Abstract
The doubling of atmospheric methane (CH4) during the twentieth century due largely to growth in anthropogenic emissions has made CH4 the second largest contributor behind carbon dioxide to anthropogenic forcing of climate change. However, the global CH4 budget and its decadal evolution remain poorly quantified despite re-evaluations that include the IPCC Fourth Assessment Report. Potentially, the aggregation of national anthropogenic emission inventories as reported to the United Nations Framework Convention on Climate Change could document the changing anthropogenic emission since 1990, as could other “bottom–up” inventories such as EDGAR. As an examination of the recent CH4 budget evolution, we compare two constructions of CH4 source history, one based on an aggregation of national emission inventories, the other version 4 of EDGAR, each in combination with alternative natural CH4 emissions, for consistency with observed atmospheric mixing ratio and carbon isotope content (δ13C(CH4)). We conclude that despite the utility of isotopic constraints on budget evolution, the level of uncertainty in sink strengths and their isotopic fractionation limits the confidence in constructing anthropogenic emission histories over recent decades.
1. Introduction
The global methane (CH4) budget remains ill-quantified, notwithstanding several decades of research. According to the recent IPCC Assessment hereafter abbreviated AR4 (Denman et al. Citation2007), the global strengths of the various processes that remove tropospheric CH4 are uncertain to within about 20%, which confers a similar uncertainty on the “top–down” estimate of global CH4 emission. However, the emission contribution from individual source categories such as wetlands, coal-mining, etc, is in general poorly determined, so that source partitioning (i.e. the global source inventory) is quite poorly constrained. This has enabled significant new source categories to be postulated or discovered and still be consistent with the top–down aggregate.
A case in point is the plant source of CH4 which its discoverers assessed to be as large as 236 Tg yr−1 (Keppler et al. Citation2006), fully 40% of the global source. However, further research has revised that upper limit downward markedly (e.g. Kirschbaum et al. Citation2006; Parsons et al. Citation2006), and led some to question that plants produce CH4 at all other than as a degradation product (e.g. Beerling et al. Citation2008; Nisbet et al. Citation2009). We here ignore “plant CH4” as a significant CH4 source that is independent of that of its host ecosystem.
Another case in point is hitherto-ignored terrestrial seepages of CH4 from geologic reservoirs which Etiope et al. (Citation2008) assess at 53 ± 15 Tg yr−1.
Stable isotope information, especially 13CH4, can be used to constrain the budget. Primarily, these constraints arise because the “isotope signature”, δ13C (defined below), of an emission depends strongly on how CH4 production is mediated. Specifically, microbially mediated (“biogenic”) CH4 is relatively depleted in 13CH4 (δ13C ≈ −60‰), CH4 as a byproduct of biomass combustion (“pyrogenic CH4”) is relatively enriched (δ13C ≈ −25‰ or −12‰ for C3 or C4 biomass), while “fossil CH4” has an intermediate signature (δ13C ≈ −40‰). While the contemporary signature in atmospheric CH4 is well known at about −47‰ through measurements at various sites (e.g. Quay et al. Citation1999; Tyler et al. Citation2007; Lassey et al. Citation2010) and its history can be reconstructed from measurements in air extracted from polar ice (e.g. Ferretti et al. Citation2005), the isotope fractionation associated with the various sink processes and the relative roles of those processes confer some uncertainty. In addition, the minor radio-isotopologue 14CH4 of which fossil CH4 is uniquely devoid has been used to assess the “fossil fraction” in the CH4 source (Quay et al. Citation1999; Lassey et al. Citation2007b).
The temporal pattern in the global source history can arguably be constructed with at least as much confidence as the budget at a time snapshot. Lassey et al. (Citation2007a) have demonstrated that this pattern together with its companion pattern in δ13C can be used to further constrain the budget history. This constraint pre-supposes that any systematic change in isotope fractionation of the combined (first-order) sink is not the principal cause of systematic variation in atmospheric δ13C. Applying mass balance to postulated CH4 and δ13C source histories yields a simulated history of atmospheric δ13C to within an additive offset. One can then examine for reasonableness both the match between the simulated and historical patterns of atmospheric δ13C and the offset between them. Lassey et al. (Citation2007a) argued that the offset should be consistent with a fractionation of magnitude 7.7 ± 1.4‰ (95% confidence interval), revised here to 7.6 ± 1.4‰ (). This approach has been used to address the reconciliation of constructed source inventories with the atmospheric record (Lassey et al. Citation2005, Citation2007a).
