Abstract
American shad Alosa sapidissima once supported large commercial fisheries along the Atlantic coast, but by the early 1980s many of these fisheries had declined precipitously. In contrast to most Atlantic coastal rivers, the abundance of American shad in the Santee–Cooper River basin of South Carolina has grown substantially; still, much of their upstream spawning habitat in this watershed has been restricted or adversely affected by impoundments. In an effort to rebuild populations in upstream river reaches of the Santee–Cooper basin, state and federal agencies developed an approach relying on hatchery augmentation, the relocation of prespawning adults, and the construction of permanent fish passage structures. Genetic monitoring was adopted to ensure that the genetic integrity of the population remains intact during the rebuilding process. Our study provided an initial assessment of genetic diversity for the genetic monitoring program. There were three components to the study: an assessment of within-basin genetic stock structure, a comparison of genetic diversity (the number of alleles and observed heterozygosity) between hatchery and wild stocks, and an evaluation of molecular tags for monitoring hatchery returns. The estimation of genetic diversity and the assessment of molecular tags were based on 10 microsatellite loci. We found no apparent stock structure in the Santee–Cooper basin and no difference in genetic diversity between broodstock and their wild counterparts. Eight microsatellite markers provided enough resolution to successfully (>95% assignment success) match returning American shad with their respective hatchery parents. Our results highlight the utility and importance of integrating genetic information in supportive breeding programs in an effort to evaluate the programs’ effectiveness (both in terms of increasing the census size and in terms of maintaining the long-term viability of a population).
Received March 18, 2011; accepted August 8, 2011
The American shad Alosa sapidissima is an indigenous clupeid inhabiting Atlantic coastal rivers from the St. Lawrence River, Canada, to the St. Johns River, Florida (it has subsequently been introduced to the Pacific coast and Kamchatka Peninsula on the Asiatic side of the North Pacific; Facey and Van Den Avyle Citation1986). This species has a complex life history similar to that of many salmonid species in that it is considered anadromous (Facey and Van Den Avyle Citation1986), phylopatric (Melvin et al. Citation1986), and for southern populations (i.e., Florida, Georgia, and South Carolina) semelparous (Leggett and Carscadden Citation1978). Spawning runs in southern populations begin as early as November in Florida (Mansueti and Kolb Citation1953), when sexual mature males (ages 3–5) and females (ages 4–6) migrate into freshwater (Facey and Van Den Avyle Citation1986). Broadcast spawning typically occurs over substrates of sand, gravel, and mud in main channels and tributaries (Jenkins and Burkhead Citation1994). Eggs, which generally hatch 4–6 d postfertilization, are nonadhesive and semibuoyant in moving water (Facey and Van Den Avyle Citation1986). Larvae develop into juveniles in freshwater and then migrate to estuaries. The oceanic movements of juveniles and adults have been extensively studied (Talbot and Sykes Citation1958; Dadswell et al. Citation1987), with adult American shad typically spending summers in northwestern Atlantic waters and beginning their southward migration to their natal spawning areas as fall approaches.
Historically, American shad supported one of the most important commercial fisheries in North America as well as sport fisheries in almost every state along the Atlantic coast (Facey and Van Den Avyle Citation1986; ASMFC Citation2007). In the late 1800s the annual shad harvest was nearly 23 million kg coastwide (ASMFC 2007); shad stocks could not sustain this rate of exploitation, however, and coupled with habitat loss (e.g., dam construction) and habitat degradation (e.g., pollution), it led to precipitous declines in the abundance of American shad along the Atlantic coast (current abundance estimates are less than 10% of historical estimates; ASMFC 2007). Dramatic declines in commercial landings prompted many states to place moratoriums on American shad harvest (Olney and Hoenig Citation2001) and ultimately led the Atlantic States Marine Fisheries Commission (a commission of U.S. states formed to coordinate and manage fishery resources along the Atlantic coast of the United States) to recommend closing the commercial fishery along much of the coast in 2007 (ASMFC 2007).
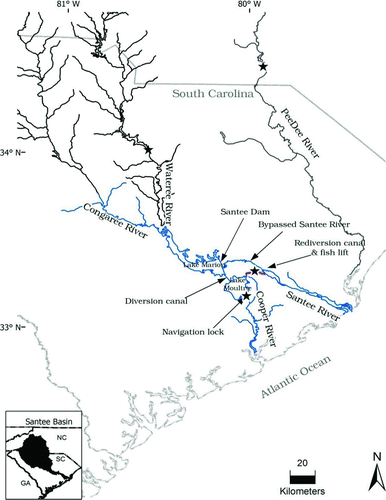
In contrast to most Atlantic coastal rivers, the abundance of American shad in the Santee–Cooper River basin of South Carolina has grown substantially over the last several decades, making this population one of the largest on the Atlantic coast (ASMFC 2007). The increase in abundance was presumably due to past human-induced, hydrological changes in the Santee and Cooper rivers (Hill Citation2009). Specifically, both the Santee and Cooper rivers were impounded in 1942, creating two reservoirs (Lake Marion and Lake Moultrie) that are connected by a diversion canal (). During this period, shad abundance was declining (i.e., from commercial gill-net landings of 24,610 kg in 1960 to an average of 2,554 kg from 1979 to 1985; ASMFC 2007) because impoundments restricted the spawning migrations of these anadromous fish to the lower portions of these rivers. In 1986, a rediversion canal (including a hydroelectric facility) was constructed to connect Lake Moultrie to the Santee River () and the construction of a fish lift at the hydroelectric facility once again provided fish passage above Lake Moultrie. Pre- and postrediversion canal assessments of American shad abundance on the Santee River showed that, on average, landings were several orders of magnitude greater after the completion of this canal and fish lift (ASMFC 2007).
FIGURE 1 Locations (stars) at which American shad were sampled in the Santee–Cooper River basin and the Pee Dee River.
While American shad abundance in the Santee–Cooper basin has appeared to increase over the last several decades, access to historical spawning and foraging habitats has remained limited because of the construction of nearly 45 other impoundments that inhibit upstream fish passage (Hill Citation2009). In an effort to reestablish populations in upstream river reaches of the Santee–Cooper basin, state and federal agencies have developed a multifaceted restoration approach relying on hatchery augmentations, relocations of adults, and the construction of fish passage structures (Hill Citation2009).
A supportive breeding program, which is defined as bringing part of a natural population into captivity for reproduction and the release of their offspring into the wild (Ryman and Laikre Citation1991), can be an important tool for restoration initiatives; however, adverse genetic change to wild populations can occur if such a program is not correctly implemented and monitored. Potentially harmful effects of hatchery augmentation on existing aquatic gene pools are well known (Meffe Citation1992; Araki et al. Citation2007b; Laikre et al. Citation2010) and have been studied for at least three decades. Genetic monitoring (i.e., quantifying the temporal changes in population genetic metrics) of hatchery releases can be used as a tool to evaluate potentially harmful genetic effects (Schwartz et al. Citation2007) and is becoming a standard part of many restoration and conservation initiatives relying on supportive breeding (Dowling et al. Citation2005; Araki et al. Citation2007a; Thériault et al. Citation2011).
