ABSTRACT
One of the most challenging tasks for formulators in the pharmaceutical industry is the formulation of poorly soluble drugs. Conventional techniques employed for improving solubility of these drugs have gained limited success. This holds true more often when dealing with drugs having poor aqueous as well as organic solubility. Nanoparticles facilitates formulation of hydrophobic drugs to improve solubility and efficacy mainly through nanosuspension approach. Nanosuspensions are submicron colloidal dispersions of pure drug particles, stabilized by surfactants. This nanobiomedicine delivery system is simple and advantageous compared to other strategies. Techniques such as media milling, high-pressure homogenization, and use of microemulsion as a template have been used for production of nanosuspensions. This green nanobiomedicine can be delivered by various routes, such as oral, parenteral, pulmonary, and ocular systems. It is also possible to convert nanosuspensions to patient-acceptable dosage forms like tablets, capsules, and lyophilized powder products. Nanosuspension technology has also been studied for active and passive targeted drug delivery systems. This review article focuses on various manufacturing and formulation perspectives and applications of nanosuspensions as a drug delivery system.
INTRODUCTION
The automation of the drug discovery process by technologies such as high-throughput screening, combinatorial chemistry, and computer-aided drug design is leading to a vast number of drug candidates possessing a very good efficacy.[ Citation 1 ] Unfortunately, many of these drug candidates (about 60%) are exhibiting poor aqueous and nonaqueous solubility and/or erratic absorption and hence require innovative formulations and drug delivery systems.[ Citation 2 ] In addition, poorly water-soluble drugs are specially challenging, as they cannot achieve dissolution and therefore they have a very difficult pass through the dissolving fluid to contact the absorbing mucosa and to be absorbed.[ Citation 3 ] If the dissolution process of the drug molecule is slow, due to the physicochemical properties of the drug molecules or formulation factors, then dissolution may be the rate-limiting step in absorption and will influence drug bioavailability.[ Citation 4 ] This is the case of Biopharmaceutical Classification System (BCS) Class II and Class IV drugs. For these kind of drugs, micronization,[ Citation 5 , Citation 6 , Citation 7 , Citation 8 ] nanonization,[ Citation 9 , Citation 10 , Citation 11 ] complexation (e.g., cyclodextrins),[ Citation 12 , Citation 13 , Citation 14 , Citation 15 , Citation 16 , Citation 17 ] preparation of liposomes,[ Citation 18 , Citation 19 , Citation 20 ] amorphous solid dispersions,[ Citation 21 , Citation 22 , Citation 23 , Citation 24 , Citation 25 ] solubilization using cosolvents, use of permeation enhancers, oily solutions, surfactant dispersions, salt formation, precipitation techniques, etc., have been proposed to increase the rate of dissolution and especially the drug bioavailability after oral administration for systemic drug absorption.[ Citation 26 ] These techniques have their own limitations and hence have limited utility.[ Citation 27 ] An alternative and promising approach is the production of drug nanosuspensions that improves drug efficacy and pharmacoeconomics.[ Citation 28 ] A nanosuspension consists of drug nanocrystals, stabilizing agents, and a liquid dispersion medium. The dispersion media can be water, aqueous solutions, or nonaqueous media. The terms drugs nanocrystals implies crystalline state of the discrete particles. The major advantages of nanosuspension technology are its general applicability to most drugs and its simplicity and are now the universal formulation approach for most drugs.[ Citation 29 ] Nanosuspension technology offers solution not only to solubility of drug but also alters the pharmacokinetic of drug and thus improves drug safety and efficacy.[ Citation 30 ] The poorly soluble and low-bioavailability drug, so called “brick dust” candidate once abandoned from formulation development, can be rescued by formulating into nanosuspension.[ Citation 31 ] The aim is “maximizing dissolution rate and boosting bioavailability” of drugs.
SOLUBILITY
One of the primary physicochemical considerations in preparing pharmaceutical solutions is the solubility of the drug in a suitable solvent. Solubility may be defined as the maximum concentration of a substance that may be completely dissolved in a given solvent at a given temperature and pressure. When both solute and solvent are liquids, the term miscibility rather than solubility may be used to describe the affinity between the liquids.[ Citation 32 ] The solubility of a substance may be described in a variety of ways. The United States Pharmacopeia–National Formulary (USP-NF) generally expresses the solubility in terms of the volume of solvent required to dissolve 1 g of the drug at a specified temperature (e.g., 1 g acetylsalicylic acid in 300 mL H2O, 5 mL ethanol at 25°C). Other references may use more subjective terms to describe solubility, such as those given in .[ Citation 32 , Citation 33 ]
TABLE 1 The descriptive terms of approximate solubility of substances
Liquids that form a homogenous system when mixed in any proportion are said to be miscible (e.g., water and ethanol). Those in which only certain volume ratios produce homogenous mixtures are said to be miscible in certain proportions (e.g., water and chloroform). Immiscible liquids will not produce a homogenous solution in any proportions (e.g., water and olive oil). The aqueous solubility of all drugs is of interest to us, because it is only in the form of an aqueous solution that a drug can be absorbed into the general circulation to exert a therapeutic effect.
Thermodynamic Solubility and Kinetic Solubility
From a thermodynamic or chemical point of view, each substance has only one solubility value at a specific temperature, pressure and volume. This value is defined as the saturated concentration of the substance in solution when it is in equilibrium with its most stable solid state structure. In the literature, the term ‘solubility’ is often used loosely to mean the metastable or kinetic solubility, rather than the solubility at this thermodynamically stable state. The apparent or kinetic solubility, i.e., the concentration of the material in solution at apparent equilibrium (supersaturation), decreases to the level of the true or thermodynamically stable solubility, after an infinite equilibrium time.[ Citation 34 , Citation 35 ]
It should be noted that the term ‘apparent solubility’ has also been used in the literature to discuss a change in solubility due to ionic interaction. The result of such interionic interactions may be that the actual or effective number of ions is no longer the same as that calculated based on the concentration of the solution. This is the reason why sometimes it is necessary to replace concentration with activity, which is the effective concentration. The term apparent solubility used here refers to a deviation from the thermodynamic stability, which is caused by the existence of disordered structure. On the other hand, the solubility of sparingly soluble salts is often expressed or discussed in terms of the solubility product, which is an equilibrium constant describing the equilibrium between a sparingly soluble compound and a saturated solution of its ions. The numerical value of the solubility product of a salt can thus be regarded as a quantitative statement of the limit of solubility of the salt.[ Citation 34 , Citation 35 ]
Solubility Enhancement
Poor water solubility of drugs is normally associated with low oral bioavailability in body. Saturation solubility enhancement is the most interesting method to overcome this problem. In order to obtain increased solubility for drugs with poor water solubility, it is necessary to alter the formulation to facilitate solubilization. There are many classical approaches for solubilization of poorly soluble drugs. These technologies are commonly used as primary strategies. The choice conventional solubilization method will depend upon how the drug can be solubilized, stability in the system, and biocompatibility of the vehicle for a given delivery route. For solid dosage form, it may be possible to modify the solid phase to enhance dissolution.
Solid-Phase Modification
It is important to always consider that for any raw drug substance or formulation, only one solid phase is thermodynamically stable for a given set environmental condition. The most stable solid will have the lowest free energy and correspondingly the lowest solubility. The crystal forms with higher free energy may exhibit an apparent solubility that is higher than the true equilibrium solubility for the system. An apparent solubility increase can occur anytime the starting solid material is not the most stable for the given system. However, a metastable crystal will produce only a transient increase in solubility. The most stable crystal will eventually precipitate and the apparent solubility gained will diminish until the thermodynamic equilibrium solubility is reached. The degree of supersaturation and of duration will depend on the characteristics of starting material and on the nucleation rate and the growth kinetics of the stable form. Consequently, an inherent difficulty in working with metastable systems is that the kinetics of conversion often cannot be predicted or controlled.[ Citation 36 , Citation 37 , Citation 38 ]
By virtue of having a higher apparent solubility, a metastable crystal will have an increased dissolution rate compared to the more stable form. The change in mass, M, as a function of time, t, for a solute is directly proportional to its apparent solubility, S app, according to
It may be practical at time to use a solid material that gives a higher apparent solubility and/or increase in the dissolution rate in order to enhance bioavailability. As an example, polymorph B of chloramphenicol palmitate, which can produce a solubility that is roughly 2 times that of polymorph A, gives higher blood levels in vivo. It showed that as the percentage of polymorph B increases in the dosage form, there is a linear increase in the blood concentration of chloramphenicol palmitate.[ Citation 38 ]
Through alteration of the solid-state form, it is possible to increase the apparent solubility of a drug. However, the practical use of a higher energy solid form is limited due to physical and chemical issues. Significant investigations must be made in order to assure that a dosage form using metastable crystals will maintain integrity throughout the product shelf life.[ Citation 38 ]
Salt Formation and pH Control
Salt Formation The aqueous solubility of nonelectrolytes is nearly always affected in some way by the addition of an electrolyte. Salting-out is the precipitation of organic solutes from aqueous solution by the addition of an electrolyte or salt. This is attributed to competition between solute molecules for the solvent and is dependent upon the size and valence of the ions. Salting-in is the increase in solubility of an organic solute upon addition of an electrolyte.[ Citation 36 , Citation 37 , Citation 38 ] The mechanism of this phenomenon is poorly understood and it is rarely encountered. An example is the group of proteins called globulins, which are more soluble in dilute salt solutions than in water.[ Citation 36 , Citation 37 , Citation 38 , Citation 39 ] Complex ion formation occurs when an insoluble solute reacts with a soluble substance to form a soluble complex. An example is the addition of the soluble potassium iodide (KI) to the insoluble iodine molecule (I2) to form a soluble triiodide complex (KI3).
pH Control According to the Henderson-Hasselbalch equation, the relationship between pH, pK a , and relative concentrations of a weak acid and its salt is
Changes in solubility brought about by alterations of the solvent pH can be predicted by the pHp equation. The pHp is the pH below which an acid or above which a base will begin to precipitate.