Table 1. Representative construction of the global contemporary methane sink and isotope fractionation.a
The aim here is to extend earlier work (Lassey et al. Citation2005) to examine the compatibility of globally-aggregated “bottom–up” anthropogenic emission inventories with the global CH4 cycle, constrained by δ13C data. Two such aggregations are examined, each in tandem with two alternative constructions of the natural emission inventory with contrasting δ13C. Further constraints by 14CH4 data are included in the formalism but their implementation is outside the present scope.
2 Modelling strategy
With the modelling strategy described in detail elsewhere (Lassey et al. Citation2007a), only a brief summary is presented here.
Both the global CH4 budget and that of each isotopologue separately (viz, 12CH4, 13CH4, 14CH4, identified by subscripts “12”, “13”, “14”) satisfy global mass balance:
A specification of any two of the time series C(t), S(t) and λ(t) plus an initial state over a time interval fully defines the budget history over that interval. An initial steady state is prescribed for 1700AD prior to the near-monotonic growth in atmospheric CH4 (e.g. Ferretti et al. Citation2005). With this long spin-up time, simulations of recent decades when national emission inventories are available are essentially independent of initial state, even allowing for the slowness of δ13C(CH4) to respond to source perturbations (Tans Citation1997; Lassey et al. Citation2000). With largely aseasonal input to the model, integration from 1700 is discretised into annual time steps. Indeed, processes within polar ice and firn limit temporal resolution available to several years. The discretised EquationEquation (1) can be solved for C(t) in forward mode or for either S(t) or λ(t) in inverse mode, and we adopt a combination of these to apply isotopic mass balance as summarised in the following subsections.
2.1 Total methane
Of the three time series in EquationEquation (1), C(t) has the highest quality through mixing ratio determinations in air extracted from polar ice (MacFarling Meure et al. Citation2006) and, since the 1980s, through direct atmospheric monitoring (e.g. Dlugokencky et al. Citation2009). We apply the MacFarling Meure (Citation2006) dataset for C(t), and construct time series of the global CH4 source to define S(t) (Section 3). The history of removal rate λ(t) is then “inverse modelled” through mass balance, EquationEquation (1).
2.2 The stable isotopologue 13CH4
With time series C(t), S(t) and λ(t) all defined from 1700, mass balance can be addressed separately for each isotopologue through “forward modelling”. By prescribing a time-independent sink fractionation ε ≡ λ13/λ12−1 both λ13(t) and λ12(t) follow immediately as time series proportional to λ(t). Further assigning δ13C to each source component allows S
13(t) and S
12(t) to be constructed. EquationEquation (1) is then solved separately for C
13(t) and C
12(t) whose sum reproduces C(t) but whose ratio allows the evolving tropospheric δ13C to be constructed. Here, δ13C follows the usual definition:
2.3 The radio-isotopologue 14CH4
The 14CH4 cycle has been studied for its value in constraining the “fossil fraction” in the global contemporary CH4 source (e.g. Quay et al. Citation1999). This constraint, which is predicated on fossil CH4 sources being devoid of 14C, is severely weakened by the need to construct two time series that are poorly guided by data: the 14C content in non-fossil CH4 sources (modified by “bomb carbon”); and the direct atmospheric release of 14CH4 by the commercial nuclear industry. Lassey et al. (Citation2007a) proposed an approach to minimise the impact of these weaknesses, while Lassey et al. (Citation2007b) applied a regression analysis that, with certain assumptions, simultaneously constrained both the fossil fraction and strength of the nuclear-industry source. While the approach of Lassey et al. (Citation2007a) can be applied to the present study, it is outside the scope and constraints of this article.