The goal of this study was to provide an initial assessment of genetic parameters for the genetic monitoring of American shad in the Santee–Cooper basin. We accomplished this by (1) evaluating the extent of American shad population genetic substructure within the Santee–Cooper basin, (2) establishing an estimate of genetic diversity for Santee–Cooper American shad prior to hatchery augmentation, (3) comparing preexisting levels of genetic diversity between 2009 wild prespawning adults and those used for broodstock, and (4) assessing the feasibility of genetic markers as a molecular tag to estimate hatchery return rates.
METHODS
The Santee–Cooper River basin consists of six major rivers: the Santee, Cooper, Wateree, Broad, Saluda, and Congaree rivers. The Broad and Saluda rivers converge to form the Congaree River, and the Congaree and Wateree rivers converge to form Lake Marion and then Lake Moultrie. From Lake Moultrie, water can flow to either the Santee or the Cooper River () and then to the Atlantic Ocean. To discern stock structure in this basin, sampling tributaries from throughout the basin would be ideal; however, sampling from throughout the basin is problematic because American shad migration is prevented by impoundments on the lower Broad, Saluda, and Wateree rivers. Specifically, the Wateree River is impounded by Wateree Hydroelectric Dam at rkm 122 (measured from its confluence with the Congaree River), the Saluda River is impounded by Saluda Dam at rkm 66, and the Broad River is impounded at rkm 3.22 by the Columbia Diversion dam (the Columbia Diversion dam has a vertical slot fishway, but it was opened to passage only recently, circa 2007). Given the limited migration capabilities of American shad in the Santee–Cooper basin and the lack of knowledge about spawning locations above Lake Moultrie, we chose to sample American shad over the course of the spawning run as they migrated through the fish lift located below St. Stephen Dam on Lake Moultrie (n = 255; 83 were sent to a hatchery to be used for broodstock; the remainder were assumed to be fish that would have spawned in the wild and thus were designated as wild). Our sampling rationale was that if genetic structure were present in the basin (i.e., above Lake Moultrie) and American shad collected over the course of the spawning run were examined genetically, the sample should deviate significantly from Hardy–Weinberg and gametic equilibriums (i.e., the Wahlund effect). Although spawning may occur in the Cooper and Santee rivers below Lake Moultrie, recruitment in these reaches is probably minimal because the high-salinity waters of the estuaries limit egg and larval development. We augmented collection efforts by sampling individuals from the Wateree (n = 23), Congaree (n = 2), Cooper (n = 88; fish passage can occur on the Cooper River through a navigation lock at Pinopolis Dam), and Pee Dee (n = 43; the next river north of the Santee–Cooper) rivers over the course of the spawning run (samples from the Congaree were excluded from the analyses due to the limited sample size). Shad samples were collected by state and federal agency biologists via boat electrofishing or taken directly from the fish lift below St. Stephen Dam (see and for specific sampling information). A tissue clip from each sample was preserved in 95% nondenatured ethanol and sent to Warm Springs Conservation Genetics Laboratory to be analyzed and archived.
TABLE 1 Summary information for American shad sampling in the Santee–Cooper and Pee Dee River basins. Fish were sampled over the course of the spawning run except for the Pee Dee River; N = the sample size.
Genomic DNA was extracted from a portion of the ethanol-preserved fin clip using the DNeasy Blood and Tissue kit (QIAGEN, Valencia, California) protocol. We used a suite of 11 microsatellite markers known to amplify in American shad (Julian and Bartron Citation2007). A multiplex polymerase chain reaction (PCR) was performed on the following three sets of primers (fluorescently labeled dyes for the forward primers are noted in parentheses): set A consisted of primers AsaD30 (6-FAM), AsaD31 (VIC), and AsaD429 (NED); set B consisted of primers AsaB20 (VIC), AsaC59 (6-FAM), AsaD312 (6-FAM), and AsaD55 (NED); and set C consisted of primers AsaC249 (6-FAM), AsaC334 (6-FAM), and AsaD42 (NED). A single-reaction PCR was performed for AsaD392 (NED) primers. All PCR amplifications were performed in 20-μL reactions using the following reaction components: 1× Taq reaction buffer (Applied Biosystems, Foster City, California), 3.75 mM MgCl2, 0.423 mM of each deoxynucleotide triphosphate, 0.25 μM of each primer, and 0.08 U Taq polymerase (Applied Biosystems). The PCR conditions were as follows: an initial denaturation at 94°C (10 min), followed by a touchdown procedure involving 33 cycles and consisting of denaturing (94°C for 30 s), annealing, and extension (74°C for 30 s) cycles, where the initial annealing temperature was 56°C but decreasing by 0.2°C per cycle for 30 s.
Prior to electrophoresis, 2 μL of a 1:100 dilution of PCR product was mixed with a 8-μL solution containing 97% formamide and 3% Genescan LIZ 500 size standard (Applied Biosystems). Microsatellite reactions were visualized with an ABI 3130 genetic analyzer (Applied Biosystems) using fluorescently labeled forward primers and analyzed using GeneMapper software version 3.7 (Applied Biosystems).
Tests for gametic disequilibrium (all pairs of loci per sampling site) and locus conformance to Hardy–Weinberg equilibrium (HWE; for each locus in the sampling site) were implemented using GENEPOP version 4.0.10 (Raymond and Rousset Citation1995). Significance levels for all simultaneous tests were adjusted using a sequential Bonferroni correction (Rice Citation1989). In cases in which the observed genotype frequencies deviated significantly from HWE expectations, the program MICRO-CHECKER version 2.2 (van Oosterhout et al. Citation2004) was used to infer the most probable cause of the departures.
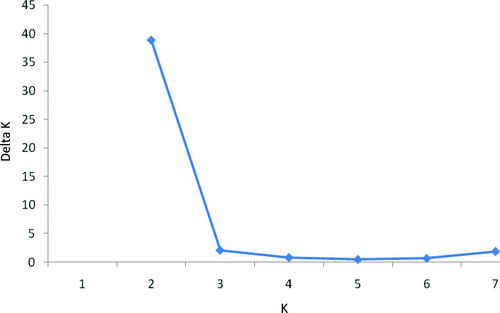
To assess the degree of population differentiation in the Santee–Cooper basin (i.e., excluding the Pee Dee River samples), we first compared the per-locus genic frequency distributions from each sampling locality using the genic differentiation option in GENEPOP version 4.0.10 (Raymond and Rousset Citation1995) with the default parameter settings. We also calculated D EST (a measure of population differentiation based on genetic polymorphism data [Jost Citation2008]) between sampling sites using the program DEMEtics (Gerlach et al. Citation2010), where confidence in the null hypothesis of no genetic differentiation among sampling sites was assessed via bootstrap resampling (500 replicates as implemented in DEMEtics). Analysis of population structure was performed using a Bayesian-based clustering algorithm implemented in the program STRUCTURE version 2.3.3 (Pritchard et al. Citation2000; Falush et al. Citation2003). The program STRUCTURE assumed no a priori sampling information; rather, individuals were probabilistically assigned to groups in such a way as to achieve Hardy–Weinberg and gametic equilibriums. The program STRUCTURE was run with three independent replicates for K (i.e., distinct populations or gene pools), with K set from one to eight. The burn-in period was 50,000 replicates, followed by 500,000 Monte Carlo simulations run under a model that assumed no admixture and independent allele frequencies. Estimates of ΔK (Evanno et al. Citation2005) and individual assignment patterns were used to determine the most likely value of K.
