
Abstract
Alternaria toxins and citrinin are mycotoxins produced by fungi growing on different raw materials and agricultural commodities. Maximum levels of these toxins in foods are currently under consideration by the European Commission as a risk management measure. In this study, a new quantitative method is described for the determination of five Alternaria toxins and citrinin in tomato and tomato juice samples based on LC-MS/MS detection. Samples were extracted with pure methanol, followed by a derivatisation step with 2,4-dinitrophenylhydrazine to improve the determination of tenuazonic acid and to decrease the wide polarity difference between the compounds of interest. Samples were purified on hydrophilic-modified styrene polymer solid-phase extraction cartridges. High-performance liquid chromatographic columns packed with different core–shell materials were tested for the separation of toxins and a C-18 phase was in the final method applied to achieve sufficient separation of all relevant analytes. A key element of this approach was to prove successful transferability of the method to three different triple quadrupole mass spectrometers. A full single laboratory method validation was performed on two LC-MS/MS systems and performance characteristics met the predefined requirements. Moreover, the method was used in an international proficiency test and the satisfactory z-scores obtained (−0.1 to 0.8 in tomato juice samples) demonstrated the reliability of the approach described. The method will be validated in an inter-laboratory collaborative study and if the criteria for method precision are met, the method will be proposed as a new Work Item to the European Committee for Standardisation.
Introduction
Mycotoxins are produced under a wide range of climatic conditions by fungi growing on agricultural raw materials and they have been considered one of the biggest public health concerns worldwide for more than half a century (Zöllner & Mayer-Helm Citation2006). A large number of mycotoxins have already been chemically characterised and classified. For those thought to be at highest risk for human and animal health maximum levels (ML) are in force or monitoring plans exist in the European Union (European Commission Citation2006). However, there are still mycotoxins for which no sufficient occurrence data exist when calculating exposure data, or for which EFSA has published opinions indicating the need for action.
Alternaria species (e.g. A. alternata) produce more than 70 secondary metabolites, but only a few of them have been structurally identified and reported as mycotoxins (EFSA Citation2011; Devari et al. Citation2014). Among these Alternaria toxins altenuene (ALT), alternariol (AOH), tentoxin (TEN), tenuazonic acid (TEA) and alternariol monomethyl ether (AME) are the main ones of concern; therefore, they are the focus of the present study. Alternaria species can occur in vegetables, cereals, fruits and oilseeds, and the continuous consumption of food infected by Alternaria mycotoxins can cause fetotoxic and teratogenic effects. Moreover, AOH and AME showed mutagenic and genotoxic properties (EFSA Citation2011; Ostry Citation2008; Paterson & Lima Citation2014; Van de Perre et al. Citation2014).
According to EFSA, agricultural commodities in Europe frequently contain ALT (73% of the analysed samples, maximum of 41 µg kg−1 in wheat grains), AOH (31% of the analysed samples, maximum of 1840 µg kg−1 in sunflower seeds), TEA (15% of the analysed samples, maximum of 4310 µg kg−1 in oats) and AME (6% of the analysed samples, maximum of 184 µg kg−1 in cereals) (EFSA Citation2011). In addition to Alternaria toxins, citrinin (CIT) is another mycotoxin of concern. CITs are produced by Aspergillus (e.g. A. niveus), Penicillium (e.g. P. citrinum, P. verrucosum) and Monascus (e.g. M. aurantiacus) fungi, and they occur mainly in grain-based products, fruit and vegetable juices, beans and herbs. Another recent EFSA opinion details the risk of CIT to human and animal health. The highest CIT concentrations detected in food (grain) and feed were 420 and 998 µg kg−1, respectively (EFSA Citation2012). Consequently, there is a great need to develop analytical methods for those mycotoxins to monitor their occurrence in food and feed.