The Henderson-Hasselbalch equation can be simplified to
When the concentrations of salt and acid are equal, the pH of the system equals the pK a of the acid. As the pH decreases, the concentration of the molecular acid increases and that of the salt decreases. This has some interesting implications regarding the aqueous solubility of the acid, because the undissociated form is much less soluble than its salt. Of further interest, therapeutically, is the fact that it is the undissociated acid (HA) that more readily penetrates biological tissues to exert a therapeutic effect.[ Citation 36 , Citation 37 , Citation 38 , Citation 39 ] Thus, in formulating the product, some balance must be made between the more soluble salt form and the biologically active acid and factors other than pK a and pH must be considered (e.g., safety and comfort).
By analogy to the weak acid, the total solubility is described by
Cosolvents
Solute molecules are held together by certain intermolecular forces (dipole-dipole, induced dipole-induced dipole, ion-ion, etc.), as are the molecules of the solvent. In order for dissolution to occur, these cohesive forces of like molecules must be broken and adhesive forces between solute and solvent must be formed.
The solubility of a drug in a given solvent is largely a function of the polarity of the solvent. Solvents may be considered polar, semipolar, or nonpolar. Polar solvents will dissolve ionic and other polar solutes (i.e., those with an asymmetric charge distribution or like dissolves like), whereas nonpolar solvents will dissolve nonpolar molecules. Semipolar solvents (e.g., alcohols and ketones) may induce a certain degree of polarity in nonpolar molecules and may thus act to improve the miscibility of polar and nonpolar liquids. The relationship between polarity and solubility may be used in practice to alter the solubility of a drug in a pharmaceutical solution.[ Citation 36 , Citation 37 , Citation 38 , Citation 39 ]
Another approach is to mix solvents of different polarities to form a solvent system of optimum polarity to dissolve the solute. Such solvents must, obviously, be miscible. This method is referred to as solvent blending or cosolvency and uses the dielectric constant as a guide to developing the cosolvent system. Because many solvents may be toxic when ingested, most solvent blends are limited to mixtures containing water, ethanol, glycerin, propylene glycol, polyethylene glycol 400, or sorbitol solution. The list is somewhat expanded for solutions for external application.
There are many pharmaceutical substances that are nonpolar or are weak acids and bases whose ionized salt forms are unstable in solution. In order to dispense solutions of these substances, we must derive a solvent of appropriate polarity (or nonpolarity). Practically speaking, this is a fairly simple problem to solve. Solutions are prepared containing varying concentrations of ethanol or acetone in water, ranging from 0% to 100%. The required concentration of drug is added to each solution and the solutions are refrigerated overnight, then viewed for precipitation.[ Citation 38 ]
From this information it is possible to formulate a vehicle, substituting other solvents, which is of the necessary polarity and is pharmaceutically elegant. These calculated values of the dielectric constant are only approximate. Interactions between multiple solutes and solvents may increase or decrease solubility. Nonetheless, the use of the dielectric constant in formulating solvent systems gives us a simple and scientific approach to estimating our needs. It is, therefore, a useful tool.[ Citation 38 , Citation 39 ]
Solubilization by Micelles
Surfactants are molecules with distinct polar and nonpolar regions. Most surfactant consists of a hydrocarbon segment (usually in the form of long aliphatic chain segment) connected to a polar group. The polar group can be anionic (such as carboxylate, sulfate, or sulfonate), cationic (such as ammonium, trialkylammonium, or pyridinium), zwitterionic (such as glycine or carnitine), or nonionic (such as polyethylene glycol, glycerol or sugar). Due to the differences in properties of the polar and nonpolar regions, surfactants tend to accumulate and orient at interfaces so that each region of the surfactant interacts with a separate phase. The polar portion of the surfactant will associate with the more polar phase (especially if it is water) and the nonpolar portion of the surfactant will remain in the more nonpolar surfactant solvent. In water, as the concentration of surfactant increases above a critical value, its molecules self-associate into soluble structures called micelles. The concentration at which they begin to form is called the critical micelle concentration (CMC).[ Citation 36 , Citation 37 , Citation 39 ]
These micelles are normally spherical, with the nonpolar regions of surfactant molecules gathered in the center (core) and surrounded by a shell of the polar region, which is in contact with the water. A nonpolar drug, which is squeezed out of water, can locate within the micelle core. A semipolar drug can locate between or partially within the core and the polar shell. Because the micelles are soluble in water, any drug that is incorporated into the micelle will also be soluble in the aqueous system. If the monomers of surfactant in solution do not affect the solubility of the solute, then the solute concentration will remain constant (at the intrinsic solubility, S W) until the CMC. After the CMC, the solute concentration will increase with increasing surfactant (micelle) concentration. A simple equation representing a solute's total solubility, S T, in a surfactant system is
Complexation
Complexation is association between two or more molecules to form a nonbonded entity with well-defined stoichiometry. Two types of complexation are most useful for increasing the solubility of drugs in aqueous media. Stacking complexes are formed by the overlap of the planar regions of aromatic molecules, whereas the inclusion complexes are formed by the insertion of the nonpolar region of one molecule into the capacity of another molecule (or group of molecules). The mathematical description for the equilibrium constant of a 1:1 complex, K 1:1, is defined by
From the above it can be seen that as the stability constant of a 1:1 complex increases, the slope will increase until the value converges to unity for a strong complex in which one ligand molecule solubilizes one solute molecule. The initial segment of the curve in illustrates this. The linear region will continue until the solubility of the complex itself is reached, at which point the total solubility of the solute remains constant as indicated by the central segment of the curve.
FIGURE 1 General solubilization profile for complexation.
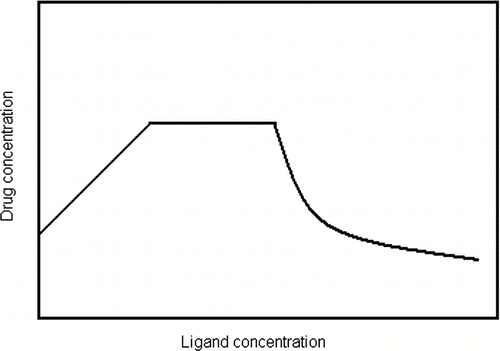
A plateau is analogous to maximum solubility of a solute. Further addition of the complexing agent can result in a reduction of the concentration of the free solute and leveling of the curve at the solubility of the pure complex, as illustrated by the final segment in the curve in . An inclusion complex is produced by inclusion of a nonpolar molecule or the nonpolar region of a molecule (known as the guest) into the nonpolar cavity of another molecule or group of molecules (known as the host). When the guest molecule enters the host molecule, the contact between water and the nonpolar region of the both is reduced. Thus inclusion phenomena are the result of the same driving force that produces micellization, self-association, and stacking, namely the squeezing out from water of nonpolar moieties.
The most commonly used host molecules are the cyclodextrins. These cyclic oligomers of glucose are relatively soluble in water and have cavities large enough to accept nonpolar portions of common drug molecules. The naturally occurring cyclodextrins contain 6, 7, and 8 glucopyranose units and are termed α, β, and γ, respectively. The size of the cavacity in the cyclodextrin is the major factor in determining which guest solute will be most acceptable for complexation. In general, alkyl groups will fit well into the cavity of the α -cyclodextrin. The β -cyclodextrin are most well suited for accepting single aromatic rings and the γ -cyclodextrin has a large enough cavity to accommodate larger hydrocarbons such as pyrene.[ Citation 37 ] The degree to which a solute molecule will be solubilized by a cyclodextrin molecule will depend on several properties. The solute molecule must have a significant nonpolar portion in order to be squeezed out of the water and into the cyclodextrin cavity. Because the interior dimensions of a given cyclodextrin are fixed, a significant part of molecule (or whole molecule) must then fit inside the cyclodextrin. The intermolecular interactions between the two molecules will determine the strength of the complex. The aliphatic compounds are preferentially solubilized by α -cyclodextrin, whereas the aromatic ring compounds are best solubilized by β -cyclodextrin. In addition, γ -cyclodextrins may increase solubilization of the fused ring compounds.[ Citation 37 ]
Solid Dispersions
Solid dispersions have been developed five decades ago. In 1961, Sekiguchi and Obi developed a practical method whereby many of the limitations of the bioavailability enhancement of poorly water-soluble drugs can be overcome. This method, which was later termed solid dispersion, involved the formation of eutectic mixtures of drugs with water-soluble carriers by the melting of their physical mixtures. Sekiguchi and Obi suggested that the drug was present in a eutectic mixture in a microcrystalline state.[ Citation 31 , Citation 40 ] Later, Goldberg et al. demonstrated that the entire drug in a solid dispersion might not necessarily be present in a microcrystalline state; a certain fraction of the drug might be molecularly dispersed in the matrix, thereby forming a solid solution. In either case, once the solid dispersion was exposed to aqueous media and the carrier dissolved, the drug was released as very fine, colloidal particles. Because of greatly enhanced surface area obtained in this way, the dissolution rate and the bioavailability of poorly water-soluble drugs were expected to be high.[ Citation 31 , Citation 41 ]
But from time to time it has been found that there are principal limitations regarding the development of solid dispersion/solution methods. Problems limiting the commercial application of solid dispersion involve its method of preparation, reproducibility of its physicochemical properties, its formulation into dosage forms, the scale up of the manufacturing processes, and the physical and chemical stabilities of drug and vehicle.