No population substructure was observed among the samples (n = 366) collected in the Santee–Cooper basin (see below); therefore, we treated the samples collected from throughout the basin as one population and calculated basic estimators of genetic diversity for American shad in the Santee–Cooper basin (i.e., prior to supportive breeding). The fixation index, along with genetic diversity in the form of the per-locus and average number of alleles, observed heterozygosity, and expected heterozygosity, were calculated using the computer program GenAIEx version 6.4 (Peakall and Smouse Citation2006). We also calculated the genetic composition for a random sample of the 2009 broodstock whose progeny were used for an initial hatchery release. Specifically, we tested for homogeneity in genic distributions, average number of alleles, observed heterozygosity, and expected heterozygosity between a random sample of the 2009 broodstock (n = 83) and wild fish collected from throughout the Santee–Cooper system during the course of the study (n = 283). Tests for significance were conducted using the Wilcoxon rank-sum test (Sokal and Rohlf Citation1995) as implemented in S-Plus version 7.0 (Insightful Corporation) except for the genic distribution test, which was implemented as above.
We also wanted to gain knowledge about the genetic diversity in the Pee Dee River (the next system to the north) and the amount population structure and genetic differentiation between the Santee–Cooper basin and the Pee Dee River. In this regard, we estimated genetic diversity for samples collected in the Pee Dee River and compared these estimates with those obtained from the Santee–Cooper basin. The estimation of genetic diversity was as described above, except that the program HP-RARE (Kalinowski Citation2005) was used to estimate allelic richness (the average number of alleles was not estimated due to differences in sample sizes). Tests for significance were conducted as outlined above. We also assessed the degree of differentiation between American shad in the Santee–Cooper basin and Pee Dee River as outlined above (i.e., comparison of genic distributions, computation of D EST, and use of STRUCTURE).
Molecular tags rely on the premise that offspring can be confidently assigned to their respective parent via parentage analysis and that type II error is minimal. We performed a simulation-based assessment of confidence using the computer program PAPA version 2.0 (Duchesne et al. Citation2002). Allocations were based on maximum likelihood using a 3% nonuniform error model distributed on the next adjacent allele (Duchesne et al. Citation2002). A genotyping error rate of 3% was estimated by regenotyping the broodstock for each locus, counting the errors for each locus, and averaging these values for a global estimate. We used the preparental procedure to simulate pseudoparent and offspring data. The estimated number of pseudo-collected males and females (314 and 255, respectively) was based on the numbers collected as broodstock from the fish lift at the rediversion canal below St. Stephen Dam. We assumed a closed system in which all broodstock were genotyped for the loci used in this study (i.e., we set the estimated number of pseudo-uncollected parents to zero). The allele frequency file used to simulate offspring and parents was generated by using a random sample of 96 individuals from the Santee–Cooper basin genotyped for all 10 loci. Parentage analysis was then performed on these files to assess assignment success. Simulations were performed using 1,000 iterations. We also explored the statistical confidence in parentage analysis for a much larger number of hatchery broodstock. In this scenario, all simulations were as above except that 5,000 males and females were used for the estimated number of pseudo-collected males and females.
Molecular tags will be used to discern hatchery offspring from their wild conspecifics; therefore, we also estimated the level of type II error for parentage analysis (i.e., the probability that a wild fish would be incorrectly assigned to hatchery parents). We estimated the type II error by first generating wild pseudo-offspring (n = 1,000) from an allele frequency file of the wild population (n = 96). Next, we generated hatchery pseudomale and -female parents based on hatchery allele frequencies (n = 83). Type II error was assessed using either 569 (314 males and 255 females) or 10,000 (5,000 males and 5,000 females) hatchery parents. We also assess the influence of mating history on type II error by performing parentage analysis with and without a mating history file. Specifically, the mating history file assumed that hatchery parents were spawned volitionally in tanks containing 25 males and females (this was similar to the tank spawning conditions over the course of this study). The genotyping error model was as above.
TABLE 2 Probability values for tests of genic differentiation among sampling sites for American shad in the Santee–Cooper basin based on 10 microsatellite loci. All comparisons were nonsignificant following sequential Bonferroni correction (Rice Citation1989).
RESULTS
A total of 409 American shad were analyzed using 11 microsatellite markers. For each sampling site, all loci conformed to per-locus HWE after sequential Bonferroni corrections (all P > 0.01 per sampling site; n = 11 comparisons per sampling site for an α = 0.005) except AsaC59 and AsaC249. Microsatellite marker AsaC249, which had a general excess of homozygotes for most allele size-classes and was suggestive of null alleles, was removed from all subsequent analyses. Marker AsaC59 deviated from HWE for only one (the rediversion canal collection comprising hatchery broodstock) of five sampling sites, indicating possible genotyping errors; however, this locus still deviated from HWE after regenotyping individuals from this sampling site (no genotyping errors were found). No sampling site showed significant evidence of gametic disequilibrium after sequential Bonferroni correction (all P > 0.01 per sampling site; n = 45 comparisons per sampling site for an α = 0.001).
There was no significant heterogeneity in genic distributions among sampling sites in the Santee–Cooper basin after sequential Bonferroni correction (all P > 0.01; 10 comparisons for an α = 0.005; ); likewise, there was no significant differentiation among pairwise comparisons of multilocus estimates of D EST, which ranged from –0.001 to +0.022 () after sequential Bonferroni correction (all P > 0.006; n = 10 comparisons for an α = 0.005; ). The program STRUCTURE revealed that the most probable number of groups was two using the Evanno et al. (Citation2005) method (); however, the proportion of sampled individuals at each sampling site was symmetrical for all K-values 2–8 (data not shown), which is an indication of no population structure (Evanno et al. Citation2005).
TABLE 3 Pairwise comparisons of D EST estimates for American shad sampling locations in the Santee–Cooper basin. The values above the diagonal are D EST values based on 10 microsatellite loci; those below the diagonal are the corresponding P-values. All comparisons were nonsignificant following sequential Bonferroni correction (Rice Citation1989).
FIGURE 2 Values of ΔK averaged across three replicate simulations versus the simulated number of groups (K) observed in the data. The simulation results indicate that the most plausible value for K represented by American shad from the Santee–Cooper basin is 2, as evidenced by the distinct reduction in ΔK from K = 2 to K = 3. Note, however, that the proportion of sampled individuals to each group was symmetrical for all K = 2–8, which is an indication of no population structure (Evanno et al. Citation2005).