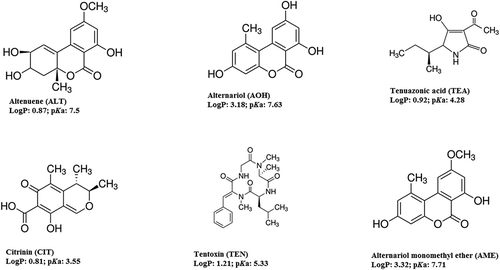
For the determination of Alternaria toxins and CIT at levels in the lower µg kg−1 range, currently only chromatographic methods are appropriate (Xu et al. Citation2006; Ostry Citation2008). These mycotoxins are medium polar or non-polar with weak acidic property (pKa = 3.55–7.71) except TEN (). Most of them show adequate LC separation on reversed-phase fully porous stationary phases, and their detection can be carried out using optical or MS detectors (Xu et al. Citation2006; Ostry Citation2008). TEA in its native form has the ability to form some tautomers and rotamers (Mikula et al. Citation2013) that makes an adequate chromatographic separation of TEA difficult. Also, TEA has poor MS properties (Siegel et al. Citation2009; Asam et al. Citation2011). Hence, LC-MS methods in the past either excluded TEA from Alternaria multi-toxin methods or focused only on TEA and its derivatisation with 2,4-dinitrophenylhydrazine (DNPH) (Lau et al. Citation2003; Magnani et al. Citation2007; Asam et al. Citation2009, Citation2013; Di Mavungu et al. Citation2009; Siegel et al. Citation2010). Recently, quantitative or semi-quantitative multi-compound methods including TEA and other Alternaria toxins were published despite the challenges as mentioned above (Prelle et al. Citation2013; Varga et al. Citation2013; Walravens et al. Citation2014). TEA is a chelating compound and forms complexes with metal ions occurring in the eluent (e.g. zinc) that can improve its LC analysis (Ostry Citation2008). However, LC-MS separation should only involve volatiles additives; consequently pre-column derivatisation of TEA with DNPH as a derivatisation agent has been introduced, since the TEA-hydrazone derivate shows better retention, enhanced peak shape on reversed-phase columns and improved MS detection with both positive and negative ionisation (Siegel et al. Citation2009; Asam et al. Citation2013; Qi et al. Citation2014). Furthermore, the derivatisation of TEA allows decreasing the differences between the polarities of the targeted toxins. Even though the TEA-hydrazone enables better LC-MS determination, there are no existing methods that report how the derivatisation influences the other Alternaria toxins in a multi-toxin LC-MS approach.
Figure 1. Structure of toxins: logP and pKa values.
This paper presents a new LC-MS/MS method for the simultaneous analysis of five Alternaria toxins (ALT, AOH, TEN, TEA, AME) and CIT utilising a derivatisation step for TEA with DNPH. The objectives of the study were (1) optimisation of the LC-MS/MS parameters to achieve enhanced performance characteristics; (2) development of a sample preparation protocol that is suitable for tomato and tomato juice samples; (3) application of the method to proficiency test samples to confirm its reliability; (4) in-house validation of the method; and (5) evaluation of method transferability from one LC-MS/MS system to systems of other brands.
Materials and methods
Reagents, solvents, equipment and samples
Dried-down analytical calibrants of AOH, TEN, TEA and AME were purchased from Sigma-Aldrich (Zwijndrecht, the Netherlands). Standards were reconstituted with 1.00 ml methanol to obtain 0.1 mg ml−1 stock solutions. Crystalline ALT and CIT were purchased from Toronto Research Chemicals (Toronto, ON, Canada) and Sigma-Aldrich, respectively. A total of 8 mg powder was dissolved in 8.00 ml methanol to obtain 1 mg ml−1 stock solutions. All stock solutions were kept at 4°C. DNPH and undecanal were purchased from Sigma-Aldrich. The derivatisation reagent (0.58% DNPH in HCl solution) was prepared as described by Siegel et al. (Citation2010). The stop reagent was 5% (v/v) undecanal in methanol. The derivatised TEA standard solution (1.91 µg ml−1, 5.071 µmol l−1 in methanol) was prepared by mixing 1 ml of the 10 µg ml−1 methanolic TEA solution with 1 ml DNPH solution. The mixture was left overnight and processed as written in the sample extraction and SPE clean-up sections. The final volume was adjusted to 10 ml with methanol. This solution was used to optimise the LC-MS/MS conditions for the derivatised TEA (TEA-hydrazone). A total of 50 mM ammonium formate buffer was prepared in water and its pH adjusted to 3.0 with formic acid. Methanol and acetonitrile were LC-MS grade obtained from Sigma-Aldrich. Ethyl acetate, n-hexane, dichloromethane, formic acid and ammonium formate were HPLC grade and purchased from Merck (Darmstadt, Germany). The Kinetex C-18 HPLC column (2.1 × 100 mm, 5 µm), Strata SPE cartridges (6 ml, 200 mg) and regenerated cellulose (RC) syringe filters (15 mm, 0.45 µm) were obtained from Phenomenex (Utrecht, the Netherland). The Supelco Ascentis Express C-18, cyano (ES-CN) and phenyl-hexyl HPLC columns (2.1 × 100 mm, 2.7 µm) were purchased from Sigma-Aldrich. Samples, used for method development, were bought in local food shops and proficiency test samples were obtained from the Federal Institute for Risk Assessment (BfR, Berlin, Germany). Samples were stored at −20°C until subjected to analysis.
