for accessing the soil microbiome in this review paper, including microscopy, nucleic acid extraction and hybridization methods, fingerprinting, PCR, and PCR-based techniques, as well as information on the development of a novel method and its application to environmental samples. Microarrays and metagenomics, for example, are new approaches that aid in the research of microbiomes that are visible and hidden in the world. The information on microbiome communities is derived from omics methods’ data. Furthermore, the roles of microorganisms in a given community may be understood. Metagenomics and metatranscriptomics aid in the in-depth investigation, and this is an interactive method that occurs in this microbial habitat. As a result of the application of molecular tools and methodologies, we have made significant progress in our knowledge of microbial communities in microbiomes. The nature of the sample, the collection, and the habitat are all important aspects of molecular identification. However, given the overall study of these techniques used to study variegated microbial communities of soil and the analytical power suggested by devouring culture microorganisms, it is strongly recommended to use a polyphasic analytical method to analyze soil and other environmental samples in these types of studies. The authors also mentioned the plant microbiome and how many different types of microbiomes exist in nature.
In addition, the positive and negative roles of microorganisms are reviewed in this review work. When studying the microbial ecology of natural habitats, ancient culture-based techniques are overpowering, but they are exceedingly unfair when studying microbial samples. Microbial ecology studies employing culture nondependent molecular procedures have ushered in a new era of microbial variety, thanks to recent advancements in the omics era and sequencing technologies.
Acknowledgments
P.C. thanks to the Department of Environmental Microbiology, BBAU, Lucknow and Enespa thank to Department of Plant Pathology, School for Agriculture, SMPDC, University of Lucknow, Lucknow for the writing of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Zhai Y, Li X, Wang T, et al. A review on airborne microorganisms in particulate matters: composition, characteristics and influence factors. Environ Int. 2018;113:74–90.
- Zhou Q, Li K, Jun X, et al. Role and functions of beneficial microorganisms in sustainable aquaculture. Bioresour Technol. 2009;100(16):3780–3786.
- Jacoby R, Peukert M, Succurro A, et al. The role of soil microorganisms in plant mineral nutrition—current knowledge and future directions. Front Plant Sci. 2017;8:1617.
- Pronk LJ, Bakker PA, Keel C, et al. The secret life of plant‐beneficial rhizosphere bacteria: insects as alternative hosts. Environ Microbiol. 2022;24:1–17.
- Xu J. Invited review: microbial ecology in the age of genomics and metagenomics: concepts, tools, and recent advances. Molecul Ecol. 2006;15:1713–1731.
- Ferone M, Gowen A, Fanning S, et al. Microbial detection and identification methods: bench top assays to omics approaches. Comprehen Rev Food Sci Food Safe. 2020;19(6):3106–3129.
- Lagier JC, Edouard S, Pagnier I, et al. Current and past strategies for bacterial culture in clinical microbiology. Clin Microbiol Rev. 2015;28(1):208–236.
- Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276(5313):734–740.
- Compant S, Cambon MC, Vacher C, et al. The plant endosphere world–bacterial life within plants. Environ Microbiol. 2021;23(4):1812–1829.
- Bodor A, Bounedjoum N, Vincze GE, et al. Challenges of unculturable bacteria: environmental perspectives. Rev Environ Sci Bio/Technol. 2020;19(1):1–22.
- Li L, Mendis N, Trigui H, et al. The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol. 2014;5:258.
- Hibbing ME, Fuqua C, Parsek MR, et al. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8(1):15–25.
- Palková Z. Multicellular microorganisms: laboratory versus nature. EMBO Rep. 2004;5(5):470–476.
- Rantsiou K, Urso R, Iacumin L, et al. Culture-dependent and-independent methods to investigate the microbial ecology of Italian fermented sausages. Appl Environ Microbiol. 2005;71(4):1977–1986.
- Su C, Lei L, Duan Y, et al. Culture-independent methods for studying environmental microorganisms: methods, application, and perspective. Appl Microbiol Biotechnol. 2012;93(3):993–1003.
- Frank DN, Spiegelman GB, Davis W, et al. Culture-independent molecular analysis of microbial constituents of the healthy human outer ear. J Clinical Microbiol. 2003;41(1):295–303.
- Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clinical Microbiol. 2007;45(9):2761–2764.
- Rosselló‐Móra R. Towards taxonomy of bacteria and archaea based on interactive and cumulative data repositories. Environ Microbiol. 2012;14(2):318–334.
- Cao Y, Fanning S, Proos S, et al. A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies. Front Microbiol. 2017;8:182.
- Schoch CL, Seifert KA, Huhndorf S, et al. Fungal barcoding consortium, fungal barcoding consortium author list nuclear ribosomal internal transcribed spacer (its) region as a universal dna barcode marker for fungi. Precede National Acad Sci. 2012;109(16):6241–6246.
- Gu X, Cheng X, Zhang J, et al. Identification of fungal community in otomycosis by internal transcribed spacer sequencing. Front Microbiol. 2022;13:820423.
- Clarridge IIIJE. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17:840–862.
- Winand R, Bogaerts B, Hoffman S, et al. Targeting the 16s rRNA gene for bacterial identification in complex mixed samples: comparative evaluation of second (illumina) and third (Oxford nanopore technologies) generation sequencing technologies. Int J Mol Sci. 2019;21(1):298.
- Jenkins C, Chapman TA, Micallef JL, et al. Molecular techniques for the detection and differentiation of host and parasitoid species and the implications for fruit fly management. Insects. 2012;3(3):763–788.
- Topalović O, Hussain M, Heuer H. Plants and associated soil microbiota cooperatively suppress plant-parasitic nematodes. Front Microbiol. 2020;11:313.
- Bez C, Esposito A, Thuy HD, et al. The rice foot rot pathogen dickeya zeae alters the in‐field plant microbiome. Environ Microbiol. 2021;23:7671–7687.
- Saeed Q, Xiukang W, Haider FU, et al. Rhizosphere bacteria in plant growth promotion, biocontrol, and bioremediation of contaminated sites: a comprehensive review of effects and mechanisms. Int J Molecul Sci. 2021;22(19):10529.
- Elshaghabee FM, Bockelmann W, Meske D, et al. Ethanol production by selected intestinal microorganisms and lactic acid bacteria growing under different nutritional conditions. Front Microbiol. 2016;7:47.
- Aguiar-Pulido V, Huang W, Suarez-Ulloa V, et al. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis: supplementary issue: bioinformatics methods and applications for big metagenomics data. Evol Bioinform. 2016;12:EBO–S36436.
- Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Molecul Bio Rev. 2004;68:669–685.
- Zhang L, Chen F, Zeng Z, et al. Advances in metagenomics and its application in environmental microorganisms. Front Microbiol. 2021;12:766364.
- Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19:1141–1152.
- Rout ME.2014. The plant microbiome. InAdvances in botanical research. Academic Press. Vol. 69. p. 279–309.
- Turner TR, James EK, Poole PS. The plant microbiome. Genome Biol. 2013;14(6):1–10.
- Babalola OO, Fadiji AE, Enagbonma BJ, et al. The nexus between plant and plant microbiome: revelation of the networking strategies. Front Microbiol. 2020;11:2128.
- Mercado-Blanco J, Abrantes I, Barra Caracciolo A, et al. Belowground microbiota and the health of tree crops. Front Microbiol. 2018;9:1006.
- Mendes R, Garbeva P, Raaijmakers JM. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev. 2013;37(5):634–663.
- Ortíz-Castro R, Contreras-Cornejo HA, Macías-Rodríguez L, et al. The role of microbial signals in plant growth and development. Plant Signal Behav. 2009;4(8):701–712.
- Chandra P, Singh E Applications and mechanisms of plant growth-stimulating rhizobacteria. In Plant-microbe interaction: an approach to sustainable agriculture. Springer, Singapore. (Ed.), 2016; (pp. 37–62).
- Mhlongo MI, Piater LA, Madala NE, et al. The chemistry of plant–microbe interactions in the rhizosphere and the potential for metabolomics to reveal signaling related to defense priming and induced systemic resistance. Front Plant Sci. 2018;9:112.
- Santos LF, Olivares FL. Plant microbiome structure and benefits for sustainable agriculture. Curr Plant Biol. 2021;26:100198.