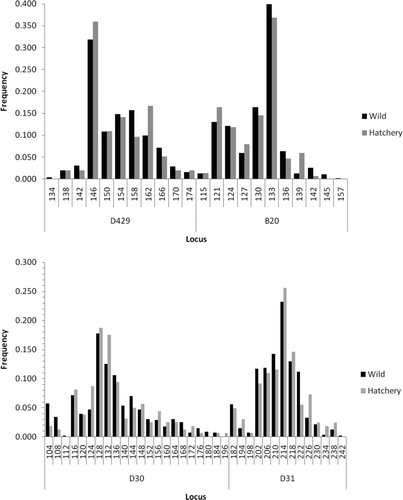
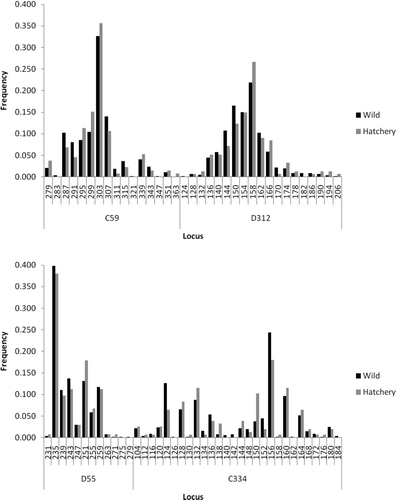
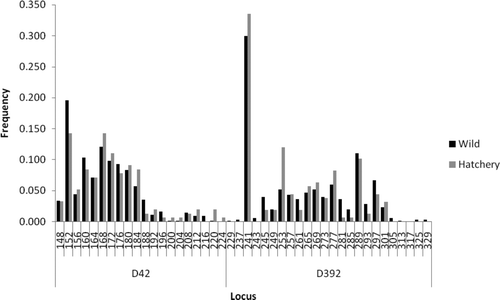
A comparison of genetic diversity between samples representing the rediversion canal collection comprising hatchery broodstock and wild fish collected from throughout the Santee–Cooper basin () showed no significant differences in the average number of alleles (15.00 versus 17.30; P = 0.36), average observed heterozygosity (0.823 versus 0.814, P = 0.63), or average expected heterozygosity (0.847 versus 0.852, P = 0.84). There was also no significant difference in the average fixation index (0.030 versus 0.045, P = 0.24) as well as in the distribution of alleles for each locus (all P > 0.06; see Appendix).
TABLE 4 Comparison of genetic diversity and the fixation index between wild and hatchery samples of American shad from the Santee–Cooper basin at various microsatellite loci. We tested for homogeneity in the average number of alleles (NA), observed heterozygosity (HO ), expected heterozygosity (HE ), and the fixation index (F) between a random sample of the 2009 broodstock (Santee River, hatchery; n = 83) and wild fish collected from throughout the Santee–Cooper system during the course of the study (n = 283). All comparisons between average values were nonsignificant.
Genic differentiation tests found no significant difference (P = 0.11) between American shad in the Santee–Cooper and Pee Dee rivers using neutral microsatellite markers. Estimates of D EST and STRUCTURE analyses corroborate the genic tests. The estimate of D EST averaged across loci was 0.020, and although it was significantly different from zero (P = 0.02) it was similar to that reported within the Santee–Cooper basin. Analyses using STRUCTURE indicated that the proportion of sampled individuals to each sampling site was symmetrical for all K-values 2–8, suggesting no population structure. We also found similar estimates of genetic diversity between American shad in the Santee–Cooper and Pee Dee rivers () with no significant differences for allelic richness (12.84 versus 13.12; P = 0.74), average observed heterozygosity (0.814 versus 0.814, P = 0.63), or average expected heterozygosity (0.852 versus 0.830, P = 0.74). The average fixation index was also similar (0.045 versus 0.019, P = 0.71).
TABLE 5 Comparison of genetic diversity and the fixation index between American shad from the Santee–Cooper (n = 283) and Pee Dee River basins (n = 43) at various microsatellite loci. The estimates of allelic richness are based on 43 individuals. All comparisons between average values are nonsignificant. See for additional information.
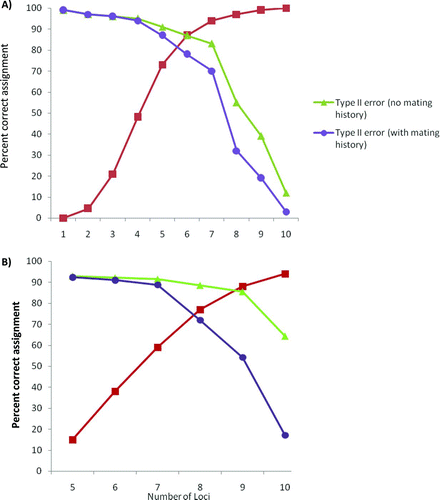
The simulation results suggested that the markers developed by Julian and Bartron (Citation2007) could be used as molecular tags. Upon regenotyping 96 individuals, our estimate of an average genotyping error rate over all loci was 3%. Simulations using this error rate and 314 males and 255 females as parents suggested that more than seven loci (in any combination) would be necessary to match progeny successfully (>95%) to their respective broodstock parent pair (). The probability of a type II error with and without assuming a mating history for the broodstock was 3% and 12%, respectively. Simulation using 5,000 males and 5,000 females as broodstock indicated a 94% parent assignment success rate would be achieved using 10 loci and a 3% genotyping error rate (); however, the type II error associated with these analyses was 17% and 64% with and without assuming a mating history, respectively ().
FIGURE 3 Percent parentage assignment success versus number of loci for simulated American shad parents and progeny (red curves), along with type II errors with and without data on mating history. The simulated numbers of broodstock males and females were (A) 314 and 255 and (B) 5,000 and 5,000. The simulations assumed a 3% genotyping error rate and that all broodstock were genotyped for all loci. Type II errors were estimated by performing parentage analysis on simulated wild progeny and hatchery broodstock. The inferred mating history allowed 25 males and 25 females to spawn together and was similar to hatchery conditions.
DISCUSSION
Declines in North American ichthyofaunal diversity have led to the widespread use of hatchery propagation to boost population sizes and recover threatened or endangered populations (Minckley Citation1995; George et al. Citation2009). Hatchery augmentation is meant to have positive demographic consequences; however, genetic risks associated with the release of hatchery fish are well known (Nelson and Soule Citation1987; Miller and Kapuscinski Citation2003). Unfortunately, monitoring and evaluation of reintroduced or augmented populations, if done at all, often involves noting the presence of (or an increase in) the numbers of the target species at a site (Ostermann et al. Citation2001); therefore, potential adverse genetic changes to wild populations (via augmentation) can go unnoticed (Araki et al. Citation2007b; Thériault et al. Citation2010, 2011). Genetic monitoring of hatchery releases is important because it can provide (1) an understanding of the present and historical levels of genetic diversity in a population or species (i.e., prior to the release of hatchery individuals), (2) an assessment of the alteration of these characteristics after release, and (3) an evaluation of the biological consequences of hatchery releases (Schwartz et al. Citation2007; Laikre et al. Citation2010). Our study was initiated as an initial assessment of the genetic diversity within the Santee–Cooper basin with the intention of estimating the native population genetic structure and level of diversity prior to hatchery release and the genetic composition of the broodstock used to initiate augmentation of the wild population.
