Abstract
In order to determine the levels of ochratoxin A (OTA) in cocoa and cocoa products available in Canada, a previously published analytical method, with minor modifications to the extraction and immunoaffinity clean-up and inclusion of an evaporation step, was initially used (Method I). To improve the low method recoveries (46–61%), 40% methanol was then included in the aqueous sodium bicarbonate extraction solvent (pH 7.8) (Method II). Clean-up was on an Ochratest™ immunoaffinity column and OTA was determined by liquid chromatography (LC) with fluorescence detection. Recoveries of OTA from spiked cocoa powder (0.5 and 5 ng g−1) were 75–84%; while recoveries from chocolate were 93–94%. The optimized method was sensitive (limit of quantification (LOQ) = 0.07–0.08 ng g−1), accurate (recovery = 75–94%) and precise (coefficient of variation (CV) < 5%). It is applicable to cocoa and chocolate. Analysis of 32 samples of cocoa powder (16 alkalized and 16 natural) for OTA showed an incidence of 100%, with concentrations ranging from 0.25 to 7.8 ng g−1; in six samples the OTA level exceeded 2 ng g−1, the previously considered European Union limit for cocoa. The frequency of detection of OTA in 28 chocolate samples (21 dark or baking chocolate and seven milk chocolate) was also 100% with concentrations ranging from 0.05 to 1.4 ng g−1; one sample had a level higher than the previously considered European Union limit for chocolate (1 ng g−1).
Introduction
Ochratoxin A (OTA, L-phenylalanylcarbonyl-5-chloro-8-hydroxy-3,4-dihydro-3-R-methylisocoumarin) is a mycotoxin formed by certain species of Aspergillus and Penicillium (Bayman and Baker Citation2006; Clark and Snedeker Citation2006). It is carcinogenic, nephrotoxic, teratogenic, immunotoxic and hepatotoxic, although the mechanism of action, whether genotoxic or epigenetic in nature, remains unclear (O’Brien and Dietrich Citation2005) and the International Agency for Research on Cancer (IARC) has classified it as possibly carcinogenic to humans (group 2B) (IARC Citation1993). OTA is found in grains and many other kinds of foodstuffs, including cocoa and cocoa products (Bayman and Baker Citation2006; Clark and Snedeker Citation2006; Codex Alimentarius Commission Citation2007, Citation2008). The European Commission had previously considered maximum limits (MLs) for OTA in cocoa products that included 2 µg kg−1 in cocoa powder (Codex Alimentarius Commission Citation2007), which may have had the potential to affect adversely the producing countries (Bonvehi Citation2004) and 1 µg kg−1 in consumer cocoa products such as chocolate (Codex Alimentarius Commission Citation2007). Recently, the European Commission stated that ‘on the basis of the information available, it does not appear necessary for the protection of public health to set a maximum level of OTA in … cocoa and cocoa products …’ (European Commission Citation2010). No limits for OTA in cocoa have been proposed in Canada, although proposals have been made for limits in many other foods (Health Canada Citation2009). A recent Canadian health risk assessment of OTA did not consider cocoa products consumed in Canada (Kuiper-Goodman et al. Citation2010), but surveillance data were still needed.
Most of the OTA present in cocoa beans is found in the shell (Amézqueta et al. Citation2005) and is linked to pod defects, climatic conditions during harvest and poor storage/processing conditions (Codex Alimentarius Commission Citation2008). Cocoa is finely ground cocoa cake that remains after most of the cocoa butter is removed. Chocolate typically consists of cocoa butter, chocolate liquor, sugar and milk solids. In this surveillance of cocoa and cocoa products obtained in Canada, alkalized cocoa, natural cocoa powder (no added alkali) and chocolate were analysed for OTA. Alkalization of cocoa (Dutch process) neutralizes the normal cocoa acidity and raises the pH into the 7–8 range.