3 Source construction
A natural and an anthropogenic global source component are combined and extrapolated back in annual time steps to a postulated steady state in 1700. These are selected from two alternative natural source inventories (Section 3.1), and two alternative anthropogenic histories (Section 3.2).
3.1 The natural source
One of two alternative natural source inventories, “Nat1” or “Nat2” of , is applied to every year since 1700. While there will have been systematic emission variations over time, such variations are poorly documented and would have been dwarfed by the growth in anthropogenic emissions during the twentieth century, and likely even by the uncertainty in that growth.
Table 2. Natural global source scenarios, Nat1 and Nat2.
Scenario Nat1 is the natural source previously adopted by Lassey et al. (Citation2005, Citation2007a) and taken from Houweling et al. (Citation2000), aside from a small change in the δ13C assignment for geologic sources after Etiope et al. (Citation2009), −40‰ to −45‰, that lightens the total natural source by 0.3‰. Nat2 embraces a reappraised geologic CH4 source that takes account of previously-overlooked terrestrial CH4 seeps (Etiope et al. Citation2008). We elect to retain the same total source as for Nat1, 222 Tg yr−1, in order that simulation differences can be related to isotopic contrasts between Nat1 and Nat2 without ambiguity from source-strength differences. The geologic source in Nat2 is therefore enlarged at the partial expense of wetland emission which nevertheless still lies within accepted bounds and is similar to that recently proposed by Kaplan et al. (Citation2006) of ∼110 Tg yr−1.
3.2 The anthropogenic source
Two globally-aggregated anthropogenic source histories are denoted “EPA06” and “Edgar4”.
EPA06 (USEPA Citation2006) comprises a consistent and comprehensive time series of CH4 emission based on emission inventories compiled and submitted to UNFCCC by over 90 individual countries. Careful scrutiny has identified data gaps, including missing countries, and filled them from authoritative reports or by applying IPCC estimation methodologies to assure completeness and consistency. While inventory compilation may be perceived as lacking validation and scientific rigour, it can nonetheless accommodate local knowledge that uncritical use of global data could miss. Inventories are constructed for every 5th year, 1990–2020, of which 2005–2020 are projections based on established mitigation programmes, sector agreements, or measures already in place, but excluding planned mitigation activities that have yet to be implemented. illustrates the dataset for eight country groupings, including projections beyond 2005 that are not considered here. illustrates EPA06 for 2000 grouped by biogenic, pyrogenic and fossil components.
Figure 1. EPA06 dataset of anthropogenic CH4 emissions by eight country groupings at 5-yearly increments from 1990 projected to 2020, along with the world total shrunk by a factor of 5.
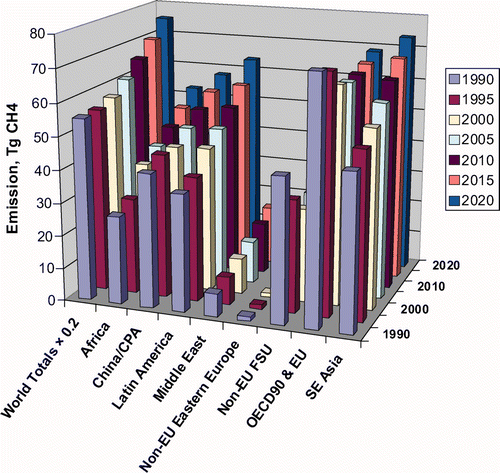
Table 3. Anthropogenic global source EPA06 for 2000.a
Edgar4 is the most recent release of EDGAR (“Emission Database for Global Atmospheric Research”) which is available and documented at http://edgar.jrc.ec.europa.eu/ (J.G.J. Olivier, personal communication). Annual emissions for 1970–2005 are reported, and display inter-annual variations that make it less smooth than EPA06. Since C(t) is inherently smoothed, these variations propogate into a removal rate λ(t) (Section 2.1) with inter-annual variations that are at least partially spurious.