DISSOLUTION VELOCITY
The dissolution process of solids in liquids has been described by Hildebrand and Scott[ Citation 42 , Citation 43 ] as involving three steps: (1) the removal of a molecule from the solute; (2) creation of a hole in the solvent; and (3) insertion of the solute molecule into the solvent (i.e., solute-solvent interaction). This interaction between the solute and the solvent is obviously dependent on the physical and chemical nature of the two participating molecules. The processes involved in the dissolution of hydrophobic materials, which have low aqueous solubility, will differ from those affecting the dissolution of hydrophilic substances. The amount of energy required to remove a molecule of a sparingly soluble drug from the solute particle is lower for a hydrophobic drug than for a hydrophilic drug (such as those composed of inorganic salts). The intermolecular bonds in inorganic salts are so strong that a large amount of energy is required to disassociate the discrete molecules. However, when the individual solute molecules are liberated, a hydrophilic salt molecule (or ion) is more likely to interact with water than a hydrophobic drug molecule. Thus, the main factor affecting the solubility and dissolution of a hydrophobic drug is the limited energy released when a drug molecule is bonded to the solvent.[ Citation 39 ] In contrast, the main barrier to dissolution of a sparingly soluble hydrophilic substance appears to be disruption of the strong intermolecular forces.[ Citation 34 ]
Theory of Dissolution
When a solid dosage form is introduced into a beaker of water or into the gastrointestinal tract, the drugs begin to pass into solution from the intact solid. Unless the solid dosage form is a continguous polymeric device, the solid matrix also disintegrates into granules and these granules deaggregate in turn into fine particles. Disintegration, deaggregation, and dissolution may occur simultaneously with the release of a drug from its delivery form. These steps are separated for clarification as depicted in .[ Citation 36 ]
FIGURE 2 Disintegration, deaggregation, and dissolution stages as a drug leaves a tablet or granular matrix.
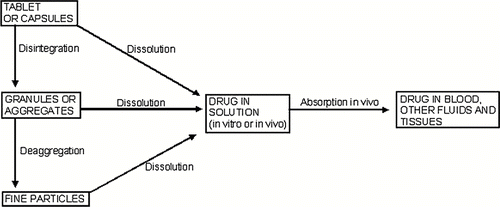
The effectiveness of a solid dosage form in releasing of its drug for systemic absorption depends somewhat on the rate of disintegration of the dosage form and deaggregation of the granules. Ordinarily of more importance, however, is the dissolution rate of the solid drugs. For class II drugs of the Biopharmaceutical Classification System (BCS), frequently dissolution is the limiting or rate-controlling step in absorption of poorly soluble drugs, because it is often the slowest of the various step involved in release of the drugs from its dosage forms and passage into systemic circulation [36]. The rate at which the solid dissolves in the solvent was proposed in quantitative terms by Noyes and Whitney in 1897 and elaborated subsequently by other coworkers.[ Citation 44 ] The equation may be written as
In dissolution or mass transfer theory, it is assumed that an aqueous diffusion layer or stagnant liquid film of thickness h exist at the surface of solid undergoing dissolution, as shown in . This thickness h represents a stationary layer of solvent in which the solute molecule exist in concentrations from C s to C. Beyond the static diffusion layer, at x greater than h, mixing occurs in the dissolution and the drug is found at a uniform concentration, C, throughout the bulk phase.[ Citation 36 ]
FIGURE 3 Dissolution of the drug from a solid matrix, showing the stagnant diffusion layer between the dosage form surface and the bulk solution.
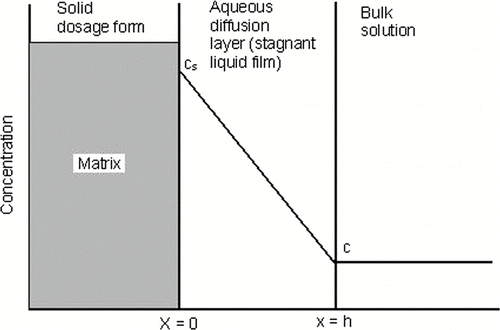
At the solid surface-diffusion layer interface, x = 0, the drug in the solid is equilibrium with the drug in the diffusion layer. The gradient or change in concentration with distance across the diffusion layer is constant as shown by the straight downward-sloping line. This is the gradient represented in Equations Equation7 and Equation8 by the term (C s−C)/h. When C is considerably less than the drug's solubility, C s, the system is represented by sink condition, and concentration C may eliminated from the equations. The equation then becomes
Biopharmaceutical Classification System (BCS)
The BCS is scientific framework for classifying drug substances based on their aqueous solubility and intestinal permeability. When combined with the dissolution of the drug product, the BCS takes into account three major factors that govern the rate and extent of drug absorption from solid oral dosage forms: dissolution, solubility, and intestinal permeability. According to the BCS, drug substances are classified as follows[ Citation 46 , Citation 47 ]:
Class I: High Solubility–High Permeability | |||||
Class II: Low Solubility–High Permeability | |||||
Class III: High Solubility–Low Permeability | |||||
Class IV: Low Solubility–Low Permeability | |||||
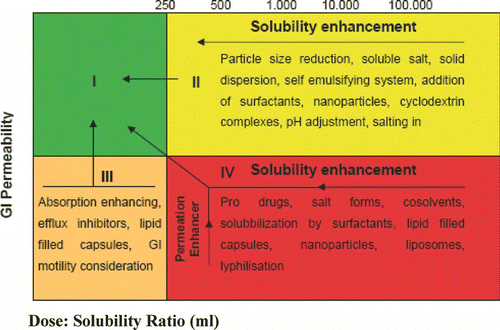
The Biopharmaceutical Classification System (BCS) groups poorly soluble compounds as Class II and IV drugs, compounds that feature poor solubility and high permeability, and poor solubility and poor permeability, respectively. Drug substances are considered highly soluble when the largest dose of a compound is soluble in < 250 mL water over a range of pH from 1.0 to 7.5; highly permeable compounds are classified as those compounds that demonstrate > 90% absorption of the administered dose. In contrast, compounds with solubilities below 0.1mg/mL face significant solubilization obstacles, and often even compounds with solubilities below 10 mg/mL present difficulties related to solubilisation during formulation.[ Citation 48 ] gives a diagramic representation of solubility-dissolution characteristics.
FIGURE 4 Possibilities of shifting the solubility-dissolution characteristics from a very poorly soluble drug to dose:solubility ratio (D:S) within the range of values encountered in the human GI tract (D:S > 250 mL).
NANOSUSPENSIONS
Theoretical Overview
Recently, many poorly soluble drugs have been nanonized to increase their dissolution rate, their saturation solubility, and in turn to enhance their oral bioavailability.[ Citation 49 , Citation 50 , Citation 51 , Citation 52 , Citation 53 , Citation 54 , Citation 55 ] The concept of oral nanosuspensions has been specifically used to increase the rate and extent of the absorption of drugs that have poor and/or erratic dissolution. In the current decade, the concept of nanosuspensions could be commercially exploited by pharmaceutical companies as micronization did in the last few decades. Therefore, drug nanocrystals represent an alternative to existing drug delivery technologies for poorly soluble compounds.[ Citation 56 , Citation 57 ]
Drug nanonization can be achieved through different techniques, where one distinguishes between the bottom-up and the top-down technologies. Bottom-up technologies are precipitation methods,[ Citation 58 , Citation 59 , Citation 60 , Citation 61 ] whereas top-down technologies start from coarse drug macrosuspensions. The diminution is achieved either by pearl/ball milling,[ Citation 9 , Citation 62 ] high-pressure homogenization, either in water[ Citation 10 ] or in water-free or water-reduced media,[ Citation 63 ] by combination technologies, e.g., precipitation and subsequent high-pressure homogenization[ Citation 64 ] or ball milling and subsequent high-pressure homogenization.[ Citation 65 ] In the literature, many data are available covering the formulation and optimization of nanosuspensions.[ Citation 66 , Citation 67 , Citation 68 ] Liversidge and Cundy have reported that if bioavailability is truly dissolution rate limited, particle size reduction can significantly improve the oral bioavailability of the drug.[ Citation 62 ] It has been also investigated that producing nanosuspensions for oral administration leads to effective therapeutic concentrations in the blood because solubility and absorption problems in the gastrointestinal tract have been overcome by extensive size reduction.[ Citation 10 , Citation 49 , Citation 56 , Citation 62 , Citation 69 , Citation 70 ]
An outstanding feature of nanosuspensions is the increase in saturation solubility and consequently an increase in the dissolution velocity of the compound. The increase in saturation solubility can be explained by the Kelvin–Gibbs and the Ostwald–Freundlich equations. The Kelvin equation describes the vapor pressure over a curved surface of a liquid. It describes droplets in a gas phase. The vapor pressure increases with increasing curvature of the droplets, which means decreasing particle size. It has also been postulated that the surface curvature of the dissolving solid particles will influence solubility in water. The basic theory derives from the classical Gibbs–Kelvin equation, which, when adapted to the solubility of solids, is known as the Ostwald–Freundlich equation:
The most important feature of nanocrystals is the increase in saturation solubility and surface area, and consequently an increase in the dissolution velocity of the compound. The law of Noyes–Whitney describes the dissolution velocity[ Citation 44 ]:
Production Process
There are several production techniques to produce drug nanocrystals. Basically, one can differentiate between top-down and bottom-up technologies. Typically, the drug nanocrystals are generated in a liquid dispersion medium (e.g., by precipitation or a disintegration process). The obtained product from this process is a suspension of drug nanocrystals in a liquid stabilized by a surfactant or polymer (so-called “nanosuspensions”). In contrast to micronized powders, the drug nanocrystals can be administered using very different administration routes. Oral administration is possible as a suspension. More patient-convenient dosage forms can be produced by transferring the liquid nanosuspensions to solid dosage forms, i.e., tablets or pellets or granulate-containing capsules. In addition, because of their small size, the nanosuspensions can be injected parenteraly, especially intravenously. Intravenous injection leads ‘per definition’ to a 100% bioavailability.[ Citation 50 ]
Bottom-up Technologies
The term “bottom-up technology” means that one starts from the molecular level, and goes via molecular association to the formation of a solid particle. That means that we are discussing classical precipitation techniques by reducing the solvent quality, for example, by pouring the solvent into a nonsolvent or changing the temperature or a combination of both. Precipitation is a classical technique in pharmaceutical chemistry and technology. The Latin terminology is via humida paratum (v.h.p.), which means being prepared via a liquid process (solutions are made to obtain a fine powder [precipitate] dispersed in a ‘wet’ environment). Because it is a long known process in pharmacy, a clear outline is necessary to document the innovative height of patent applications based on this process.[ Citation 54 ]
The particles generated by precipitation, as for example by Sucker, are in most cases crystalline in nature; in contrast to this, the company Knoll (nowadays owned by Abbott, the new name is “Soliqs”) created amorphous particles by a precipitation technique.[ Citation 23 ] The product is NanoMorph™. As outlined above, an additional positive feature is a further increase in the dissolution velocity due to the amorphous character of the product. The precipitation in the amorphous form is achieved by an aqueous polymer solution. However, for a commercial product, it is necessary to preserve the amorphous character during the shelf life to avoid changes in bioavailability caused by a reduction in the dissolution velocity due to the transfer of the amorphous drug to a crystalline drug.[ Citation 54 ]
The advantage of the precipitation techniques is that relatively simple equipment can be used. For example, the solvent can be poured into the nonsolvent with a constant velocity in the presence of a high-speed stirrer. Very elegant approaches are the use of static mixers or micromixers, which simulates the precipitation conditions in a small volume (i.e., simulating lab-scale conditions), but being simultaneously a continuous process. In the case of micromixers, scaling up can be performed in a simple way just by so-called numbering up, which means arranging many micromixers in parallel. The equipment is relatively simple, and in the case of large conventional static mixers, of relatively low cost (this is not necessarily valid for the micromixers). However, there are some major general disadvantages of the precipitation techniques.