The evaluation of genetic stock structure is an essential first step in choosing an appropriate donor population to use in supportive breeding because an inappropriate choice of broodstock may reduce the overall fitness of the population by disrupting locally adapted gene complexes (Miller and Kapuscinski Citation2003). Fish population genetic structure typically coincides with differences among major river basins; however, anadromous fish such as Chinook salmon Oncorhynchus tshawytscha normally migrate to their natal river to spawn and often demonstrate fine-scale genetic structure in a river or river system (Neville et al. Citation2006; Narum et al. Citation2008). Like salmon, American shad migrate to their natal river to spawn (Melvin et al. Citation1986; Hasselman et al. Citation2010); therefore, unrecognized population structure could exist within the Santee–Cooper river system. Our data provided no evidence of fine-scale population structure with in the Santee–Cooper basin. Microsatellite markers were generally in accordance with HWE and gametic disequilibrium, tests for genetic differentiation indicated no differentiation between sampling sites, and while Bayesian cluster analysis revealed the potential for two distinct groups, the proportion of sampled individuals to each sampling site was symmetrical for all K-values, which was an indication of no population structure (Evanno et al. Citation2005). Our findings were consistent with those of previous genetic (Brown et al. Citation2000; Waters et al. Citation2000; Hasselman Citation2010) and otolith microchemistry studies (Hendricks et al. Citation2002; Walther et al. Citation2008) that showed limited site fidelity in American shad spawning at fine spatial scales, presumably because of a high level of straying or gene flow among tributaries.
Although there was a lack of observed fine-scale genetic structure in the Santee–Cooper basin, can the same be said for larger spatial scales? The observed patterns of genetic variation and population structure indicated very little, if any, genetic differentiation between American shad at a larger spatial scale (i.e., the drainage level), at least between the Santee–Cooper and Pee Dee rivers. Genic homogeneity was observed between American shad in the Santee–Cooper and Pee Dee rivers, and the level of genetic differentiation (D EST), while significant between river systems, was similar to the values found among sampling sites in the Santee–Cooper. A lack of genetic differentiation was surprising given the life history of this species (philopatric); however, it is consistent with Hasselman (Citation2010), who found nonsignificant genic heterogeneity and weak genetic differentiation among American shad from the Waccamaw, Cooper, and Edisto rivers (the Waccamaw is the next river system north of the Pee Dee, and the Edisto is the river system directly south of the Santee–Cooper). Hasselman (Citation2010) offered several hypotheses for the observed lack of differentiation in southern spawning runs of American shad (e.g., stock transfer); however, testing competing hypotheses was beyond the scope of our study.
The observed lack of differentiation between American shad in the Santee–Cooper and Pee Dee systems should be treated with caution because a lack of differentiation at presumably neutral markers does not necessarily constitute proof of no differentiation between these spawning runs. It is important to understand that discrepancies in genetic differentiation can be found between neutral and quantitative markers, especially in recently diverged populations (Bekessy et al. Citation2003; Leinonen et al. Citation2008). Recently diverged populations that have a large effective size can show a lack of differentiation at neutral loci even if they are demographically and reproductively independent—having accumulated genetic differentiation at nonneutral loci via natural selection (Laikre et al. Citation2005). Genetic differences may still exist between the Pee Dee and Santee–Cooper populations of American shad but were simply not observed with the neutral markers used in our study; therefore, the use of a local broodstock (in this case from the Santee–Cooper basin) is recommended because this practice will minimize the risk of outbreeding depression (Miller and Kapuscinski Citation2003).
Once the choice of the donor population has been made for a supportive breeding program, the actual collection of hatchery broodstock will require careful consideration. To maximize genetic and ecological (adaptive) diversity, a primary goal for any supportive breeding program should be the maintenance of similar genetic resources and life history patterns between the hatchery broodstock and wild populations (Lynch and O’Hely Citation2001; Wedekind Citation2002). Therefore, both collection procedures (i.e., collecting broodstock from over the course of the spawning run) and the numbers of broodstock should be important considerations in minimizing the risks of domestication selection and inbreeding depression (Allendorf Citation1993; Miller and Kapuscinski Citation2003). The American shad supportive breeding program for the Santee–Cooper basin outlined two adaptive scenarios for producing juvenile hatchery-reared shad: collect spawning shad and manually strip and fertilize the eggs, or induce the shad to spawn by means of hormone injections. To reach stocking targets (Hill Citation2009), the plan indicated that approximately 400–800 ripe females and 200–400 males would be necessary for the first spawning scenario and that 1,000–2,000 females (and a comparable number of males) would be necessary for the second scenario. For both scenarios, the plan recommended that shad collections occur during a 6-week period when water temperatures were approximately 15–25°C (roughly March–May and representative of the spawning run). In 2009, which was the initial year of the supportive breeding program, approximately 314 males and 255 females were collected for broodstock at the rediversion canal over the course of a 2-month period corresponding to the spawning run (March–April). While approximately 100–200 parents (equal number of males and females) have been suggested as the minimum number of broodstock needed to maintain most alleles in a population (Kincaid Citation1983; Allendorf and Luikart Citation2007), we assessed this recommendation by testing whether estimates of genetic variation differed between the sampled broodstock and the wild population. Our data indicated that the recommended number was sufficient to maintain similar levels of genetic variation (number of alleles and observed heterozygosity) between the broodstock and wild populations. What is not known, however, is whether this similarity will still be maintained in the progeny used for augmentation.