Model integration requires that the source be extrapolated in annual steps back to the initial state in 1700. The detailed realism of the entire extrapolation is less important than the requirement of a plausible transition through the mid-late twentieth century. We follow Lassey et al. (Citation2005, Citation2007a) by applying Edgar-Hyde (E-H) v1.4, an emission time series for 1890–1995. E-H v1.4, available from http://www.mnp.nl/edgar/, updates v1.3 (van Aardenne et al. Citation2001) to match EDGAR v3.2 (Olivier and Berdowski 2001). EPA06 and Edgar4 are each extrapolated back to 1890 by scaling the biogenic, fossil and pyrogenic components of E-H v1.4 to match those of EPA06 in 1990 and of Edgar4 in 1970. Each scaled dataset is extrapolated linearly from 1890 back to 30 Tg yr−1 assumed as part of a steady source of 252 Tg yr−1 for 1700 (Houweling et al. Citation2000; Lassey et al. Citation2007a).
For application herein, each source history is interpolated onto an annualised series, which ensures a reasonably smoothly changing time series other than for Edgar4 after 1970.
4 Results
and show the model inputs, C(t) and S(t), respectively. The turnover time required by mass balance is shown in , and the simulated atmospheric δ13C evolution in . Only the period 1950 to 2005 is highlighted in the absence of unexpected features in the pre-1950 period which in any case could be unduly influenced by pre-1990 (EPA06) or pre-1970 (Edgar4) back-extrapolation of the source. The turnover times in are not fully compatible in magnitude because they depend on the strength of the particular sink process or aggregation of processes being considered, recognising that turnover time is the ratio of atmospheric burden to sink strength.
Figure 2. The upper two panels show the 1950–2005 sub-intervals of the model inputs, and the lower panels the corresponding modelled output, with atmospheric data in the left-hand panels. is the globally-adjusted MacFarling Meure (Citation2006) mixing ratio dataset, C(t), with illustrative annual global-mean data from the NOAA/ESRL network (E.J. Dlugokencky, personal communication), and Southern Hemisphere datasets from Baring Head, New Zealand (Lassey et al. Citation2010) and from archived samples collected at Cape Grim, Australia (Francey et al. Citation1999). shows the back-extrapolated EPA06 and Edgar4 constructions of the anthropogenic source history, each of which is augmented by a natural source of 222 Tg yr−1 to give S(t). shows the corresponding turnover times, λ(t)−1, along with modelled estimates of turnover time based on OH chemistry alone (Karlsdóttir and Isaksen Citation2000; Dentener et al. Citation2003). shows the modelled atmospheric δ13C evolution for four combinations of natural and anthropogenic source histories, each for a value of ε (cited in square brackets in the legend) chosen to provide an adequate fit to δ13C data from the Southern Hemisphere (Craig et al. Citation1988, Francey et al. Citation1999, Ferretti et al. Citation2005).
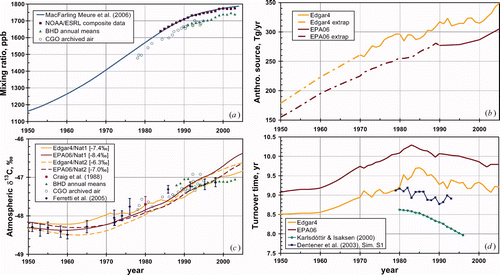
The choice of isotope fractionation, ε, determines the position on the δ13C axis in . The value chosen to optimally account for data in recent decades is indicated in the legend. Since those data all originate from the Southern Hemisphere (SH), each choice of ε requires adjustment for an inter-hemispheric gradient in δ13C(CH4) of ∼0.3‰ (heavier in the SH atmosphere) (Lassey et al. Citation2007a). That adjustment would decrease the magnitude of each ε reported in the legend to by ∼0.15‰. Thus, only for Edgar4 with Nat2 is the adjusted ε value outside the 95% confidence interval for εtotal =−7.6 ± 1.4‰ (), and even then only by ∼0.05‰.