Materials and methods
The first method used (Method I) was based on that of Brera et al. (Citation2003) and applied to analysis of cocoa powder samples; minor modifications were made to the extraction and immunoaffinity clean-up and an evaporation step was included. Later, the method was further optimized and 40% methanol was included in the aqueous sodium bicarbonate extraction solvent (Method II); this method was applied to both cocoa and chocolate. The methods are described in detail below.
Chemicals
Ochratoxin A (>98%) was purchased from Sigma-Aldrich (Oakville, ON, Canada) and stored at −20°C. Toluene, acetonitrile and methanol (≥99.9%) were high-performance liquid chromatography (HPLC) grade. Acetic acid (glacial), NaHCO3, 0.1 M HCl, 0.1 M NaOH, Na2HPO4, KH2PO4, KCl, and NaCl were ACS grade. Polyethylene glycol (PEG 8000) was Ultra grade. MilliQ water was ultra-pure (>18 megohm.cm). BF3, 14% in methanol, was from Sigma-Aldrich and stored at approximately 4°C. Phosphate-buffered saline (PBS) (pH 7.4) was prepared by mixing 1.16 g Na2HPO4, 0.2 g KH2PO4, 0.2 g KCl and 8 g NaCl and making up to 1 L with MilliQ water (solution adjusted to pH 7.4 with 0.1 M HCl if necessary).
Sixty samples of cocoa products were provided by the Canadian Food Inspection Agency or purchased in local stores.
Standard solutions
Solutions were stored in silanized glass vials at −18°C. Stock OTA standard solution was 10 µg ml−1 OTA in toluene–acetic acid (99 : 1, v/v); intermediate solution 1 was 2 µg ml−1 OTA in toluene–acetic acid (99 : 1, v/v); intermediate solution 2 (prepared from intermediate solution 1) was 40 ng ml−1 OTA in injection solvent water–methanol–acetic acid (70 : 30 : 1, v/v/v); LC calibration standards were 0.2–5.0 ng ml−1 OTA in injection solvent, filtered through a 0.45 µm polytetrafluoroethylene (PTFE) filter. The spiking solution was 50 ng ml−1 OTA in methanol (prepared from intermediate solution 1).
Apparatus
Cocoa products were extracted using a Kinematica Polytron® Homogenizer (Bohemia, NY, USA), Model PT 10/35 with generator PTA-10TS (12 mm OD). OchraTest WB columns were from Vicam (product number G1034, Watertown, MA, USA) and stored at approximately 4°C. LC was performed with an Agilent 1100 series HPLC (Agilent Technologies, Mississauga, ON, Canada) equipped with a quaternary pump (G1311A), autosampler (G1313A), fluorescence detector (G1321A), degasser (G1322A), using an Opti-guard C18 (Optimize Technologies, Oregon City, OR, USA) guard column, and analytical column: 4.6 × 50 mm × 1.8 µ Zorbax Eclipse plus C18 Rapid Resolution High Throughput (RRHT) column.
Extraction and clean-up
Samples (5 g) were extracted with 100 ml of 0.1% NaHCO3 + 0.3% PEG in water (adjusted to pH 7.8 with 0.1 M HCl or 0.1 M NaOH) (Method I) or 0.1% NaHCO3 + 0.3% PEG in water (adjusted to pH 7.8 with 0.1 M HCl or 0.1 M NaOH) and methanol (60 : 40, v/v) (Method II) in a 250-ml Nalgene centrifuge bottle for approximately 3 min at Polytron speed 5. After centrifuging for 15 min at 2000 rpm (15°C), the extract was filtered through No. 4 filter paper into a 125-ml glass Erlenmeyer flask. A 5 ml aliquot was transferred to a 50-ml polypropylene Falcon tube. A total of 20 ml of PBS was added, mixed and then the mixture was filtered through a 1 µm GFM Acrodisc filter fitted on a 30-ml syringe.