| • | The drug needs to be soluble in at least one solvent (thus excluding all new drugs that are simultaneously poorly soluble in aqueous and inorganic media). | ||||
| • | The solvent needs to be miscible with at least one nonsolvent. | ||||
| • | Solvent residues need to be removed, thus increasing production costs. | ||||
| • | It is a little bit tricky to preserve the particle character (i.e., size, especially the amorphous fraction). | ||||
In general, it is recommended that a second consecutive process be performed for particle preservation, that is, spray drying or lyophilization.[ Citation 54 ]
Top-down Technologies
There are basically two top-down techniques, which means starting from a large-sized powder and performing a size diminution.[ Citation 50 , Citation 54 ]
Pearl Milling Liversidge and co-workers developed the milling process to yield the so-called NanoCrystals®. The mills used by the Elan Company are basically containers with small pearls, beads, or balls that can have different sizes, a typical size being 1–2 mm. The drug powder is dispersed in a surfactant solution, and the obtained suspension is poured into the mill. In the milling process, the pearls are moved, either by using a stirrer or by moving the milling container itself. The drug particles are disintegrated between the moving pearls. This process is accompanied by the erosion of milling material from the pearls, a phenomenon well known for these mills and documented in the literature.[ Citation 54 ] The milling technique clearly has some advantages:
| • | Simple technology | ||||
| • | Low-cost process regarding the milling itself | ||||
| • | Large-scale production possible to some extent (batch process) | ||||
As disadvantages, we can see:
| • | Potential erosion from the milling material leading to product contamination | ||||
| • | Duration of the process not being very production friendly | ||||
| • | Potential growth of germs in the water phase when milling for a longtime | ||||
| • | Time and costs associated with the separation procedure of the milling material from the drug nanoparticle suspension, especially when producing parenteral sterile products | ||||
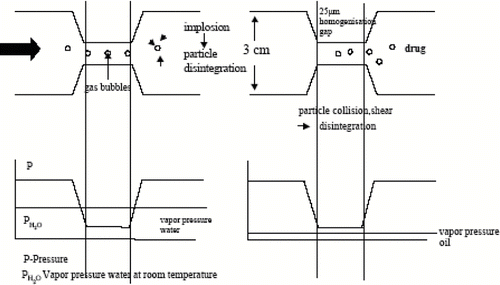
Cavitation was employed as the most important effect to diminute particles in a piston-gap homogenizer. In this homogenizer types, the dispersion (emulsion or suspension) passes a very tiny gap with an extremely high velocity. Prior to entering the gap, the suspension is contained in a cylinder with a relatively large diameter compared to the width of the following gap. In the APV LAB 40, the diameter of the cylinder is about 3 cm, it narrows to about roughly 3–25 μ m (varies with applied pressure) when the suspension enters the homogenization gap. The cavitation and shear forces in the gap were sufficiently high to break particles that were distinctly larger than the gap width. Therefore it is possible to disrupt relatively large sized powders (up to 200 μ m).[ Citation 50 , Citation 54 , Citation 77 ]
According to the law of Bernoulli, the flow volume of a liquid in a closed system per cross-section is constant. That means the reduction in the diameter leads to a tremendous increase in the dynamic pressure and simultaneously a decrease of the static pressure when the liquid is in the homogenizer gap. A liquid boils when its vapor pressure is equal to the air/static pressure of the environment. In the gap, the static pressure drops below the vapor pressure of the liquid at room temperature (). Consequently, the liquid starts boiling, forms gas bubbles that implode after leaving the homogenization gap, and being again under normal air pressure conditions.[ Citation 50 , Citation 54 ]
FIGURE 5 Change of the diameter of the streaming dispersion in a piston-gap homogenizer from the cylinder containing the bulk suspension to then narrow homogenization gap. The actual range of the static pressure as a function of the location inside the homogenizer is given in the diagrams below (left: situation for homogenization in water; right: homogenization in water-mixtures or water-free media).
There are advantages of the piston-gap homogenization technique:
| • | The effective particle diminution | ||||
| • | Production lines can be qualified and validated | ||||
| • | Production lines already exist in industry, for example, production of parenterals, and so on | ||||
| • | No contamination, for example, iron content is below 1 ppm[ Citation 78 ] | ||||
| • | Off-shelf equipment | ||||
| • | Simple process | ||||
| • | Low-cost process/low-cost equipment | ||||
To summarize: homogenization with piston-gap homogenizers has good potential to be used as a production technique because it fulfills the key industrial features such as large-scale production and various regulatory aspects.[ Citation 50 , Citation 54 ]
Homogenization in Water–Liquid Mixtures and Nonaqueous Media The DissoCubes® patent describes homogenization in pure water as a dispersion medium; the rationale behind—as explained in detail in the patent—is that the cavitation is seen as being responsible for the diminution of the particles. To obtain cavitation in the dispersion medium, one needs to have a high vapor pressure of the dispersion fluid at room temperature. This is fulfilled with water, but not with other liquids such as oils or liquid polyethyleneglycol (PEG) 600. In addition, homogenization at higher temperatures (e.g., 80°C) should be much more efficient than at room temperature because the vapor pressure of water is much higher at 80°C, thus leading to more extensive cavitation.[ Citation 79 ] The basic of the Nanopure® technology is that homogenization was performed against these teachings, leading to a comparable or even improved product. Under the Nanopure® technology, not only drugs, but also polymers can be processed, leading to a product with a mean diameter in the nanometer range (nanoparticles) or a size of a few micrometers (both particle ranges are covered).
Nonaqueous dispersion media can be used to produce oral formulations, for example, drug nanocrystals dispersed in liquid PEG 600 or Miglyol (MCT) oil, that can be directly filled into soft gelatin capsules, sealed hard gelatine capsules, or hydroxypropylmethyl cellulose (HPMC) capsules. Nanosuspensions in mixtures of water with water-miscible liquids (e.g., ethanol, isopropanol) can be used in the granulation process for tablet production; the solvent mixture evaporates much better than water. Drugs that are susceptible to hydrolysis in water can be prepared as nonaqueous suspensions, for example ethanol, glycerol, or propylene glycol.[ Citation 80 ] Prior to intravenous injection, the glycerol drug nanosuspensions can be diluted with water for injection to yield an isotonic suspension. Alternatively, isotonic water glycerol mixtures can be directly homogenized. Previously cavitation was considered as the dominating factor for particle diminution; therefore, homogenization was only performed in pure water, and glycerol was added afterwards to avoid an impairment of particle diminution efficiency. Another option of the Nanopure® technology is the production of aqueous polymer particle dispersions with a diameter of a few micrometers or in the nanometer range. This could be an alternative to polymer dispersions (e.g., surelease, ethyl cellulose) for the coating of tablets that are now produced by using solvents. Aqueous shellac dispersions produced with Nanopure® technology have already been described.[ Citation 54 ] gives a comparison of the three techniques.