5 Discussion and conclusions
The mass-balancing sink appears to have slowly weakened through the industrial era until the 1980s (), noting that inter-annual sink variations may be partially an artefact of a smooth C(t). This is qualitatively consistent with model simulations of OH chemistry that suggest such weakening (Lelieveld et al. Citation1998) followed by 1980s strengthening (Karlsdóttir and Isaksen Citation2000; Dentener et al. Citation2003). Recognising the inverse relationship between turnover time and sink strength, the IPCC AR4 with larger sink and source than considered here (581 Tg yr−1 ± 15%, excluding the chlorine sink) also requires a longer turnover time (8.7 ± 1.3 yr).
Of the four combinations of natural and anthropogenic source constructions, all give a reasonable portrayal of the recent atmospheric δ13C pattern observed in the SH, once a reconciling ε value () is chosen. Edgar4 even partially captures the levelling off in δ13C since the late 1990s at Baring Head, probably due to the inclusion in Edgar4 of growing biogenic emissions such as from livestock. For all combinations of anthropogenic and natural inventory except Edgar4 with Nat2 the chosen ε value is within the acceptable range defined by . Given the quite different fossil components of Nat1 and Nat2 (), simulations of 14CH4 could potentially be more discriminating.
The available atmospheric δ13C(CH4) data, dominantly from the SH, are adequately simulated within uncertainty bounds by three of the four combinations of natural and anthropogenic source history. These uncertainties are strongly influenced by poor knowledge of the relative roles of the various sink processes and their isotopic fractionations (). With the additional uncertainty inherent in constructing inventories of natural emissions, anthropogenic source inventories remain poorly constrained by available data.
Acknowledgements
Funding support from the NZ Foundation for Research, Science and Technology to develop the model, and from the US Environmental Protection Agency to apply it is acknowledged. Useful discussions with Bill Allan (NIWA) and Jos Olivier (PBL, Netherlands) are also acknowledged.
Additional information
Notes on contributors
K.R. Lassey
The views of the authors do not necessarily represent the views of the U.S. Government or the Environmental Protection Agency.Related Research Data
References
- Allan , W , Struthers , H and Lowe , D C . 2007 . Methane carbon isotope effects caused by atomic chlorine in the marine boundary layer: Global model results compared with southern hemisphere measurements . J Geophys Res. , 112 : D04306 doi:10.1029/2006JD007369
- Beerling , D J , Gardiner , T , Leggett , G , McLeod , A and Quick , W P . 2008 . Missing methane emissions from leaves of terrestrial plants . Global Change Biol. , 14 ( 8 ) : 29 – 35 .
- Cantrell , C A , Shetter , R E , McDaniel , A H , Calvert , J G , Davidson , J A , Lowe , D C , Tyler , S C , Cicerone , R J and Greenberg , J P . 1990 . Carbon kinetic isotope effect in the oxidation of methane by the hydroxyl radical . J Geophys Res. , 95 ( D13 ) : 22455 – 22462 .
- Craig , H , Chou , C C , Welhan , J A , Stevens , C M and Engelkemeir , A . 1988 . The isotopic composition of methane in polar ice cores . Science. , 242 : 1535 – 1539 .
- Crowley , J N , Saueressig , G , Bergamaschi , P , Fischer , H and Harris , G W . 1999 . Carbon kinetic isotope effect in the reaction CH4 + Cl: a relative rate study using FTIR spectroscopy . Chem Phys Lett. , 303 : 268 – 274 .
- Denman , K L , Brasseur , G , Chidthaisong , A , Ciais , P , Cox , P M , Dickinson , R E , Hauglustaine , D , Heinze , C , Holland , E Jacob , D . 2007 . “ Couplings between changes in the climate system and biogeochemistry ” . In Climate change 2007: the physical science basis. Contribution of Working Group I to the fourth assessment report of the Intergovernmental Panel on Climate Change , Edited by: Solomon , S , Qin , D , Manning , M , Marquis , M , Averyt , K , Tignor , M MB , Miller , H L and Chen , Z . 499 – 587 . Cambridge, , UK : Cambridge University Press .