The immunoaffinity column (IAC) placed on an SPE manifold (Supelco, Bellefonte, PA, USA) was conditioned with 5 ml of PBS at approximately one to two drops per second, maintaining approximately 1 cm of solvent above the IAC antibodies at all times. The sample extract/PBS (25 ml) was transferred into a reservoir above the IAC and loaded at approximately one to two drops per second. A total of 10 ml of MilliQ water was added to the Falcon tube, mixed, transferred to the reservoir and loaded on to the IAC at approximately one to two drops per second. The reservoir was removed and the IAC drained, applying slight vacuum to drain fully. (The IAC should not go dry.) The first portion of elution solvent was immediately added followed by a 5-min wait. OTA was eluted with two portions of 0.75 ml methanol into a 2 ml silanized autosampler vial, using gravity flow and applying slight positive pressure to drain the IAC fully. The eluates were evaporated to dryness with nitrogen at 40°C, 500 µl of injection solvent (water–methanol–acetic acid (70 : 30 : 1, v/v/v) were added, and the solution was vortexed then filtered into a silanized autosampler vial by syringe pressure through a 0.45 µm Teflon 4-mm filter. Sample concentration in the final extract was 0.5 g ml−1. For each new lot of IACs, a 5 ml water blank was extracted.
LC determination
Determination of OTA was performed by reversed-phase LC, using fluorescence detection with an excitation wavelength of 330 nm and an emission wavelength of 460 nm. The column heater was set at 40 ± 0.5°C. The isocratic mobile phase used was acetonitrile–water–acetic acid (50 : 49 : 1, v/v/v) (filtered) with a flow rate of 0.5 ml min−1 (Method I) or water–acetonitrile–acetic acid (54 : 45 : 1, v/v/v) (filtered) with a flow rate of 0.6 ml min−1 (Method II). A five-point standard curve covering 0.2–5 ng OTA ml−1 was prepared (r 2 > 0.9997), and 50 µl were injected into the LC. The instrumental limit of detection (LOD) from standard in injection solvent (3 × the signal-to-baseline noise (S/N)) was 0.01 ng OTA ml−1 (0.5 pg) and the instrumental limit of quantification (LOQ) was 0.03 ng OTA ml−1 (10 × S/N). Typical retention times of OTA were about 4.2 min (Method I) and 5.2 min (Method II).
A typical instrument sequence consisted of a blank (injection solvent), three calibration standards (0.2, 0.5 and 1 ng ml−1 OTA), six sample extracts, two calibration standards (2 and 5 ng ml−1 OTA) and a methanol–water wash before LC shutdown.
Confirmation of results
Confirmation by formation of the methyl ester with BF3-methanol (Bonvehi Citation2004) was performed when OTA levels above 2 ng g−1 in cocoa and 1 ng g−1 in chocolate were found.
A total of 300 µl of extract in a 2 ml autosampler vial were evaporated to dryness under nitrogen at 60°C then cooled to room temperature. A total of 300 µl of 14% BF3 in methanol were added, vortexed, kept at 60°C for 20 min, then evaporated to dryness under nitrogen at 60°C. After cooling, 300 µl of injection solvent were added, and the vial was capped and vortexed. The mixture was filtered into a silanized autosampler vial by syringe pressure through a 0.45 µm Teflon 4-mm filter, then transferred to a 250 µl flat bottom vial insert for LC analysis under the same conditions as for OTA.
Results and discussion
Initially the method of Brera et al. (Citation2003) with some minor modifications (Method I) was used for the analysis of cocoa for OTA, but recoveries were lower than expected (). However, when 40% methanol was included in the aqueous NaHCO3 extraction solvent (pH 7.8) (Method II), recoveries of OTA from cocoa improved considerably (). Additional single recoveries from alkalized cocoa at 0.5 and 5 ng g−1 spiking levels were 75% and 84%, respectively. It was necessary to modify the LC mobile phase to resolve the OTA because the original LC mobile phase for Method I did not separate co-extracted chemicals that eluted near the OTA. This is illustrated in for baking chocolate. The optimized method is sensitive (LOQ = 0.07–0.08 ng g−1), accurate (recovery = 75–94%) and precise (CV < 5%). It is applicable to both cocoa and chocolate. Traces of OTA, equivalent to 0.05 ng g−1 sample concentration, were detected from the IAC column when solvent blanks were run. These levels are below the LOQ for cocoa and chocolate and were not subtracted from reported values for cocoa and chocolate.