- Cirvilleri G, Spina S, Iacona C, et al. Study of rhizosphere and phyllosphere bacterial community and resistance to bacterial canker in genetically engineered phytochrome A cherry plants. J Plant Physiol. 2008;165(10):1107–1119.
- Bakker PA, Berendsen RL, Doornbos RF, et al. The rhizosphere revisited: root microbiomics. Front Plant Sci. 2013;4:165.
- Hu J, Wei Z, Kowalchuk GA, et al. Rhizosphere microbiome functional diversity and pathogen invasion resistance build up during plant development. Environ Microbiol. 2020;22(12):5005–5018.
- Barea JM, Pozo MJ, Azcon R, et al. Microbial co-operation in the rhizosphere. J Exp Bot. 2005;56(417):1761–1778.
- Mahmud K, Missaoui A, Lee K, et al. Rhizosphere microbiome manipulation for sustainable crop production. Curr Plant Biol. 2021;27:100210.
- Chandra P, Enespa. Soil–microbes–plants: interactions and ecological diversity. In Varma, A., Tripathi, S., Prasad, R. (eds). Plant microbe interface. Cham: Springer; 2019. p. 145–176.
- Enespa CP.2017. Microbial Volatiles as Chemical Weapons Against Pathogenic Fungi. In Choudhary, D., Sharma, A., Agarwal, P., Varma, A., Tuteja, N. (eds.). Volatiles and Food Security. Singapore: Springer. p. 227–254.
- Miranda‐Sánchez F, Rivera J, Vinuesa P. Diversity patterns of rhizobiaceae communities inhabiting soils, root surfaces and nodules reveal a strong selection of rhizobial partners by legumes. Environ Microbiol. 2016;18(8):2375–2391.
- Kudoyarova G, Arkhipova T, Korshunova T, et al. Phytohormone mediation of interactions between plants and non-symbiotic growth promoting bacteria under edaphic stresses. Front Plant Sci. 2019;10 (1368):1–11.
- Starr MP. Bacteria as plant pathogens. Annual Rev Microbiol. 1959;13:211–238.
- Lindow SE, Brandl MT. Microbiology of the phyllosphere. Appl Environ Microbiol. 2003;69(4):1875–1883.
- Bringel F, Couée I. Pivotal roles of phyllosphere microorganisms at the interface between plant functioning and atmospheric trace gas dynamics. Front Microbiol. 2015;6:486.
- Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30(6):492–506.
- Braga RM, Dourado MN, Araújo WL. Microbial interactions: ecology in a molecular perspective. Brazilian J Microbiol. 2016;47:86–98.
- Gottel NR, Castro HF, Kerley M, et al. Distinct microbial communities within the endosphere and rhizosphere of populus deltoides roots across contrasting soil types. Appl Environ Microbiol. 2011;77(17):5934–5944.
- Nongkhlaw FM, Joshi SR. Distribution pattern analysis of epiphytic bacteria on ethno medicinal plant surfaces: a micrographical and molecular approach. J Microscopy Ultrastruct. 2014;2(1):34–40.
- Der Wal A V, Tecon R, Kreft JU, et al. Explaining bacterial dispersion on leaf surfaces with an individual-based model (PHYLLOSIM). PloS one. 2013;8(10):e75633.
- Carvalho SD, Castillo JA. Influence of light on plant–phyllosphere interaction. Front Plant Sci. 2018;9:1482.
- Afzal I, Shinwari ZK, Sikandar S, et al. Plant beneficial endophytic bacteria: mechanisms, diversity, host range and genetic determinants. Microbiol Res. 2019;221:36–49.
- Partida-Martínez LP, Heil M. The microbe-free plant: fact or artifact? Front Plant Sci. 2011;2:100.
- Vishwakarma K, Kumar N, Shandilya C, et al. Revisiting plant–microbe interactions and microbial consortia application for enhancing sustainable agriculture: a review. Front Microbiol. 2020;11:560406.
- Baldrian P. Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol Rev. 2017;41(2):109–130.
- Grilli J. Macroecological laws describe variation and diversity in microbial communities. Nat Commun. 2020;11(1):1.
- Franco-Duarte R, Černáková L, Kadam S, et al. Advances in chemical and biological methods to identify microorganisms—from past to present. Microorganism. 2019;7(5):130.