- Dentener , F , Peters , W , Krol , M , van Weele , M , Bergamaschi , P and Lelieveld , J . 2003 . Interannual variability and trend of CH4 lifetime as a measure for OH changes in the 1979–1993 time period . J Geophys Res. , 108 ( D15 ) : 4442 doi:10.1029/2002JD002916
- Dlugokencky , E J , Bruhwiler , L , White , J WC , Emmons , L K , Novelli , P C , Montzka , S A , Masarie , K A , Lang , P M , Crotwell , A M Miller , J B . 2009 . Observational constraints on recent increases in the atmospheric CH4 burden . Geophy Res Lett. , 36 : L18803 doi:10.1029/2009GL039780
- Etiope , G , Feyzullayev , A and Baciu , C L . 2009 . Terrestrial methane seeps and mud volcanoes: a global perspective of gas origin . Marine Petrol Geol. , 26 : 333 – 344 .
- Etiope , G , Lassey , K R , Klusman , R W and Boschi , E . 2008 . Reappraisal of the fossil methane budget and related emission from geologic sources . Geophys Res Lett. , 35 : L09307 doi:10.1029/GL2008033623
- Ferretti , D F , Miller , J B , White , J WC , Etheridge , D M , Lassey , K R , Lowe , D C , MacFarling Meure , C M , Dreier , M F , Trudinger , C M van Ommen , T D . 2005 . Unexpected changes to the global methane budget over the past 2000 years . Science. , 309 ( 5741 ) : 1714 – 1717 .
- Francey , R J , Manning , M R , Allison , C E , Coram , S A , Etheridge , D M , Langenfelds , R L , Lowe , D C and Steele , L P . 1999 . A history of δ13C in atmospheric CH4 from the Cape Grim Air Archive and Antarctic firn air . J Geophys Res. , 104 ( D19 ) : 23631 – 23643 .
- Houweling , S , Dentener , F and Lelieveld , J . 2000 . Simulation of preindustrial methane to constrain the global source strength of natural wetlands . J Geophys Res. , 105 ( D13 ) : 17243 – 17255 .
- Kaplan , J O , Folberth , G and Hauglustaine , D A . 2006 . Role of methane and biogenic volatile organic compound sources in late glacial and Holocene fluctuations of atmospheric methane concentrations . Global Biogeochem Cycl. , 20 : GB2016 doi:10.1029/2005GB002590
- Karlsdóttir , S and Isaksen , I SA . 2000 . Changing methane lifetime: possible cause for reduced growth . Geophys Res Lett. , 27 ( 1 ) : 93 – 96 .
- Keppler , F , Hamilton , J TG , Braß , M and Röckmann , T . 2006 . Methane emissions from terrestrial plants under aerobic conditions . Nature. , 439 ( 7073 ) : 187 – 191 .
- Kirschbaum , M UF , Bruhn , D , Etheridge , D M , Evans , J R , Farquhar , G D , Gifford , R M , Paul , K I and Winters , A J . 2006 . A comment on the quantitative significance of aerobic methane release by plants . Funct Plant Biol. , 33 : 521 – 530 .
- Lassey , K R , Brailsford , G W , Bromley , A M , Martin , R J , Moss , R C , Gomez , A J , Sherlock , V , Allan , W , Nichol , S E Schaefer , H . 2010 . Recent changes in methane mixing ratio and its 13C content observed in the southwest Pacific region . J Integr Environ Sci. (this issue) ,
- Lassey , K R , Etheridge , D M , Lowe , D C , Smith , A M and Ferretti , D F . 2007a . Centennial evolution of the atmospheric methane budget: what do the carbon isotopes tell us? . Atmos Chem Phys. , 7 : 2119 – 2139 .
- Lassey , K R , Lowe , D C and Manning , M R . 2000 . The trend in atmospheric methane δ13C and implications for isotopic constraints on the global methane budget . Global Biogeochem Cycl. , 14 ( 1 ) : 41 – 49 .
- Lassey , K R , Lowe , D C and Smith , A M . 2007b . The atmospheric cycling of radiomethane and the “fossil fraction” of the methane source . Atmos Chem Phys. , 7 : 2141 – 2149 .