Instrumentation and equipment
The method development was carried out using an Agilent 1100 HPLC system (Agilent Technologies, Waldbronn, Germany) coupled to a Micromass Quattro Ultima PT triple quadrupole MS detector (Waters, Milford, MA, USA). Data acquisition and evaluation were performed with MassLynx version 4.0. The final method was also transferred to a Thermo TSQ Quantum Ultra LC-MS/MS system (Thermo Finnigan, San Jose, CA, USA) that involved a Shimadzu LC-20AD binary pump, an Accela autosampler, an Accela column thermostat and a TSQ Quantum Ultra triple quadrupole MS detector. Data acquisition and evaluation were performed using Xcalibur software 2.0.7. SP1. Both systems were equipped with an electrospray (ESI) interface in which negative ionisation alone was used during acquisition. Nitrogen was used as drying and collision gas. The ion source parameters are summarised in .
Table 1. Ion source settings on Ultima PT and Thermo TSQ Quantum instruments.
Table 2. MS/MS ion transitions, settings, and ion ratios on Ultima PT and TSQ Quantum instruments.
Further, the method transferability was investigated with an LC-MS/MS system that consisted of an Agilent 1100 HPLC coupled to an AB Sciex 4000 triple quadrupole MS (Framingham, MA, USA).
A Sartorius ME36S balance (Sartorius AG, Goettingen, Germany) was used for weighing standards. For sample extraction and centrifugation, a CAT S50 flask shaker (Zipperer GmbH, Staufen, Germany) and an Eppendorf 5810R centrifuge was used (Eppendorf AG, Hamburg, Germany), respectively. During the derivatisation, a GFL 3018 (Labortechnik mbH, Burgwedel, Germany) reciprocating shaker was applied. Samples were evaporated using a Techne Dri-Block DB-3D evaporator (Biostep, Jahnsdorf, Germany).
Sample extraction
Test portions (1 g) (recorded to two decimal places) were weighed into 50 ml polypropylene (PP) centrifuge tubes. A total of 5 ml methanol was added to the samples and tubes were capped and vortex mixed for 5 s. Next, the samples were shaken at 600 min−1 for 40 min at ambient temperature and centrifuged at 2700g for 10 min at 22°C after the extraction. The whole supernatant was then collected in new 50 ml PP centrifuge tubes. A total of 100 µl derivatisation reagent was added to the decanted supernatant and samples were vortex mixed for 5 s and let to react for 1 h at ambient temperature (approximately 22°C) while shaking at 200 min−1. The reaction was stopped after 1 h by adding 500 µl stop reagent to the whole derivatised sample extract in the tube. Tubes were then vortex mixed for 5 s and shaken for 30 min. The complete derivatised extracts were diluted in the PP tubes up to 40 ml with ammonium formate buffer (50 mM, pH 3), shortly shaken by hand and subjected to SPE clean-up.