- Ritchie NJ, Schutter ME, Dick RP, et al. Use of length heterogeneity PCR and fatty acid methyl ester profiles to characterize microbial communities in soil. Appl Environ Microbiol. 2000;66(4):1668–1675.
- Portillo MC, Villahermosa D, Corzo A. Microbial community fingerprinting by differential display-denaturing gradient gel electrophoresis. Appl Environ Microbiol. 2011;77(1):351–354.
- Keyser M, Witthuhn RC, Lamprecht C, et al. PCR-based DGGE fingerprinting and identification of methanogens detected in three different types of UASB granules. Syst Appl Microbiol. 2006;29(1):77–84.
- Wertz S, Degrange V, Prosser JI, et al. Decline of soil microbial diversity does not influence the resistance and resilience of key soil microbial functional groups following a model disturbance. Environ Microbiol. 2007;9(9):2211–2219.
- Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol. 2009;68(1):1–3.
- Chen X, Zheng B, Liu H. Optical and digital microscopic imaging techniques and applications in pathology. Anal Cell Pathol. 2011;34(1–2):5–18.
- Wisse E, Braet F, Duimel H, et al. Fixation methods for electron microscopy of human and other liver. World J Gastroenterol. 2010;16(23):2851.
- Goldsmith CS, Miller SE. Modern uses of electron microscopy for detection of viruses. Clin Microbiol Rev. 2009;22(4):552–563.
- Berg G, Rybakova D, Fischer D, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiom. 2020;8(1):1–22.
- Romano I, Ventorino V, Pepe O. Effectiveness of plant beneficial microbes: overview of the methodological approaches for the assessment of root colonization and persistence. Front Plant Sci. 2020;11:6.
- Wu CH, Bernard SM, Andersen GL, et al. Developing microbe–plant interactions for applications in plant‐growth promotion and disease control, production of useful compounds, remediation and carbon sequestration. Microbial Biotechnol. 2009;2(4):428–440.
- Hannig C, Follo M, Hellwig E, et al. Visualization of adherent micro-organisms using different techniques. J Medical Microbiol. 2010;59(1):1–7.
- Scott TM, Rose JB, Jenkins TM, et al. Microbial source tracking: current methodology and future directions. Appl Environ Microbiol. 2002;68(12):5796–5803.
- Ali N, Rampazzo RD, Costa AD, et al. Current nucleic acid extraction methods and their implications to point-of-care diagnostics. BioMed Res Int. 2017;1–17.
- Sirakov IN. Nucleic acid isolation and downstream applications. Nucleic Acids-From Basic Aspects to Laboratory Tools. 2016;1–26.
- Collins ML, Zayati C, Detmer JJ, et al. Preparation and characterization of RNA standards for use in quantitative branched DNA hybridization assays. Analyt Biochem. 1995;226(1):120–129.
- Olson ND, Morrow JB. DNA extract characterization process for microbial detection methods development and validation. BMC Res Note. 2012;5(1):1–4.
- Hafner M, Katsantoni M, Köster T, et al. CLIP and complementary methods. Nat Rev Met Primers. 2021;1(1):1–23.
- Wintzingerode F, Göbel UB, Stackebrandt E. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev. 1997;21(3):213–229.
- Li Q, Luan G, Guo Q, et al. A new class of homogeneous nucleic acid probes based on specific displacement hybridization. Nucleic Acids Res. 2002;30(2):e5.
- Pagano JM, Clingman CC, Ryder SP. Quantitative approaches to monitor protein–nucleic acid interactions using fluorescent probes. Rna. 2011;17(1):14–20.
- Volpi EV, Bridger JM. FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques. 2008;45(4):385–409.
- Ratan ZA, Zaman SB, Mehta V, et al. Application of fluorescence in situ hybridization (FISH) technique for the detection of genetic aberration in medical science. Cureus. 2017;9(6):e1325.
- Ramette A, Tiedje JM. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proceed National Acad Sci. 2007;104(8):2761–2766.
- Gabor EM, de Vries Ej, Janssen DB. Efficient recovery of environmental DNA for expression cloning by indirect extraction methods. FEMS Microbiol Ecol. 2003;44(2):153–163.