The effective population size (Ne ), which refers to the size of an ideal population experiencing the same rate of random genetic change over time as the real population under consideration (Wright Citation1938), is a key parameter in conservation biology because it measures the rate of loss of genetic variation and increase in inbreeding (Wright Citation1938). General hatchery conservation goals based on genetic considerations are frequently established at Ne = 50 to minimize inbreeding depression and Ne = 500 to maintain sufficient evolutionary potential (Franklin Citation1980; Franklin and Frankham Citation1998; Miller and Kapuscinski Citation2003). It is often assumed that the Ne of a hatchery group can be estimated from the number of males and females used as broodstock to produce the hatchery group. However, large differences in reproductive success among broodstock can reduce Ne to a level below the one that was expected and increase the effects of artificial selection in captivity (Allendorf Citation1993). In 2009, American shad females were hormone-implanted and then allowed to spawn volitionally in tanks with approximately equal numbers of males. If a high proportion of the hatchery broodstock produced progeny in fairly equal numbers, we would expect that similar genetic resources would be maintained in their progeny. However, parentage analyses in a pilot study of American shad from the Roanoke River indicated that of the 39 females and 35 males allowed to tank spawn, only 4 females (mean number of offspring = 0.82; variance = 6.41) and 15 males (mean number of offspring = 0.91; variance = 2.26) produced the progeny that were randomly sampled (n = 66) for parentage analysis (G. R. Moyer, U.S. Fish and Wildlife Service, unpublished data). Thus, the observed value of Ne (8) was much less than that which was expected (73). The cause of such a discrepancy is often due to hatchery spawning protocols. As in most American shad hatchery programs, the Roanoke study relied on the volitional tank spawning of a large number of males and females because of space limitations in the hatchery. In such circumstances, not all females and males will produce progeny; thus, the observed value of Ne will often be less than that expected (Crow and Denniston Citation1988). Further, equalizing reproductive success among the broodstock after tank spawning is arduous and will also contribute to a lower observed value of Ne (Crow and Denniston Citation1988). In this type of situation, one possible strategy is to estimate the number of contributing males and females and their reproductive success (via parentage analysis of a subset of offspring collected from each tank) so that these parameters are known prior to hatchery release (as was done in the Roanoke River study above). In this way, the potential effects of family-correlated survival can be closely monitored (Moyer et al. Citation2007).
As shown above, supportive breeding can have important genetic consequences for the broodstock. It can also accelerate the loss of genetic variation and reduce the fitness of the augmented population (Ryman and Laikre Citation1991; Araki et al. Citation2007b; Thériault et al. Citation2011). Unfortunately, the effects of supportive breeding on natural populations are difficult to predict (Wang and Ryman Citation2001) and highlight the importance of genetic monitoring and adaptive management. To assess the effects of supportive breeding on the wild population requires knowledge of the census and effective population sizes for both the returning hatchery progeny and the wild population (Ryman and Laikre Citation1991; Araki et al. Citation2007c; Moyer et al. Citation2007) and an understanding of the relative reproductive success of hatchery fish and their wild counterparts (Araki et al. Citation2007a; Thériault et al. Citation2011). Molecular markers can be used to estimate such demographic parameters and monitor whether captive individuals are being recruited to the natural population (Schwartz et al. Citation2007). Essentially, molecular markers can be utilized just like conventional tags but are not subject to failure because of battery life, tissue regeneration, or loss of an external or internal tag (Guy et al. Citation1996). Molecular markers also serve as a means to tag relatively small organisms and are a part of the fish until death—a considerable advantage over many conventional tags. Molecular tags are increasingly being used in lieu of physical tags to identify individuals (Schwartz et al. Citation2007); yet there are technical aspects to this approach that must be assessed prior to field implementation.
The premise of a molecular tag is that offspring can be confidently matched to their respective parents via parentage analysis. Mutations, scoring errors, relatedness, and gametic disequilibrium can lead to circumstances in which all hypothesized parent–offspring pairings possess finite probabilities of being true (Jones and Ardren Citation2003). Therefore, a major goal prior to the implementation of a molecular tagging study should be to assess the statistical confidence in parentage assignments. Our simulation-based assessment of confidence found that eight microsatellite markers were sufficient to confidently (>95% assignment success of all progeny to their respective mating pairs) discriminate progeny from 569 (314 males and 255 females) contributing broodstock (i.e., assuming an overall genotyping error rate of ≤3% and that all broodstock had been genotyped for all loci). Further simulations indicated that 10 loci can be used to perform parentage analysis of broodstocks of at least 10,000 individuals (5,000 males and 5,000 females) with 94% success in matching progeny to their respective parents (again assuming a 3% genotyping error rate and that all broodstock have been genotyped). However, for the latter scenario, the probability that a wild fish would be incorrectly assigned to hatchery parents was high (>60%) unless the mating history was incorporated into the analysis. Knowledge of mating history greatly improved assignment success by reducing the number of possible parent pairs, a finding similar to that of Olsen et al. (Citation2001). Therefore, when working with large numbers of broodstock, it will be imperative to keep accurate records on mating history so that type II errors can be minimized. The degree of assignment success is a testament to the diverse allelic variation present in these markers and indicates that these markers, when used as a molecular tag, can estimate the demographic parameters essential for monitoring the effects of supportive breeding on the wild population of American shad in the Santee–Cooper basin.
In conclusion, the importance of genetic variation as a basis for future biological evolution and the long-term viability of populations, species, and ecosystems is well established (Frankel and Soule Citation1981; Frankham Citation1995). Therefore, identifying and monitoring processes that are likely to have adverse impacts on the conservation of natural populations are becoming increasingly important. Unfortunately, most monitoring programs do not take full advantage of the potential afforded by molecular genetic markers (Schwartz et al. Citation2007; Laikre Citation2010). The genetic data collected in this study will serve as a point of reference in the ongoing effort to monitor temporal changes in the population-genetic metrics and other population data generated at regular intervals as part of the Santee–Cooper American shad restoration initiative. In this way, these data will provide a understanding of the effectiveness (both in terms of increasing the census size and in terms of maintaining the long-term viability of the population) of hatchery augmentation for American shad in the Santee–Cooper basin.
FIGURE A1 Comparison of allele frequencies at various microsatellite loci between hatchery and wild American shad from the Santee–Cooper River basin.
ACKNOWLEDGMENTS
We would like to thank W. Booker, M. Cantrell, D. Coughlan, R. Heise, A. Hill, B. Post, M. Quattlebaum, L. Rose, F. Sessions, and W. Starnes for sample collection. We also thank M. Bartron and S. Julian for providing initial multiplex PCR reaction conditions. was provided by M. Cantrell. Under the Santee Cooperative Accord, Duke Energy Carolinas, LLC, and South Carolina Electric and Gas Company provided funding to G.R.M. for this project. Use of trade names throughout the manuscript does not constitute endorsement by the United States government.
Subject editor: Kristina Miller, Fisheries and Oceans Canada, Pacific Biological Station, Nanaimo, British Columbia, Canada
REFERENCES
- Allendorf , F. W. 1993 . Delay of adaptation to captive breeding by equalizing family size . Conservation Biology , 7 : 416 – 419 .
- Allendorf , F. W. and Luikart , G. 2007 . Conservation and the genetics of populations , Cambridge , Massachusetts : Blackwell Scientific Publications .
- Araki , H. , Ardren , W. R. , Olsen , E. , Cooper , B. and Blouin , M. S. 2007a . Reproductive success of captive-bred steelhead trout in the wild: evaluation of three hatchery programs in the Hood River . Conservation Biology , 21 : 181 – 190 .
- Araki , H. , Cooper , B. and Blouin , M. S. 2007b . Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild . Science , 318 : 100 – 103 .
- Araki , H. , Waples , R. S. , Ardren , W. R. , Cooper , B. and Blouin , M. S. 2007c . Effective population size of steelhead trout: influence of variance in reproductive success, hatchery programs, and genetic compensation between life-history forms . Molecular Ecology , 16 : 953 – 966 .