TABLE 2 Advantages and disadvantages of different methods for the production of nanocrystals
Combination Technology
In 2001, the Baxter Healthcare Company presented their NANOEDGE® technology at the annual meeting of the American Association of Pharmaceutical Scientists (AAPS) in Denver. NANOEDGE® is an aqueous suspension of drug nanoparticles suitable for intravenous injection. The particles are prepared basically by a high-pressure homogenization process, which means either by microfluidization or homogenization in water using piston-gap homogenizers, or alternatively, by a new production process developed by Baxter.[ Citation 54 ] This process is called the “microprecipitation method,” and is a combination of precipitation followed by a process with high-energy input (e.g., homogenization). The process includes three steps:
| • | dissolving the organic compound in a water-miscible first solvent to form a solution; | ||||
| • | mixing this solution with the second solvent to obtain a “presuspension” (= precipitate); and | ||||
| • | adding energy to this “presuspension” to form particles having an average effective particle size of 400 nm to 2.0 μ m. | ||||
The advantages of the Baxter method are that the problem of the precipitation technique—continuing crystal growth after precipitation—has been overcome because the particles are only an intermediate product to undergo subsequent diminution/annealing. In addition, by optimal choice of precipitation conditions, more friable particles (semicrystalline, amorphous) can be produced which allow more effective diminution in the subsequent high-pressure homogenization step. This is of special importance when the available jet milled drug is highly crystalline and simultaneously possesses a very hard crystal structure (high Mohs degree). In such cases, the direct homogenization of the jet-milled powder can require a higher number of homogenization cycles, which means longer production times. In the case of very hard crystals, the maximum homogenization pressure applicable during production (e.g., 2000 bar) might potentially lead to a maximum dispersitivity being rather close to 1 μ m, and not in the preferred nanometer range.[ Citation 50 , Citation 54 ] Another combination technology has been developed by Petersen using ball milling and subsequent high-pressure homogenization (CT).[ Citation 65 ] With the CT technology, the performance (e.g., physical stability) of nanocrystals could be distinctly improved, as for example for dermal application. Nanocrystals can increase the penetration of poorly soluble cosmetic and pharmaceutical actives into the skin. The increased saturation solubility leads to an increased concentration ingredient, thus promoting passive penetration. Molecules penetrated into the skin are very fast replaced by new molecules dissolving from the nanocrystal depot in the cream. The first four cosmetic nanocrystal products with rutin were launched by Juvena. Compared to the water-soluble rutin glucoside, the original rutin molecule as smartCrystal formulation possesses a 500 times higher bioactivity (as measured by sun protection factor [SPF]).[ Citation 65 , Citation 81 ] Of course the same principle can be applied to poorly soluble pharmaceutical drugs of interest for dermal application. Dermal application of nanocrystals is protected by a US and Patent Cooperation Treaty (PCT) patent application.[ Citation 65 , Citation 81 ]
Other Techniques for Production of Drug Nanoparticles
The intensive researches of new technologies lead to many other approaches to produce nanocrystals. There are many alternative technologies, as discussed below.
Precipitation with Compressed Fluid Antisolvent This technique uses an antisolvent-based recrystallization process. CO2 is used as antisolvent. The substances are atomized into a chamber containing compressed CO2. As two liquids collide, intense atomization into micronized droplets occurs, subsequently drying of microdroplets occurs as the solvent(s) and CO2 mix. Nanoparticles are formed because the two way mass transfer: extraction of organic solvent and CO2 diffusion into the droplets.[ Citation 82 ]
High mass transfers rates have been successfully achieved in the solution enhanced dispersion by the supercritical fluids (SEDS). The SEDS process was patented by Hanna and York and owned by University of Bradford.[ Citation 83 ] This technique has produced, e.g., a new polymorph of flucticasonepropionate, it also enabled control over the particle size and shape of formed particles. This polymorph exhibited improved drug delivery characteristics in a metered dose inhaler (MDI) formulation compared to conventional and micronized drugs.[ Citation 83 , Citation 84 ]
Rapid Expansion from Liquified-Gas Solution Young et al. developed a process based on supercritical fluids, rapid expansion from the supercritical to the aqueous solution (RESAS). This technique[ Citation 85 ] could produce stable nanosuspensions of poorly water-soluble drugs. The principle of this process is to induce rapid nucleation of in supercritical fluids dissolved drugs in the presence of surface-modifying agents resulting in particles formulation with a desirable size distribution in a very short time. The surface-modifying agents serve to stabilize the formed small particles and suppress any tendency towards particle agglomeration or particle growth while they are being formed. The rapid intimate contact with the surface modifier is achieved by having the surface modifiers dissolved in the supercritical fluid containing the dissolved drugs. A rapid intimate contact between the surface modifier and the newly formed particles inhibits the crystal growth of the newly formed particle.[ Citation 84 , Citation 86 ]
This technique successfully produced cyclosporine with a size of 500–700 nm. Cyclosporine nanocrystals could be stabilized at drug concentrations as high as 6.2 and 37.5 mg/mL in 1.0% and 5.0% (w/w) Tween 80 solution, respectively.[ Citation 84 , Citation 87 ]
Rapid Expansion of Supercritical Solution into Aqueous Solution In contrast to RESAS, the rapid expansion of supercritical solution into aqueous solution (RESSAS) process utilizes a supercritical fluid that is expanded into an aqueous solution containing a stabilizer. This technique was used by Turk et al. to produce phytosterol particles with a diameter less than 500 nm. The surfactants or stabilizers are dissolved in the aqueous phase, not in the supercritical fluid.[ Citation 88 ]
Cryogenic Spray Processes The cryogenic spray process is an attractive alternative to obtain microparticles and nanoparticles of poorly soluble drugs. Halocarbon refrigerants and liquid nitrogen have been used as cryogenic media in conventional spray-freezing into the vapor processes. The feed solution is atomized through a nozzle positioned at a distance above the boiling refrigerant. The droplets gradually solidify while passing through the cold halocarbon vapor, and freeze completely as contact is made with the boiling refrigerant liquid. Unfortunately, this process may result in broad particle size distributions and nonmicronized particles because agglomerates of the solution droplets are solidified while passing through the vapor phase and settle onto the surface of the cryogenic liquid.[ Citation 84 , Citation 89 ]
Spray-Freezing into Liquid (SFL) Process To overcome agglomerated particles, spray-freezing into liquid was developed and patented by The University of Texas at Austin in 2003[ Citation 90 ] and commercialized by the Dow Chemical Company. The SFL process utilizes the atomization of feed solution containing drugs directly into a cryogenic liquid to produce frozen nano-structured particles. The frozen particles are then lyophilized to obtain dry, freely flowing micronized powders. The advantages of the SFL process result from intense atomization and rapid freezing rates. A high degree of atomization is achieved by spraying directly into cryogenic liquid and the ultrarapid freezing rates prevent the phase separation of solutes within the feed solution and induce formation of amorphous structures. The intense atomization and ultrarapid freezing rates lead to amorphous nanostructures with enhanced wetting and significantly enhanced dissolution rates.[ Citation 84 , Citation 90 ]
Solvent Evaporation Process-Evaporative Precipitation into Aqueous Solution Process The evaporative precipitation into aqueous solution (EPAS) applies rapid phase separation to nucleate and grow nanoparticles and microparticles of poorly water-soluble drugs. The EPAS was developed and patented by the University of Texas at Austin in 2001 and commercialized by the Dow Chemical Company.[ Citation 84 ] A drug solution in organic solvent is pumped through a tube where it is heated under pressure and then spraying through a nozzle into heated aqueous solution. This process results in an amorphous suspension. The stable aqueous drug suspension is dried by lyophilization or spray drying. A variety of hydrophilic surfactants are added to the solution to diffuse to surface of the growing particles rapidly enough to prevent growth of particles. The EPAS technique has produced cyclosporin A nanosupensions with particle a size ranging from 130 to 460 nm.[ Citation 84 , Citation 91 ]
Drug Nanosuspension Administration
Oral Administration of the Nanosuspensions
First choice of application is oral administration. When a drug is given orally, the bioavailability and finally its efficacy depends on the solubility and absorption in the gastrointestinal tract. In vitro, active compounds have failed in the past because their poor solubility has limited in vivo absorption and did not lead to effective therapeutic concentrations. Simple examples reflecting this problem of poor solubility combined with low absorption are the experimental compounds buparvaquone and atovaquone. A way to improve bioavailability of atovaquone or buparvaquone would be to increase the absorption rate by formulating them as a nanosuspension. Oral administration of nanosuspensions can overcome this problem because of the high adhesiveness of drug particles on biological surfaces—here the epithelial gut wall—and prolonging the absorption time.
In comparison to Wellvone-treated mice, containing a micronized drug, nanosuspensions of atovaquone at equivalent doses reduced infectivity from 40% to 15% at a reduced drug concentration of only 7.5 mg/kg after oral administration.[ Citation 72 ] These results reflects the potency of the nanosuspension technique, reducing drug load from 22.5 to 7.5 mg/kg (Wellvone®), but increasing activity 2.5-fold at the same time.[ Citation 72 ] Buparvaquone nanosuspensions provide some advantages, and in T-cell receptor (TCR)-α –deficient mice infected with Cryptosporidium parvum oocysts, the significance of the nanosuspensions is documented. In comparison to micronized drug powder, the infectivity score was reduced from 2.0 (negative control group, untreated) to 1.47 for micronized buparvaquone and even to 1.02 for equivalent nanosuspensions.[ Citation 72 , Citation 94 ] Despite the substantial amount of literature data in support of nanoparticles formulations, especially for BCS Class II compounds, there are relatively limited number of current drug products in the market that utilize nanoparticles for oral application ().