- Huber D, von Voithenberg Lv, Kaigala GV. Fluorescence in situ hybridization (FISH): history, limitations and what to expect from micro-scale FISH? Micro Nano Engineer. 2018;1:15–24.
- Wang T, Chen C, Larcher LM, et al. Three decades of nucleic acid aptamer technologies: lessons learned, progress and opportunities on aptamer development. Biotechnol Adv. 2019;37(1):28–50.
- Garibyan L, Avashia N. Research techniques made simple: polymerase chain reaction (PCR). J Investigat Dermatol. 2013;133(3):e6.
- Mirmajlessi SM, Destefanis M, Gottsberger RA, et al. PCR-based specific techniques used for detecting the most important pathogens on strawberry: a systematic review. Systemat Rev. 2015;4(1):1–11.
- Chen L, Cai Y, Zhou G, et al. Rapid Sanger sequencing of the 16S rRNA gene for identification of some common pathogens. PloS one. 2014;9(2):e88886.
- Barghouthi SA. A universal method for the identification of bacteria based on general PCR primers. Indian J Microbiol. 2011;51:430–444.
- Giglio S, Monis PT, Saint CP. Demonstration of preferential binding of SYBR green I to specific DNA fragments in real‐time multiplex PCR. Nucleic Acid Res. 2003;31(22):e136.
- Espy MJ, Uhl JR, Sloan LM, et al. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19(1):165–256.
- Kralik P, Ricchi M. A basic guide to real time PCR in microbial diagnostics: definitions, parameters, and everything. Front Microbiol. 2017;8:108.
- Overbergh L, Giulietti A, Valckx D, et al. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech. 2003;14(1):33.
- Del Mar Lleò M, Pierobon S, Tafi MC, et al. mRNA detection by reverse transcription-PCR for monitoring viability over time in an Enterococcus faecalis viable but nonculturable population maintained in a laboratory microcosm. Appl Environ Microbiol. 2000;66(10):4564–4567.
- Yang S, Rothman RE. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis. 2004;4(6):337–348.
- Sharkey FH, Banat IM, Marchant R. Detection and quantification of gene expression in environmental bacteriology. Appl Environ Microbiol. 2004;70(7):3795–3806.
- Khan S, Ullah MW, Siddique R, et al. Role of recombinant DNA technology to improve life. Int J Genom. 2016;1–15.
- Rasmussen HB. Restriction fragment length polymorphism analysis of PCR-amplified fragments (PCR-RFLP) and gel electrophoresis-valuable tool for genotyping and genetic fingerprinting. Techopen. 2012;1–22.
- Johnston‐Monje D, Lopez Mejia J. Botanical microbiomes on the cheap: inexpensive molecular fingerprinting methods to study plant‐associated communities of bacteria and fungi. Appl Plant Sci. 2020;8(4):e11334.
- Osborn AM, Moore ER, Timmis KN. An evaluation of terminal‐restriction fragment length polymorphism (T‐RFLP) analysis for the study of microbial community structure and dynamics. Environ Microbiol. 2000;2(1):39–50.
- DeAngelis KM, Wu CH, Beller HR, et al. PCR amplification-independent methods for detection of microbial communities by the high-density microarray phyloChip. Appl Environ Microbiol. 2011;77(18):6313–6322.
- Weinstock GM. Genomic approaches to studying the human microbiota. Nature. 2012;489(7415):250–256.
- Al‐Mailem DM, Kansour MK, Radwan SS. Capabilities and limitations of DGGE for the analysis of hydrocarbonoclastic prokaryotic communities directly in environmental samples. Microbiol Open. 2017;6(5):e00495.
- Urashima Y, Sonoda T, Fujita Y, et al. Application of PCR-denaturing-gradient gel electrophoresis (DGGE) method to examine microbial community structure in asparagus fields with growth inhibition due to continuous cropping. Microbe Environ. 2009;27:43–48.
- Douterelo I, Boxall JB, Deines P, et al. Methodological approaches for studying the microbial ecology of drinking water distribution systems. Water Res. 2014;65:134–156.
- Rastogi G, Sani RK.2011. Molecular tTechniques to Assess Microbial Community Structure, Function, and Dynamics in the Environment. In Ahmad, I., Ahmad, F., Pichtel, J. (eds). Microbes and Microbial Technology. New York NY: Springer. p. 29–57.