- ASMFC (Atlantic States Marine Fisheries Commission) . 2007 . American shad stock assessment report , Washington , D.C. : ASMFC . Report 07-01
- Bekessy , S. A. , Ennos , R. A. , Burgman , M. A. , Newton , A. C. and Ades , P. K. 2003 . Neutral DNA markers fail to detect genetic divergence in an ecologically important trait . Biological Conservation , 110 : 267 – 275 .
- Brown , B. L. , Gunter , T. P. , Waters , J. M. and Epifanio , J. M. 2000 . Evaluating genetic diversity associated with propagation-assisted restoration of American shad . Conservation Biology , 14 : 294 – 303 .
- Crow , J. F. and Denniston , C. 1988 . Inbreeding and variance effective population numbers . Evolution , 42 : 482 – 495 .
- Dadswell , M. J. , Melvin , G. D. , Williams , P. J. and Themelis , D. E. 1987 . “ Influences of origin, life history, and chance on the Atlantic coast migration of American shad ” . In Common strategies of anadromous and catadromous fishes , Edited by: Dadswell , M. J. , Klauda , R. J. , Moffitt , C. M. , Saunders , R. L. , Rulifson , R. A. and Cooper , J. E. 313 – 330 . Bethesda , Maryland : American Fisheries Society, Symposium 1 .
- Dowling , T. E. , Marsh , P. C. , Kelsen , A. T. and Tibbets , C. A. 2005 . Genetic monitoring of wild and repatriated populations of endangered razorback sucker (Xyrauchen texanus, Catostomidae, Teleostei) in Lake Mohave, Arizona-Nevada . Molecular Ecology , 14 : 123 – 135 .
- Duchesne , P. , Godbout , M. H. and Bernatchez , L. 2002 . PAPA (package for the analysis of parental allocation): a computer program for simulated and real parental allocation . Molecular Ecology Notes , 2 : 191 – 193 .
- Evanno , G. , Regnaut , S. and Goudet , J. 2005 . Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study . Molecular Ecology , 14 : 2611 – 2620 .
- Facey , D. E. and Van Den Avyle , M. J. 1986 . “ Species profiles: life histories and environmental requirements of coastal fishes and invertebrates (South Atlantic)—American shad ” . Washington , D.C. : Army Corps of Engineers . U.S. Fish and Wildlife Service, Biological Report 82 (11.45), U.S, TR EL-82-4
- Falush , D. , Stephens , M. and Pritchard , J. K. 2003 . Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies . Genetics , 164 : 1567 – 1587 .
- Frankel , O. H. and Soule , M. E. 1981 . Conservation and evolution , Cambridge , , UK : Cambridge University Press .
- Frankham , R. 1995 . Conservation genetics . Annual Review of Genetics , 29 : 305 – 327 .
- Franklin , I. R. 1980 . “ Evolutionary changes in small populations ” . In Conservation biology: an evolutionary-ecological perspective , Edited by: Soule , M. E. and Wilcox , B. A. 135 – 149 . Sunderland , Massachusetts : Sinauer .
- Franklin , I. R. and Frankham , R. 1998 . How large must populations be to retain evolutionary potential? . Animal Conservation , 1 : 69 – 70 .
- George , A. L. , Kuhajda , B. R. , Williams , J. D. , Cantrell , M. A. , Rakes , P. L. and Shute , J. R. 2009 . Guidelines for propagation and translocation for freshwater fish conservation . Fisheries , 34 : 529 – 545 .
- Gerlach , G. , Jueterbock , A. , Kraemer , P. , Deppermann , J. and Harmand , P. 2010 . Calculations of population differentiation based on GST and D: forget GST but not all of statistics! . Molecular Ecology , 19 : 3845 – 3852 .
- Guy , C. S. , Blankenship , H. L. and Nielsen , L. A. 1996 . “ Tagging and marking ” . In Fisheries techniques, , 2nd edition , Edited by: Murphy , B. R. and Willis , D. W. 353 – 383 . Bethesda , Maryland : American Fisheries Society .
- Hasselman , D. J. 2010 . Spatial distribution of neutral genetic variation in a wide ranging anadromous clupeid, American shad (Alosa sapidissima) , Halifax , Nova Scotia : Doctoral dissertation. Dalhousie University .
- Hasselman , D. J. , Bradford , R. G. and Bentzen , P. 2010 . Taking stock: defining populations of American shad (Alosa sapidissima) in Canada using neutral genetic markers . Canadian Journal of Fisheries and Aquatic Sciences , 67 : 1021 – 1039 .
- Hendricks , M. L. , Hoopes , R. L. , Arnold , D. A. and Kaufmann , M. L. 2002 . Homing of hatchery-reared American shad to the Lehigh River, a tributary to the Delaware River . North American Journal of Fisheries Management , 22 : 243 – 248 .
- Hill , A. K. 2009 . “ The Santee River Basin Accord: restoring diadromous fish through prioritization of subbasins ” . In Challenges for diadromous fishes in a dynamic global environment , Edited by: Haro , A. J. , Smith , K. L. , Rulifson , R. A. , Moffitt , C. M. , Klauda , R. J. , Dadswell , M. J. , Cunjak , R. A. , Cooper , J. E. , Beal , K. L. and Avery , T. S. 769 – 782 . Bethesda , Maryland : American Fisheries Society, Symposium 69 .
- Jenkins , R. E. and Burkhead , N. M. 1994 . Freshwater fishes of Virginia , Bethesda , Maryland : American Fisheries Society .
- Jones , A. G. and Ardren , W. R. 2003 . Methods of parentage analysis in natural populations . Molecular Ecology , 12 : 2511 – 2523 .
- Jost , L. 2008 . GST and its relatives do not measure differentiation . Molecular Ecology , 17 : 4015 – 4026 .
- Julian , S. and Bartron , M. 2007 . Microsatellite DNA markers for American shad (Alosa sapidissima) and cross-species amplification within the family Clupeidae . Molecular Ecology Notes , 7 : 805 – 807 .
- Kalinowski , S. T. 2005 . HP-Rare: a computer program for performing rarefaction on measures of allelic diversity . Molecular Ecology Notes , 5 : 187 – 189 .
- Kincaid , H. L. 1983 . Inbreeding in fish populations used for aquaculture . Aquaculture , 33 : 215 – 227 .
- Laikre , L. 2010 . Genetic diversity is overlooked in international conservation policy implementation . Conservation Genetics , 11 : 349 – 354 .
- Laikre , L. , Palm , S. and Ryman , N. 2005 . Genetic population structure of fishes: implications for coastal zone management . Ambio , 34 : 111 – 119 .
- Laikre , L. , Schwartz , M. K. , Waples , R. S. and Ryman , N. 2010 . Compromising genetic diversity in the wild: unmonitored large-scale release of plants and animals . Trends in Ecology and Evolution , 25 : 520 – 529 .