Figure 1. Chromatogram of baking chocolate (0.35 ng OTA g−1) extracted with 40% methanol and analysed with mobile phase conditions from Method I (upper curve) and Method II (lower curve).
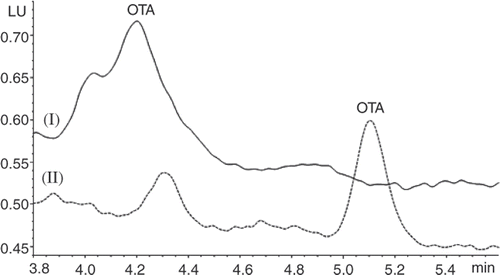
Table 1. Validation data for cocoa (Method I).
Table 2. Validation data for natural cocoa and chocolate (Method II).
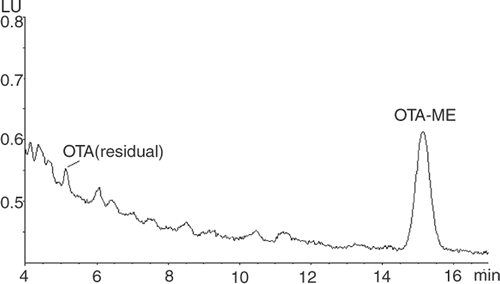
All positive results above 2 ng g−1 in cocoa and 1 ng g−1 for chocolate were confirmed. The retention time of OTA methyl ester was approximately 15 min () and the yield of the methyl ester was >90% (based on residual OTA).
Figure 2. Chromatogram of OTA methyl ester for baking chocolate sample (1.4 ng g−1).
Concentrations and incidences of OTA found in cocoa powder and chocolate in this survey () are similar to those reported in most other studies (Codex Alimentarius Commission Citation2007, 2008; Aoyama et al. Citation2010). Analysis of 32 samples of cocoa powder (16 alkalized and 16 natural) for OTA showed an incidence of 100%, with concentrations ranging from 0.25 to 7.8 ng g−1; six samples exceeded 2 ng g−1, the previously considered European limit for cocoa. We found more OTA in cocoa prepared using alkali (Dutch process) than in natural cocoa (). This difference has not previously been reported and further investigation is desirable. The incidence of OTA in 28 chocolate samples (21 dark or baking chocolate and seven milk chocolate) was also 100%, with concentrations from 0.05 to 1.4 ng g−1 (); this latter value is greater than the previously considered European limit for chocolate (1 ng g−1). There are no Canadian guidelines for OTA in cocoa, although Health Canada has proposed maximum limits for other foods (Health Canada Citation2009).
Figure 3. OTA in cocoa. The dotted line is the previously considered European Commission limit.
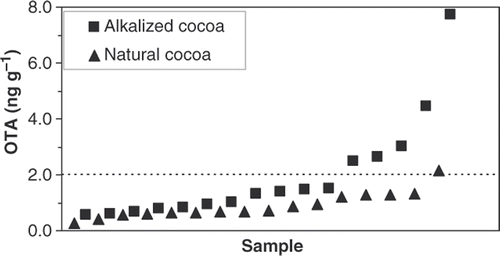
Figure 4. OTA in chocolate. The dotted line is the previously considered European Commission limit.