TABLE 3 Overview of current state of development of drugs using nanotechnology[ Citation 3 , Citation 50 , Citation 92 , Citation 93 ]
Furthermore, the nanosuspension was able to overcome the significant food effect (∼ 3.2×) observed with the microsuspension formulation. It is worth mentioning that, in the dog model, the Nanocrystal® suspension (120 nm) significantly outperformed a suspension of wet-milled drugs with a size of 480 nm in terms of fasted state exposure. The results from the dog model were confirmed in the clinic, with the EMEND formulation exhibiting no food effect. It is also worth mentioning that a prepitant is classified as a BCS Class II and IV compound, emphasizing that nanonizing is not only confined to high-permeability compounds but may also be applicable to low-permeability drugs, provided dissolution is the slower step in the absorption process.[ Citation 93 , Citation 95 ]
Parenteral Nanosuspension—Intravenous Administration Route
Administration of drugs via the parenteral route is critical, i.e., usually accompanied by physical and biological problems, such as production under aseptic conditions, a sophisticated protocol for safety issues, last but not least, biological problems, such as endotoxins, allergic reactions, and inconvenience for the patient. Formulation of atovaquone as a nanosuspension for intravenous (i.v.) use to treat murine toxoplasmosis showed a significant reduction of Toxoplasma gondii at a dose of 0.3 mg/kg in comparison to 30 mg/kg when given orally.[ Citation 72 ]
The nanoparticle-based product Abraxane® was approved by the Food and Drug Administration (FDA) in 2006 for intravenous administration. Abraxane® is a novel formulation consisting of lyophilized particles with 10% (w/w) paclitaxel and 90% (w/w) albumin. The particle size of the suspension is about 130 nm. In a Phase I trial, 39 patients with advanced nonhematologic malignancies were treated at dose levels from 80 to 200 mg/mL in multiple cycles of weekly 30-min intravenous infusions. In contrast to Taxol treatment, no premedication was used in this study. The maximum tolerated dose observed from this study was higher than the commercial Taxol® formulation. Furthermore, this study confirmed that the nanoparticle formulation of paclitaxel eliminates the need for premedication (because the toxic excipient Cremophor EL was not used in the formulation).[ Citation 96 ] Other investigators reported that particle size played an important role in pharmacokinetics and tissue distribution of oridonin nanosuspension.[ Citation 97 ] The pharmacokinetics and tissue distribution of oridonin nanosuspensions A (particle size of about 100 nm) and nanosuspension B (particle size of about 900 nm) were studied after intravenous administration using New Zealand rabbits and Kunming mice as experimental animals, respectively. An Oridonin control solution was studied in parallel. The results showed that oridonin nanosuspension A exhibited pharmacokinetic and biodistribution properties similar to the solution due to its rapid dissolution in the blood circulation. Oridonin nanosuspension B, however, showed a high uptake in reticuloendothelial system (RES) organs, thus exhibited a markedly different pharmacokinetic property compared to nanosuspension A.[ Citation 97 ]
Pulmonary Drug Delivery
Many water-insoluble drugs were delivered to the respiratory tract for local or systemic treatment of diseases. Unfortunately, many of these drugs are poorly soluble in aqueous solution and simultaneously insoluble in nonaqueous media. A number of years ago chlorofluorocarbon (CFC) aerosols was the best choice as drugs delivery for respiratory tract. But in compliance with the Montreal protocol of 1987, the use of chlorofluorocarbons (CFCs) must be avoided. Therefore, as alternative drugs delivery without CFCs, metered dose inhalers (MDIs) or dry powder inhalers (DPIs) were developed. The nebulized nanosuspensions produce aerosol droplets loaded with a large number of drugs nanocrystals. The respirable fraction is distinctly increased using nebulized nanosuspensions compared to nebulized microcrystals (conventional MDIs). The smaller particle the size of the drugs nanocrystals, the more droplets are loaded with drug. In addition, the muco-adhesive property of nanoparticles leads to a prolonged residence time at mucosal surface of the lung.[ Citation 76 ]
Yang et al. reported that nanosuspensions of fluctasone exhibited good physical/chemical properties for pulmonary delivery. The pharmacokinetic studies after the intra tracheal administration of nanosuspensions showed deep lung deposition and fast lung absorption, with solubility playing an important role in lung retention and duration of action. Overall, these studies have demonstrated that nanosuspensions can be used for pulmonary drug delivery in preclinical animal studies.[ Citation 98 ] Superiority of the drug nanosusupension was also shown by Hernandez-Kirstein using buparvaquone nanosuspensions for pulmonary delivery system.[ Citation 99 ]
Transdermal Delivery of Nanosuspensions
It has been reported to use diclofenac sodium nanosuspension for trans dermal delivery. The basic trans dermal characteristics of the nanosuspension were evaluated using a Yucatan micropig (YMP) skin model. Diclofenac sodium nanosuspension was successfully dispersed into isopropylmyristate as a nanosized suspension via complex formation with sucroseerucate. The resultant nanosuspension increased the permeability flux of diclofenac sodium across the skin by up to 3.8-fold compared to the control. The optimal weight ratio for the highest diclofenac sodium permeation was 8.8, at which point the mean diameter of the nanosuspension was 14.4 nm.[ Citation 100 ]
Ocular Delivery of Nanosuspensions
Drug nanosuspensions for ocular drug delivery system has been developed by Pignatello et al.[ Citation 101 , Citation 102 , Citation 103 , Citation 104 ] The codispersion of cloricromenehydrochloride (AD6) in Eudragit RS or RL polymers resulted in nanosuspensions that showed good mean sizes for ophthalmic applications and a positive surface charge. The suspensions allowed for improved corneal adhesion and stability upon storage, particularly at low temperatures. When preparation was performed in an isotonic saline solution, the dispersion of AD6 in the polymer network protected the ester drug from the hydrolytic cleavage into the inactive and insoluble acid form. According to preliminary biological evaluation of the nanosuspensions that showed a higher drug availability in the rabbit aqueous humor after the drug's administration in Eudragit RL nanosuspensions, AD6-loaded Eudragit Retard nanosuspensions appear to offer a promising means of improving the shelf life and bioavailability of this drug after ophthalmic application.[ Citation 101 ]
Nanosuspension as an ophthalmic delivery system has been also investigated by Kassem et al.[ Citation 105 ] The effect of particle size in the micron and nano size ranges as well as the effect of viscosity of the nanosuspensions on the ocular bioavailability was studied by measuring the intraocular pressure of normotensive Albino rabbits using a tonometer. The results show that compared to solution and microcrystalline suspensions, it is a common feature of the three drugs that the nanosuspensions always enhance the rate and extent of ophthalmic drug absorption as well as the intensity of drug action. In the majority of cases, nanosuspensions extend the duration of the drug effect to a significant extent. The data presented confirm that nanosuspensions differ from microcrystalline suspensions and solutions as ophthalmic drug delivery systems and that the differences are statistically highly to very highly significant. The results confirm also the importance of viscosity of nanosuspensions especially in increasing the duration of drug action.[ Citation 105 ]
CONCLUSION
As the examples have shown, nanosuspension technology offers great benefits with only a few minor drawbacks. It is clearly ideally suited for drugs with solubility problems. Particle size diminution and the resulting increase in particle surface, curvature, saturation solubility, and consequently the increased dissolution velocity are important factors. Solubility enhancement alone is not the only important factor. It becomes even more important when a drug has a narrow therapeutic window where it can be absorbed. In these cases, the increased solubility and dissolution velocity lead to an acceptable bioavailability. In addition, the nanosuspension technology enables formulations to be developed without the need of problematic surfactants (e.g., Cremophor EL), which may cause enhanced side effects or adverse reactions. Furthermore, nanosuspensions allow for a fast action onset, as the drug is absorbed quickly due to the fast dissolution of the nanoparticles. This is an advantage, especially for drugs that need to work fast (e.g., naproxen for headache relief). The enhanced solubility also leads to an identical or very similar absorption in fed and fasted conditions. Drugs that normally require food to become soluble will be bioequivalent as nanosuspensions in fed and fasted states. If it is necessary to give a large dosage in order to achieve reasonable blood levels for poorly soluble drugs resulting in increased side effects, the nanosuspension technology allows for smaller doses and thus decreased side effects. Disadvantages are the sometimes long production times, e.g., in pearl milling. However, new patents to accelerate large scale production have just been filed (e.g., H42 Müller and Moeschwitzer 2005). In addition, improved technologies are required to be able to produce tablets with high drug nanosuspension loads to formulate high-dose drugs in—preferentially—one single tablet. In the future, more drugs will be poorly soluble and thus require smart formulation technologies to make them soluble and bioavailable. An increased awareness in patients not willing to suffer from unnecessary side effects will lead to an increased number of products using nanosuspensions to reduce these risks. By modifying the nanocrystal surface it is possible to achieve a prolonged or a targeted release. This will be an important part of the work for the nanoparticles in future.
REFERENCES
- Nielloud , F. and Marti-Mestres , G. 2008 . Drugs and the Pharmaceutical Sciences, , 2nd ed. , New York : Informa Healthcare .
- Gao , L. , Zhang , D. and Chen , M. 2008 . Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system . J. Nanoparticle Res. , 10 : 845 – 862 .
- Ravichandran , R. 2009 . Nanotechnology based drug delivery systems . NanoBiotechnology , http://www.springerlink.com/content/j6440717w6232125in press. DOI: 10.1007/s12030-009-9028-2
- Liu , R. 2008 . Water-Insoluble Drug Formulation: Pharmaceutical Emulsions and Suspensions, , 2nd ed. , Baco Raton , FL : CRC Press .
- Rasenack , N. , Steckel , H. and Muller , B. W. 2003 . Micronization of anti-inflammatory drugs for pulmonary delivery by a controlled crystallization process . J. Pharm. Sci. , 92 : 35 – 44 .