- Lassey , K R , Scheehle , E A and Kruger , D . 2005 . Towards reconciling national emission inventories for methane with the global budget . Environ Sci. , 2 ( 2–3 ) : 193 – 204 .
- Lelieveld , J , Crutzen , P J and Dentener , F J . 1998 . Changing concentration, lifetime and climate forcing of atmospheric methane . Tellus. , 50B ( 2 ) : 128 – 150 .
- MacFarling Meure , C , Etheridge , D , Trudinger , C , Steele , P , Langenfelds , R , van Ommen , T , Smith , A and Elkins , J . 2006 . Law dome CO2, CH4 and N2O ice core records extended to 2000 years BP . Geophys Res Lett. , 33 : L14810 doi:10.1029/2006GL026152
- Nisbet , R ER , Fisher , R , Nimmo , R H , Bendall , D S , Crill , P M , Gallego-Sala , A V , Hornibrook , E RC , López-Juez , E , Lowry , D Nisbet , P BR . 2009 . Emission of methane from plants . Proc Royal Soc B. , 276 : 1347 – 1354 .
- Olivier , J GJ and Berdowski , J JM . 2001 . “ Global emission sources and sinks ” . In The climate system , Edited by: Berdowski , J , Guicherit , R and Heij , B J . 33 – 78 . Lisse, , The Netherlands : A. A. Balkema Publishers/Swets & Zeitlinger Publishers .
- Parsons , A J , Newton , P CD , Clark , H and Kelliher , F M . 2006 . Scaling methane emissions from vegetation . Trends Ecol Evol. , 21 ( 8 ) : 423 – 424 .
- Quay , P D , Stutsman , J , Wilbur , D , Snover , A , Dlugokencky , E J and Brown , T . 1999 . The isotopic composition of atmospheric methane . Global Biogeochem Cycl. , 13 ( 2 ) : 445 – 461 .
- Saueressig , G , Crowley , J N , Bergamaschi , P , Brühl , C , Brenninkmeijer , C AM and Fischer , H . 2001 . Carbon 13 and D kinetic isotope effects in the reaction of CH4 with O(1D) and OH: new laboratory measurements and their implications for the isotopic composition of stratospheric methane . J Geophys Res. , 106 ( D19 ) : 23127 – 23138 .
- Snover , A K and Quay , P D . 2000 . Hydrogen and carbon kinetic effects during soil uptake of atmospheric methane . Global Biogeochem Cycl. , 14 ( 1 ) : 25 – 39 .
- Tans , P P . 1997 . A note on isotope ratios and the global atmospheric methane budget . Global Biogeochem Cycl. , 11 ( 1 ) : 77 – 81 .
- Tyler , S C , Ajie , H O , Rice , A L , Cicerone , R J and Tuazon , E C . 2000 . Experimentally determined kinetic isotope effects in the reaction of CH4 with Cl: implications for atmospheric CH4 . Geophys Res Lett. , 27 ( 12 ) : 1715 – 1718 .
- Tyler , S C , Crill , P M and Brailsford , G W . 1994 . 13C/12C fractionation of methane during oxidation in a temperate forested soil . Geochim Cosmochim Acta. , 58 : 1625 – 1633 .
- Tyler , S C , Rice , A L and Ajie , H O . 2007 . Stable isotopes in atmospheric CH4: implications for seasonal sources and sinks . J Geophys Res. , 112 : D03303 doi:10.1029/2006JD007231
- USEPA . 2006 . Global anthropogenic non-CO2 greenhouse gas emissions: 1990–2020 Report 430-R-06-005, U.S. Environmental Protection Agency, Washington DC
- van Aardenne , J A , Dentener , F J , Olivier , J GJ , Klein Goldewijk , C GM and Lelieveld , J . 2001 . A 1° × 1° resolution data set of historical anthropogenic trace gas emissions for the period 1890–1990 . Global Biogeochem Cycl. , 15 ( 4 ) : 909 – 928 .