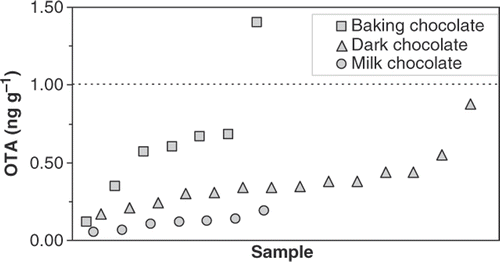
Table 3. Results for OTA in cocoa and chocolate.
Related Research Data
References
- Amézqueta , S , González-Peñas , E , Murillo , M and López de Cerain , A . 2005 . Occurrence of ochratoxin A in cocoa beans: effect of shelling . Food Addit Contam A , 22 : 590 – 596 .
- Aoyama , K , Nakajima , M , Tabata , S , Ishikuro , E , Tanaka , T , Norizuki , H , Itoh , Y , Fujita , K , Kai , S , Tsutsumi , T , Takahashi , M , Tanaka , H , Iizuka , S , Ogiso , M , Maeda , M , Yamaguchi , S , Sugiyama , K , Sugita-Konishi , Y and Kumagai , S . 2010 . Four-year surveillance for ochratoxin A and fumonisins in retail foods in Japan . J Food Protect , 73 : 344 – 352 .
- Bayman , P and Baker , JL . 2006 . Ochratoxins: a global perspective . Mycopathologia , 162 : 215 – 223 .
- Bonvehi , JS . 2004 . Occurrence of ochratoxin A in cocoa products and chocolate . J Agric Food Chem , 52 : 6347 – 6352 .
- Brera , C , Grossi , S , De Santis , B and Miraglia , M . 2003 . High performance liquid chromatography method for the determination of ochratoxin A in cocoa powder . J Liq Chromatogr Rel Technol , 26 : 585 – 598 .
- Clark , HA and Snedeker , SM . 2006 . Ochratoxin A: its cancer risk and potential for exposure . J Toxicol Environ Health B , 9 : 265 – 296 .
- Codex Alimentarius Commission. 2007. Discussion Paper on ochratoxin A in cocoa. Joint FAO/WHO Food Standards Programme. Codex Committee on Contaminants in Foods. First Session, Beijing, China. CX/CF 07/1/19. Available from: ftp://ftp.fao.org/codex/cccf1/cf01_19e.pdf/; and CX/CF 07/1/19 Add. 2. Available from: ftp://ftp.fao.org/Codex/cccf1/cf0119be.pdf/
- Codex Alimentarius Commission. 2008. Discussion Paper on ochratoxin A in cocoa. Joint FAO/WHO Food Standards Programme. Codex Committee on Contaminants in Foods. Second Session, The Hague (the Netherlands). CX/CF 08/2/15. Available from: ftp://ftp.fao.org/codex/cccf2/cf02_15e.pdf/
- European Commission. 2010. Commission Regulation (EU) No 105/2010 of 5 February 2010 amending Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuffs as regards ochratoxin A. Off J Eur Union L35:7–8
- Health Canada. 2009. Information document on Health Canada's proposed maximum limits (standards) for the presence of the mycotoxin ochratoxin A in foods. Ottawa (ON, Canada): Health Canada. Available from: http://www.hc-sc.gc.ca/fn-an/consultation/_limits-max-seuils/myco_consult_ochra-eng.php/
- International Agency for Research on Cancer (IARC). 1993. Some naturally occurring substances: Food items and constituents, heterocyclic aromatic amines and mycotoxins. Monographs on the Evaluation of Carcinogenic Risks to Humans Number 56. p. 489–521. Lyon (France): IARC Press
- Kuiper-Goodman , T , Hilts , C , Billiard , SM , Kiparissis , Y , Richard , IDK and Hayward , S . 2010 . Health risk assessment of ochratoxin A for all age–sex strata in a market economy . Food Addit Contam A , 27 : 212 – 240 .
- O’Brien , E and Dietrich , DR . 2005 . Ochratoxin A: the continuing enigma . Crit Rev Toxicol , 35 : 33 – 60 .