- Steckel , H. , Rasenack , N. and Muller , B. W. 2003 . In-situ-micronization of disodium cromoglycate for pulmonary delivery . Eur. J. Pharm. Biopharm. , 55 : 173 – 180 .
- Steckel , H. 2003 . In vitro characterization of jet-milled and in-situmicronizedfluticasone-17-propionate . Int. J. Pharm. , 258 : 65 – 75 .
- Rasenack , N. and Muller , B. W. 2004 . Micron-size drug particles: common and novel micronization techniques . Pharm. Dev. Technol. , 9 : 1 – 13 .
- Liversidge , G. G. Surface modified drugs nanoparticles . US Patent 5,145,684 . 1992 . Sterling Drug, New York; USA
- Muller , R. H. Pharmaceutical nanosuspensions for medical administration as systems with increased saturation solubility and rate of solution . US Patent 5,858,410 . 1999 . USA
- Ostrander , K. D. , Bosch , H. W. and Bondanza , D. M. 1999 . An in-vitro assessment of a NanoCrystal beclomethasone dipropionate colloidal dispersion via ultrasonic nebulization . Eur. J. Pharm. Biopharm. , 48 : 207 – 215 .
- Sri , K. V. 2007 . Preparation and characterization of quercetin and rutin cyclodextrin inclusion complexes . Drug Dev. Ind. Pharm. , 33 : 245 – 253 .
- Calabro , M. L. 2005 . The rutin/beta-cyclodextrin interactions in fully aqueous solution: spectroscopic studies and biological assays . J. Pharm. Biomed. Anal. , 36 : 1019 – 1027 .
- Brewster , M. E. and Loftsson , T. 2007 . Cyclodextrins as pharmaceutical solubilizers . Adv. Drug Deliv. Rev. , 59 : 645 – 666 .
- Loftsson , T. 2005 . Cyclodextrins in drug delivery . Expert Opin. Drug Deliv. , 2 : 335 – 351 .
- Loftsson , T. and Brewster , M. E. 1996 . Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization . J. Pharm. Sci. , 85 : 1017 – 1025 .
- Loftsson , T. , Brewster , M. E. and Másson , M. 2004 . Role of cyclodextrins in improving oral drug delivery . Am. J. Drug. Deliv. , 2 : 1 – 15 .
- Mu , X. and Zhong , Z. 2006 . Preparation and properties of poly(vinyl alcohol)-stabilized liposomes . Int. J. Pharm. , 318 : 55 – 61 .
- Johnston , M. J. 2007 . Characterization of the drug retention and pharmacokinetic properties of liposomal nanoparticles containing dihydrosphingomyelin . Biochim. Biophys. Acta , 1768 : 1121 – 1127 .
- Kreuter , A. 2005 . Liposomal pegylated doxorubicin versus low-dose recombinant interferon Alfa-2a in the treatment of advanced classic Kaposi's sarcoma; retrospective analysis of three German centers . Cancer Invest. , 23 : 653 – 659 .
- Dannenfelser , R. M. 2004 . Development of clinical dosage forms for a poorly water soluble drug I: Application of polyethylene glycolpolysorbate 80 solid dispersion carrier system . J. Pharm. Sci. , 93 : 1165 – 1175 .
- Joshi , H. N. 2004 . Bioavailability enhancement of a poorly water-soluble drug by solid dispersion in polyethylene glycol-polysorbate 80 mixture . Int. J. Pharm. , 269 : 251 – 258 .
- Karavas , E. 2007 . Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactions . Eur. J. Pharm. Biopharm. , 66 : 334 – 347 .
- Overhoff , K. A. 2007 . Solid dispersions of itraconazole and enteric polymers made by ultra-rapid freezing . Int. J. Pharm. , 336 : 122 – 132 .
- Serajuddin , A. T. 1999 . Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs . J. Pharm. Sci. , 88 : 1058 – 1066 .
- Torchilin , V. P. 2008 . Nanotechnology in Drugs, , 2nd ed. , London : Imperial College Press .
- Sahoo , S. K. and Labhasetwar , V. 2008 . Nanotech approaches to drug delivery and imaging . Drug Discov. Today , 8 : 1112 – 1120 .
- Salata , O. V. 2004 . Applications of nanoparticles in biology and medicine . J Nanobiotechnol. , 2 : 1 – 6 .
- Suri , S. S. , Fenniri , H. and Singh , B. 2007 . Nanotechnology-based drug delivery systems . J Occup. Med. Toxicol. , 1 : 2 – 16 .
- Kirupakar , B. R. 2009 . Nanosuspension drug delivery: Technology and application . Express Pharma Pulse , : 1 – 6 . http://www.expresspharmaonline.com./20050224/nanotechol.shtml
- Chingunpituk , J. 2007 . Nanosuspension technology for drug delivery. Walailak . J. Sci, Tech. , 4 : 139 – 153 .
- US Pharmacopoeia Conventional . 2000 . The United States Pharmacopoeia, , 24th rev. ed , 8 Rockville , MD : Rockville: The United States Pharmacopoeia Conventional Inc. .
- Remington , J. P. 1980 . Remington's Pharmaceutical Sciences, , 16th ed. , 176 – 178 . Easton , PA : Mack .
- Mosharraf , M. , Sebhatu , T. and Nystrom , C. 1999 . The effects of disordered structure on the solubility and dissolution rates of some hydrophilic, sparingly soluble drugs . Int. J. Pharm. , 177 : 29 – 51 .
- Mosharraf , M. and Nystrom , C. 1999 . The effect of dry mixing on the apparent solubility of hydrophobic, sparingly soluble drugs . Eur. J. Pharm. Sci. , 9 : 145 – 156 .
- Martin , A. , Swarbrick , J. and Cammarata , A. 1993 . Physical Pharmacy: Physical Chemical Principles in the Pharmaceutical Sciences, , 4th subed. , 125 – 142 . 212 – 250 . 329 – 334 . Philadelphia : Lippincott Williams & Wilkins . Philadelphia
- Myrdal , P. B. and Yalkowsky , S. H. 2002 . Solubilization of Drugs in Aqueous Media, 2nd ed., Encyclopedia of Pharmaceutical Technology , Edited by: Swarbick , J. and Boylan , J. C. Vol. 3 , 2458 – 2580 . New York : Marcel Dekker .
- Wells , J. I. 1988 . Pharmaceutical Preformulation: The Physicochemical Properties of Drug Substances, , 1st ed. , 21 – 85 . 94 – 100 . Chichester : Ellis Horwood . Chichester
- Florence , A. T. and Attwood , D. 1981 . Physicochemical Principles of Pharmacy, , 2nd ed. , 89 – 90 . 131 – 172 . 199 – 208 . London : Macmillan Publishers .
- Sekiguchi , K. and Obi , N. 1961 . Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man . Chem. Pharm. Bull. , 9 : 866 – 872 .
- Goldberg , A. H. , Gibaldi , M. and Kanig , J. L. 1966 . Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures II. Experimental evaluation of eutectic mixture: urea acetaminophen system . J. Pharm. Sci. , 55 : 482 – 487 .
- Hildebrand , J. H. and Scott , R. L. 1950 . Solubility of Nonelectrolytes, , 3rd ed. , 11 – 13 . 47 160 175 – 197 . New York : Reinhold .
- Scatchard , G. 1931 . Equilibria in non-electrolyte solutions in relation to the vapor pressure and densities of the components . Chem. Rev. , 8 : 321 – 333 .
- Noyes , A. A. and Whitney , W. R. 1897 . The rate of solution of solid substances in their own solutions . J. Am. Chem. Soc. , 19 : 930 – 934 .
- Hixon , A. W. and Crowell , J. H. 1931 . Dependence of reaction velocity upon surface and agitation. I. Theoretical consideration . Ind. Eng. Chem. , 23 : 923 – 931 .
- Amidon , G. L. 1995 . A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability . Pharm. Res. , 12 : 413 – 420 .
- http://en.wikipedia.org/wiki/Biopharmaceutics_Classification_System
- Hite , M. , Turner , S. and Cathy Federici , R. C. 2002 . Part 1: Oral Delivery of Poorly Soluble Drugs , 1 – 3 . Bothwell , WA : Research and Product Development Group at SCOLR® Pharma Inc. .
- Hecq , J. 2005 . Preparation and characterization of nanocrystals for solubility and dissolution rate enhancement of nifedipine . Int. J. Pharm. , 299 : 167 – 177 .
- Keck , C. M. and Muller , R. H. 2006 . Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation . Eur. J. Pharm. Biopharm. , 62 : 3 – 16 .
- Merisko-Liversidge , E. , Liversidge , G. G. and Cooper , E. R. 2003 . Nanosizing: a formulation approach for poorly-water-soluble compounds . Eur. J. Pharm. Sci. , 18 : 113 – 120 .
- Muller , R. H. , Böhm , B. H. L. and Grau , M. J. 2000 . “ Nanosuspensions—a formulation approach for poorly soluble and poorly bioavailable drugs ” . In Handbook of Pharmaceutical Controlled Release , Edited by: Wise , D. L. 345 – 357 . New York : Marcel Dekker .
- Muller , R. H. , Jacobs , C. and Kayser , O. 2003 . “ DissoCubes—a novel formulation for poorly soluble and poorly bioavailable drugs ” . In Modified-Release Drug Delivery Systems , Edited by: Rathbone , M. J. , Hadgraft , J. and Roberts , M. S. 135 – 149 . New York : Marcel Dekker .
- Muller , R. H. and Akkar , A. 2004 . “ Drug nanocrystals of poorly soluble drugs ” . In Encyclopedia of Nanoscience and Nanotechnology; , Edited by: Nalwa , H. S. 627 – 638 . Valencia , CA : American Scientific Publishers .