- Das S, Dash HR, Mangwani N, et al. Understanding molecular identification and polyphasic taxonomic approaches for genetic relatedness and phylogenetic relationships of microorganisms. J Microbiol Methods. 2014;103:80–100.
- Schabereiter‐Gurtner C, Saiz‐Jimenez C, Piñar G, et al. Phylogenetic 16S rRNA analysis reveals the presence of complex and partly unknown bacterial communities in tito bustillo cave, Spain, and on its palaeolithic paintings. Environ Microbiol. 2002;4(7):392–400.
- Dı́ez B, Pedrós-Alió C, Marsh TL, et al. Application of denaturing gradient gel electrophoresis (DGGE) to study the diversity of marine picoeukaryotic assemblages and comparison of DGGE with other molecular techniques. Appl Environ Microbiol. 2001;67(7):2942–2951.
- Muyzer G, De Waal EC, Uitterlinden A. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59(3):695–700.
- Morales SE, Cosart TF, Johnson JV, et al. Extensive phylogenetic analysis of a soil bacterial community illustrates extreme taxon evenness and the effects of amplicon length, degree of coverage, and DNA fractionation on classification and ecological parameters. Appl Environ Microbiol. 2009;75(3):668–675.
- Schloter M, Nannipieri P, Sørensen SJ, et al. Microbial indicators for soil quality. Biol Fertil Soil. 2018;54(1):1–10.
- Hoekstra HE, Coyne JA. The locus of evolution: evo devo and the genetics of adaptation. Evol Int J Organic Evol. 2007;61(5):995–1016.
- Goldfarb KC, Karaoz U, Hanson CA, et al. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol. 2011;2:94.
- Tan B, Ng CM, Nshimyimana JP, et al. Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front Microbiol. 2015;6:1027.
- Priemé A, Braker G, Tiedje JM. Diversity of nitrite reductase (nirK and nirS) gene fragments in forested upland and wetland soils. Appl Environ Microbiol. 2002;68(4):1893–1900.
- Braker G, Zhou J, Wu L, et al. Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in Pacific Northwest marine sediment communities. Appl Environ Microbiol. 2000;66(5):2096–2104.
- Raman R. The impact of Genetically Modified (GM) crops in modern agriculture: a review. GM Crops Food. 2017;8(4):195–208.
- Frapolli M, Moënne-Loccoz Y, Meyer J, et al. A new DGGE protocol targeting 2, 4-diacetylphloroglucinol biosynthetic gene phlD from phylogenetically contrasted biocontrol pseudomonads for assessment of disease-suppressive soils. FEMS Microbiol Ecol. 2008;64(3):468–481.
- Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
- Lu F, Jiang H, Ding J, et al. cDNA sequences reveal considerable gene prediction inaccuracy in the Plasmodium falciparum genome. BMC Genomics. 2007;8(1):1.
- Smalla K, Jechalke S, Top EM. Plasmid detection, characterization, and ecology. Microbiol Spectr. 2015;3(1):3–21.
- Shintani M, Sanchez ZK, Kimbara K. Genomics of microbial plasmids: classification and identification based on replication and transfer systems and host taxonomy. Front Microbiol. 2015;6:242.
- Blainey PC. The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev. 2013;37:407–427.
- Hayat R, Ali S, Amara U, et al. Soil beneficial bacteria and their role in plant growth promotion: a review. Annal Microbiol. 2010;60(4):579–598.
- Trevino V, Falciani F, Barrera-Saldaña HA. DNA microarrays: a powerful genomic tool for biomedical and clinical research. Mol Med. 2007;13(9):527–541.
- Bumgarner R. Overview of DNA microarrays: types, applications, and their future. Curr Protoc Mol Biol. 2013;101:1–22.
- Lucito R, Healy J, Alexander J, et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 2003;13(10):2291–2305.
- Miller MB, Tang YW. Basic concepts of microarrays and potential applications in clinical microbiology. Clin Microbiol Rev. 2009;22(4):611–633.
- Thomsen PF, Willerslev E. Environmental DNA–an emerging tool in conservation for monitoring past and present biodiversity. Biol Conserv. 2015;183:4–18.