- Leggett , W. C. and Carscadden , J. E. 1978 . Latitudinal variation in reproductive characteristics of American shad (Alosa sapidissima): evidence for population specific life history stages in fish . Journal of the Fisheries Research Board of Canada , 35 : 1469 – 1478 .
- Leinonen , T. , O’Hara , R. B. , Cano , J. M. and Merilä , J. 2008 . Comparative studies of quantitative trait and neutral marker divergence: a meta-analysis . Journal of Evolutionary Biology , 21 : 1 – 17 .
- Lynch , M. and O’Hely , M. 2001 . Captive breeding and the genetic fitness of natural populations . Conservation Genetics , 2 : 363 – 378 .
- Mansueti , R. J. and Kolb , H. 1953 . A historical review of the shad fishery of North America , Solomons , Maryland : Chesapeake Biological Laboratory . Publication 97
- Meffe , G. K. 1992 . Techno-arrogance and halfway technologies: salmon hatcheries on the Pacific Coast of North America . Conservation Biology , 6 : 350 – 354 .
- Melvin , G. D. , Dadswell , M. J. and Martin , J. D. 1986 . Fidelity of American shad, Alosa sapidissima (Clupeidae), to its river of previous spawning . Canadian Journal of Fisheries and Aquatic Sciences , 46 : 640 – 646 .
- Miller , L. M. and Kapuscinski , A. R. 2003 . “ Genetic guidelines for hatchery supplementation programs ” . In Population genetics: principles and applications for fisheries scientists , Edited by: Hallerman , E. M. 329 – 355 . Bethesda , Maryland : American Fisheries Society .
- Minckley , W. L. 1995 . Translocation as a tool for conserving imperiled fishes: experiences in western United States . Biological Conservation , 72 : 297 – 309 .
- Moyer , G. R. , Blouin , M. S. and Banks , M. A. 2007 . The influence of family-correlated survival on Nb/N for progeny from integrated multi- and single-generation hatchery stocks of coho salmon (Oncorhynchus kisutch) . Canadian Journal of Fisheries and Aquatic Sciences , 64 : 1258 – 1265 .
- Narum , S. R. , Banks , M. , Beacham , T. D. , Bellinger , M. R. , Campbell , M. R. , Dekoning , J. , Elz , A. , Guthrie , C. M. III , Kozfkay , C. , Miller , K. M. , Moran , P. , Phillips , R. , Seeb , L. W. , Smith , C. T. , Warheit , K. , Young , S. F. and Garza , J. C. 2008 . Differentiating salmon populations at broad and fine geographical scales with microsatellites and single nucleotide polymorphisms . Molecular Ecology , 17 : 3464 – 3477 .
- Nelson , K. and Soule , M. E. 1987 . “ Genetical conservation of exploited fishes ” . In Population genetics and fishery management , Edited by: Ryman , N. and Utter , F. 345 – 368 . Seattle : University of Washington Press .
- Neville , H. M. , Isaak , D. J. , Dunham , J. B. , Thurow , R. F. and Rieman , B. E. 2006 . Fine-scale natal homing and localized movement as shaped by sex and spawning habitat in Chinook salmon: insights from spatial autocorrelation analysis of individual genotypes . Molecular Ecology , 15 : 4589 – 4602 .
- Olney , J. E. and Hoenig , J. M. 2001 . Managing a fishery under moratorium: assessment opportunities for Virginia's stocks of American shad . Fisheries , 26 ( 2 ) : 6 – 12 .
- Olsen , J. B. , Busack , C. , Britt , J. and Bentzen , P. 2001 . The aunt and uncle effect: an empirical evaluation of the confounding influence of full sibs of parents on pedigree reconstruction . Journal of Heredity , 92 : 243 – 247 .
- Ostermann , S. D. , Deforge , J. R. and Edge , W. D. 2001 . Captive breeding and reintroduction evaluation criteria: a case study of peninsular bighorn sheep . Conservation Biology , 15 : 749 – 760 .
- Peakall , R. and Smouse , P. E. 2006 . GENALEX 6: genetic analysis in Excel—population genetic software for teaching and research . Molecular Ecology Notes , 6 : 288 – 295 .
- Pritchard , J. K. , Stephens , M. and Donnelly , P. 2000 . Inference of population structure using multilocus genotype data . Genetics , 155 : 945 – 959 .
- Raymond , M. and Rousset , F. 1995 . GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism . Journal of Heredity , 86 : 248 – 249 .
- Rice , W. R. 1989 . Analyzing tables of statistical tests . Evolution , 43 : 223 – 225 .
- Ryman , N. and Laikre , L. 1991 . Effects of supportive breeding on the genetically effective population size . Conservation Genetics , 5 : 325 – 329 .
- Schwartz , M. K. , Luikart , G. and Waples , R. S. 2007 . Genetic monitoring as a promising tool for conservation and management . Trends in Ecology and Evolution , 22 : 25 – 33 .
- Sokal , R. R. and Rohlf , F. J. 1995 . Biometry: the principles and practice of statistics in biological research, , 3rd edition , New York : Freeman .
- Talbot , G. B. and Sykes , J. E. 1958 . Atlantic coast migrations of American shad . U.S. Fish and Wildlife Service Fishery Bulletin , 58 : 473 – 480 .
- Thériault , V. , Moyer , G. R. and Banks , M. A. 2010 . Survival and life-history characteristics among wild and hatchery coho salmon returns: how do unfed fry differ from smolt releases? . Canadian Journal of Fisheries and Aquatic Sciences , 67 : 486 – 497 .
- Thériault , V. , Moyer , G. R. , Jackson , L. , Blouin , M. S. and Banks , M. A. 2011 . Reduced reproductive success of hatchery coho salmon in the wild: insights into most likely mechanisms . Molecular Ecology , 20 : 1860 – 1869 .
- van Oosterhout , C , Hutchinson , W. F. and Wills , D. M. P. 2004 . Micro-checker: software for identifying and correcting genotyping errors in microsatellites data . Molecular Ecology Notes , 4 : 535 – 538 .
- Walther , B. D. , Thorrold , S. R. and Olney , J. E. 2008 . Geochemical signatures in otoliths record natal origins of American shad . Transactions of the American Fisheries Society , 137 : 57 – 69 .
- Wang , J. L. and Ryman , N. 2001 . Genetic effects of multiple generations of supportive breeding . Conservation Biology , 15 : 1619 – 1631 .
- Waters , J. M. , Epifanio , J. M. , Gunter , T. and Brown , B. L. 2000 . Homing behaviour facilitates subtle genetic differentiation among river populations of Alosa sapidissima: microsatellites and mtDNA . Journal of Fish Biology , 56 : 622 – 636 .
- Wedekind , C. 2002 . Sexual selection and life-history decisions: implications for supportive breeding and the management of captive populations . Conservation Biology , 16 : 1204 – 1211 .
- Wright , S. 1938 . Size of population and breeding structure in relation to evolution . Science , 87 : 430 – 431 .