- Muller , R. H. and Keck , C. M. 2007 . “ Improvement of delivery & solubility of poorly soluble drugs by nanonisation ” . In Konferenzdokumentation LTS Academy
- Liversidge , G. G. and Conzentino , P. 1995 . Drug particle size reduction for decreasing gastric irritancy and enhancing absorption of naproxen in rats . Int. J. Pharm. , 125 ( 2 ) : 309 – 313 .
- Muller , R. H. , Jacobs , C. and Kayser , O. 2000 . Nanosuspensions for the Formulation of Poorly Soluble Drugs, Pharmaceutical Emulsions and Suspensions , Edited by: Nielloud , F. and Marti-Mestres , G. 383 – 407 . Marcel Dekker .
- Wu , T. H. 2008 . Preparation, physicochemical characterization, and antioxidant effects of quercetin nanoparticles . Int. J. Pharm. , 346 : 160 – 168 .
- Auweter , H. Precipitated water-insoluble colorants in colloid disperse form . US Patent 6,494,924 . 2002 . BASF Aktiengesellschaft; USA
- List , M. and Sucker , H. Pharmaceutical colloidal hydrosols for injection . GB Patent 2200048 . 1988 . Sandoz Ltd., Switzerland; UK
- Sucker , H. and Gassmann , P. Improvements in pharmaceutical compositions . GB Patent 2269536A . 1994 . Sandoz Ltd., Switzerland; UK
- Liversidge , G. G. and Cundy , K. C. 1995 . Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs . Int. J.Pharm. , 125 : 91 – 97 .
- Muller , R. H. , Mäder , K. and Krause , K. Verfahren zur schonenden Herstellung von hochfeinen Micro-/Nanopartikeln . PCT Application PCT/EP00/06535 . 2000 . Germany
- Kipp , J. E. Microprecipitation method for preparing submicron suspensions . US Patent 6,607,784 . 2003 . Baxter International Inc., Deerfield, IL; USA
- Petersen , R. D. Nanocrystals for use in topical fomulations and method of production thereof . PCT/EP2007/009943 . 2006 . Abbott GmbH; Germany
- Weder , H. G. and Hoogevest , V. Nanosuspensions for intravenous administration . US Patent 5,726,164 . 1998 . USA
- Muller , R. H. , Benita , S. and Bohm , B. 1998 . Emulsions and Nanosuspensions for the Formulation of Poorly Soluble Drugs , 396 Stuttgart : Medpharm Scientific Publishers . Stuttgart
- Muller , R. H. , Möschwitzer , J. and Bushrab , F. N. 2006 . “ Manufacturing of nanoparticles by milling and homogenisation techniques ” . In Nanoparticle Technology for Drug Delivery , Edited by: Gupta , R. B. and Kompella , U. B. 21 – 52 . New York : Tayler & Francis .
- Jinno , J. 2006 . Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs . J Control Release , 111 : 56 – 64 .
- Rao , Y. M. , Pavan Kumar , M. and Apte , S. 2008 . Formulation of nanosuspensions of albendazole for oral administration . Curr. Nanosci. , 4 : 53 – 58 .
- Buckton , G. and Beezer , A. E. 1992 . The relationship between particle size and solubility . Int. J. Pharm. , 82 : R7 – R10 .
- Muller , R. H. , Jacobs , C. and Kayser , O. 2001 . Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future . Adv. Drug Deliv. Rev. , 47 : 3 – 19 .
- Mukerjee , P. 1972 . Thermodynamic aspects of solubility of small particles . J. Pharm. Sci. , 61 : 478 – 479 .
- Muller , R. H. 1998 . “ Nanosuspensionen—eine neue Formulierung fürschwerlösliche Arzneistoffe ” . In Pharmazeutische Technologie: Moderne Arzneiformen, , 2nd ed. , Edited by: Müller , R. H. and Hildebrand , G. E. 393 – 400 . Stuttgart : Wissenschaftliche Verlagsgesellschaft . Stuttgart
- Mosharraf , M. and Nystrom , C. 1995 . The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs . Int. J. Pharm. , 122 : 35 – 47 .
- Moschwitzer , J. 2006 . Drug Nanocrystals Prepared by High Pressure Homogenisation—The Universal Formulation Approach for Poorly Soluble Drugs , Dissertation Institut für Pharmazeutische Technologie, Freie Universität Berlin .
- Muller , R. H. and Peters , K. 1998 . Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique . Int. J. Pharmaceut. , 160 : 229 – 237 .
- Krause , K. P. 2000 . Heavy metal contamination of nanosuspensions produced by high-pressure homogenisation . Int. J. Pharm. , 196 : 169 – 172 .
- Muller , R. H. , Krause , K. and Mader , K. Method for controlled production of ultrafine microparticles and nanoparticles . PCT Application PCT/EP2000/006535 . 2000 . Germany
- Bushrab , N. F. and Muller , R. H. 2003 . Nanocrystals of poorly soluble drugs for oral administration . New Drugs , 5 : 20 – 22 .
- Keck , C. M. , Kobierski , S. , Mauludin , R. and Muller , R. H. 2008 . Second generation of drug nanocrystals for delivery of poorly soluble drugs: smartCrystals technology . DOSIS , 24 : 125 – 130 .
- Hu , J. 2002 . Improvement of dissolution rates of poorly water soluble APIs using novel spray freezing into liquid technology . Pharm. Res. , 19 : 1278 – 1284 .
- Hanna , M. H. and York , P. Method and apparatus for the formulation of particles . US Patent 5,851,453 . 1998 . USA
- Hu , J. , Johnston , K. P. and Williams , R. O. 3rd . 2004 . Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs . Drug. Dev. Ind. Pharm. , 30 : 233 – 245 .
- Young , T. J. 1999 . Encapsulation of lysozyme in a biodegradable polymer by precipitation with a vapor-over-liquid antisolvent . J. Pharm. Sci. , 88 : 640 – 650 .
- Pace , G. W. Process to generate submicron particles of water insoluble compounds . US Patent 6, 103 . 2001 . USA
- Young , T. J. 2000 . Rapid expansion from supercritical to aqueous solution to produce submicron suspensions of water-insoluble drugs . Biotechnol. Prog. , 16 : 402 – 407 .
- Turk , M. and Lietzow , R. 2004 . Stabilized nanoparticles of phytosterol by rapid expansion from supercritical solution into aqueous solution . AAPS Pharm. Sci. Techol. , 5 ( 4 ) http://www.aapspharmscitech.org/view.asp?art=pt050456article 56
- Rogers , T. L. , Johnston , K. P. and Williams , R. O. 3rd . 2001 . Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies . Drug Dev. Ind. Pharm. , 27 : 1003 – 1015 .
- Williams , R. O. Process for production of nanoparticles and microparticles by spray freezing into liquid . US Patent 20030041602 . 2003 . USA
- Chen , X. 2002 . Preparation of cyclosporine A nanoparticles by evaporative precipitation into aqueous solution . Int. J. Pharm. , 242 : 3 – 14 .
- Elan , P. Drug technologies nanocrystals—technology overview . Presented at the Annual Meeting of Controlled Release Sociaty (CRS) . Long Beach , CA : CRS .
- Kesisoglou , F. , Panmai , S. and Wu , Y. 2007 . Nanosizing–oral formulation development and biopharmaceutical evaluation . Adv. Drug Deliv. Rev. , 59 : 631 – 644 .
- Jacobs , C. , Kayser , O. and Muller , R. H. 2001 . Production and characterization of mucoadhesive nanosuspensions for the formulation of bupravaquone . Int. J. Pharm. , 214 : 3 – 7 .
- Scholer , N. 2001 . Atovaquone nanosuspensions show excellent therapeutic effect in a new murine model of reactivated toxoplasmosis . Antimicrobial. Agents Chemother. , 45 : 1771 – 1779 .
- Wong , J. 2008 . Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects . Adv. Drug Deliv. Rev. , 60 : 939 – 954 .
- Gao , L. 2008 . Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions . Int. J. Pharm. , 355 : 321 – 327 .
- Yang , J. Z. 2008 . Fluticasone and budesonide nanosuspensions for pulmonary delivery: preparation, characterization, and pharmacokinetic studies . J. Pharm. Sci. , 97 : 4869 – 4878 .
- Hernandez-Trejo , N. 2005 . Characterization of nebulized buparvaquonenano suspensions—effect of nebulization technology . J. Drug Target , 13 : 499 – 507 .
- Piao , H. 2008 . A novel solid-in-oil nanosuspension for transdermal delivery of diclofenac sodium . Pharm. Res. , 25 : 896 – 901 .
- Pignatello , R. 2006 . Preparation and characterization of eudragit retard nanosuspensions for the ocular delivery of cloricromene . AAPS Pharm. Sci. Techol. , 7 ( 1 ) http://www.aapspharmscitech.orgarticle 27
- Pignatello , R. , Bucolo , C. and Puglisi , G. 2002 . Ocular tolerability of EudragitRS100 and RL100 nanosuspensions as carriers for ophthalmic controlled drug delivery . J. Pharm. Sci. , 91 : 2636 – 2641 .
- Pignatello , R. 2002 . Eudragit RS100 nanosuspensions for the ophthalmic controlled delivery of ibuprofen . Eur. J. Pharm. Sci. , 16 : 53 – 61 .
- Pignatello , R. 2002 . Flurbiprofen-loaded acrylate polymer nanosuspensions for ophthalmic application . Biomaterials , 23 : 3247 – 3255 .
- Kassem , M. A. 2007 . Nanosuspension as an ophthalmic delivery system for certain glucocorticoid drugs . Int. J. Pharm. , 340 : 126 – 133 .