- Li Q, Birkbak NJ, Gyorffy B, et al. Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics. 2011;12(1):1–7.
- Koltai H, Weingarten-Baror C. Specificity of DNA microarray hybridization: characterization, effectors and approaches for data correction. Nucleic Acid Res. 2008;36(7):2395–2405.
- Butte A. The use and analysis of microarray data. Nat Rev Drug Discov. 2002;1:951–960.
- Tu Q, Yu H, He Z, et al. GeoChip 4: a functional gene‐array‐based high‐throughput environmental technology for microbial community analysis. Mol Ecol Resour. 2014;14(5):914–928.
- Xie J, He Z, Liu X, et al. GeoChip-based analysis of the functional gene diversity and metabolic potential of microbial communities in acid mine drainage. Appl Environ Microbiol. 2011;77(3):991–999.
- Zhang Y, Lu Z, Liu S, et al. Geochip-based analysis of microbial communities in alpine meadow soils in the qinghai-tibetan plateau. BMC Microbiol. 2013;13(1):1–9.
- Escobar-Zepeda A, Vera-Ponce de Leon A, Sanchez-Flores A. The road to metagenomics: from microbiology to DNA sequencing technologies and bioinformatics. Front Genet. 2015;6:348.
- Ronaghi M, Uhlén M, Nyrén P. A sequencing method based on real-time pyrophosphate. Science. 1998;281(5375):363–365.
- Van Nieuwerburgh F, Thompson RC, Ledesma J, et al. Illumina mate-paired DNA sequencing-library preparation using Cre-Lox recombination. Nucleic Acid Res. 2012;40(3):e24.
- Van Elsas JD, Boersma FG. A review of molecular methods to study the microbiota of soil and the mycosphere. European J Soil Biol. 2011;47(2):77–87.
- Head SR, Komori HK, LaMere SA, et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 2014;56(2):61–77.
- Martin-Laurent F, Philippot L, Hallet S, et al. DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl Environ Microbiol. 2001;67(5):2354–2359.
- Bashir A, Klammer AA, Robins WP, et al. A hybrid approach for the automated finishing of bacterial genomes. Nat Biotechnol. 2012;30(7):701–707.
- Reuter JA, Spacek DV, Snyder MP. High-throughput sequencing technologies. Molecul Cell. 2015;58(4):586–597.
- Marchev AS, Vasileva LV, Amirova KM, et al. Metabolomics and health: from nutritional crops and plant-based pharmaceuticals to profiling of human biofluids. Cell Mol Life Sci. 2021;78:6487–6503.
- Razzaq A, Sadia B, Raza A, et al. Metabolomics: a way forward for crop improvement. Metabolites. 2019;9(12):303.
- Salem MA, Perez de Souza L, Serag A, et al. Metabolomics in the context of plant natural products research: from sample preparation to metabolite analysis. Metabolites. 2020;10(1):37.
- Lucaciu R, Pelikan C, Gerner S, et al. A bioinformatics guide to plant microbiome analysis. Front Plant Sci. 2019;10:1313.
- Emwas AH, Roy R, McKay RT, et al. NMR spectroscopy for metabolomics research. Metabolites. 2019;9(7):123.
- Xiao JF, Zhou B, Ressom HW. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends Anal Chem. 2012;32:1–14.
- Scalbert A, Brennan L, Fiehn O, et al. Mass-spectrometry-based metabolomics: limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics. 2009;5(4):435–458.
- Dona Anthony C, Michael K, Flora S, et al. A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Comput Struct Biotechnol J. 2016;14:135–153.
- Vladimir V, Schriemer David C. Supporting metabolomics with adaptable software: design architectures for the end-user. Curr Opin Biotechnol. 2017;43:110–117.
- Martin M, Legat B, Leenders J, et al. PepsNMR for 1H NMR metabolomic data pre-processing. Anal Chim Acta. 2018;1019:1–13.
- Sumner LW, Amberg A, Barrett D, et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics. 2007;3(3):211–221.
- Emwas AH, Roy R, McKay RT, et al. NMR spectroscopy for metabolomics research. Metabolites. 2019;9(7):123.
- Reck M, Tomasch J, Deng Z, et al. Stool metatranscriptomics: a technical guideline for mRNA stabilisation and isolation. BMC Genomics. 2015;16:494.
- Song L, Sabunciyan S, Yang G, et al. A multi-sample approach increases the accuracy of transcript assembly. Nat Commun. 2019;10(1):5000.
- Hettich RL, Pan C, Chourey K, et al. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal Chem. 2013;85(9):4203–4214.
- Wang DZ, Kong LF, Li YY, et al. Environmental microbial community proteomics: status, challenges and perspectives. Int J Mol Sci. 2016;17(8):1275.
- Benndorf D, Reichl U. Proteomics in environmental and technical microbiology. Eng Life Sci. 2014;14(1):27–46.
- Lambais MR, Barrera SE, Santos EC, et al. Phyllosphere metaproteomics of trees from the Brazilian Atlantic forest show high levels of functional redundancy. Microb Ecol. 2017;73(1):123–134.
- Levy A, Conway JM, Dangl JL, et al. Elucidating bacterial gene functions in the plant microbiome. Cell Host Microbe. 2018;24(4):475–485.
- Turner TR, James EK, Poole PS. The plant microbiome. Genome Biol. 2013;14(6):1-10.
- Maron PA, Ranjard L, Mougel C, et al. Metaproteomics: a new approach for studying functional microbial ecology. Microb Ecol. 2007;53(3):486–493.
- Kleiner M. Metaproteomics: much more than measuring gene expression in microbial communities. mSystems. 2019;4(3):e00115–19.
- Blakeley-Ruiz JA, Kleiner M. Considerations for constructing a protein sequence database for metaproteomics. Comput Struct Biotechnol J. 2022;20:937–952.
- Timmins-Schiffman E, May DH, Mikan M, et al. Critical decisions in metaproteomics: achieving high confidence protein annotations in a sea of unknowns. ISME J. 2017;11(2):309–314.
- Bastida F, Hernández T, García C. Metaproteomics of soils from semiarid environment: functional and phylogenetic information obtained with different protein extraction methods. J Proteomics. 2014;101:31–42.
- Hettich RL, Pan C, Chourey K, et al. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal Chem. 2013;85(9):4203–4214.
- Gavrilaș S, Ursachi CȘ, Perța-Crișan S, et al. recent trends in biosensors for environmental quality monitoring. Sensors. 2022;22(4):1513. (Basel, Switzerland).
- Mokhtarzadeh A, Eivazzadeh-Keihan R, Pashazadeh P, et al. Nanomaterial-based biosensors for detection of pathogenic virus. Trends Anal Chem. 2017;97:445–457.
- Mohajerani A, Burnett L, Smith JV, et al. Nanoparticles in construction materials and other applications, and implications of nanoparticle use. Materials (Basel). 2019;12(19):3052.
- Liu L, Song B, Ma J, et al. Bioinformatics approaches for deciphering the epitranscriptome: recent progress and emerging topics. Comput Struct Biotechnol J. 2020;18:1587–1604.
- Boon E, Meehan CJ, Whidden C, et al. Interactions in the microbiome: communities of organisms and communities of genes. FEMS Microbiol Rev. 2014;38(1):90–118.
- Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18(1):1–5.
- Bisanz JE, Soto-Perez P, Noecker C, et al. A genomic toolkit for the mechanistic dissection of intractable human gut bacteria. Cell Host Microb. 2020;27(6):1001–1013.
- Thomas T, Gilbert J, Meyer F. Metagenomics-a guide from sampling to data analysis. Microb Inform Exp. 2012;2(1):1–2.
- Pinu FR, Villas-Boas SG, Aggio R. Analysis of intracellular metabolites from microorganisms: quenching and extraction protocols. Metabol. 2017;7(4):53.
- Hartmann M, Frey B, Mayer J, et al. Distinct soil microbial diversity under long-term organic and conventional farming. Isme J. 2015;9(5):1177–1194.
- Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533(7602):255–259.
- Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Briefing Bioinform. 2021;22:178–193.
- Buermans HPJ, Den Dunnen JT. Next generation sequencing technology: advances and applications. Biochim Biophysica Acta. 2014;1842:1932–1941.