SPE clean-up
Strata-XL, a hydrophilic modified styrene polymer, SPE cartridges (200 mg, 6 ml, 100 µm) were conditioned with 6 ml methanol, followed by 6 ml water and 6 ml 50 mM ammonium formate buffer (pH 3). A total of 75-ml reservoirs were connected onto the cartridges and samples were loaded into them. The samples were then passed through the SPE cartridge at a rate of 1 drop per second. Afterwards, SPE columns were washed with 6 ml methanol–water (15/85, v/v) mixture and subsequently, with 6 ml n-hexane at the same speed. Cartridges had been vacuum dried for 5 min before the samples were eluted with 5 ml methanol into glass tubes. The eluates were evaporated to dryness at 45°C under a gentle stream of nitrogen and redissolved in 1.0 ml methanol by vortex-mixing for 20 s. As a final step, samples were filtered through RC filters into HPLC vials.
LC-MS/MS analysis
Alternaria toxins and CIT were separated on an Ascentis Express C-18 (2.1 × 100 mm, 2.7 µm) HPLC column equipped with a C-18 (0.5 mm) guard column using binary linear gradient elution. Two solvents (A and B) were mixed by the binary pump. Solvent A contained 10 mM ammonium formate and 0.05% formic acid in water (pH 3), while solvent B was pure methanol. The flow rate was 0.3 ml min−1. The methanol composition of the mobile phase was 10% at 0 min, 100% at 10 min, 100% at 22 min and 10% at 22.5 min. A sufficient long washing step in the gradient programme was necessary to remove the accumulated lipophilic matrix solutes that influenced the ionisation of CIT. The total run time was 25 min. The column thermostat was maintained at 30°C and the injection volume was 5 µl. The autosampler was operated at 20°C.
The MRM mode was applied in the MS/MS detector and two ion transitions (quantifier and qualifier) were recorded for each target compound. The selected ion transitions with the optimised voltages (cone or tube lens), collision energies (CE) and dwell times are summarised in .
Preparation of matrix-matched calibration solutions
In order to take into account the derivatisation during sample preparation, the derivatisation of calibration samples must be done in matrix-matched solutions. Therefore, the derivatisation of calibration samples was done in fortified sample extracts. This allowed for the same conversion to be obtained in all samples and to compensate the matrix effects (ME) adequately.
Blank samples were subjected to extraction as described above. The extracts were then spiked with different volumes of working standard solution and they were derivatised and further processed. Hence, the same conversion could be achieved in all kind of samples. Concentration levels are described in detail below. Use of isotope-labelled internal standards (ISTDs) could have further improved quantification; however, they are currently not commercially available for Alternaria toxins.
Method validation
The method was validated for tomato samples involving two different LC-MS/MS systems (TSQ Quantum and Ultima PT). During the method development, the LOQ was calculated for all compounds on both instruments. The LOQ was calculated as 10× the signal-to-noise ratio (SNR) of measurements from fortified samples considering both ion transitions and their ion ratio. Since the selected mycotoxins are not regulated yet, the selection of spiking levels in the validation plan was based on these calculated LOQs. Calculated LOQs varied between the instruments due to the differences in sensitivity. Therefore, the higher LOQs were chosen for validation levels in order to obtain acceptable results with both systems. These levels were designated as ‘estimated LOQ set for validation’ (). The confirmation of LOQs was done at the end of the validation exercise.
Table 3. Calculated and confirmed analytical limits: validation levels, linearity parameters and matrix effects.
The evaluated method performance characteristics were as follows: selectivity, identification, linearity, working range, ME, LOD, LOQ, recovery, repeatability, intermediate precision and robustness.
Selectivity was confirmed by comparing chromatograms of blank and fortified samples for the absence of interfering peaks. The identification was based on the ion ratio of qualifier and quantifier ion traces. Linearity was checked with matrix-free standard solutions containing derivatised TEA. A five-point calibration function was generated near the ‘estimated LOQ levels’ reflecting 1, 2, 5, 10 and 20× the assumed LOQ. Each solution was injected three times. The working range was evaluated using the same calibration levels in matrix-matched samples. The effect of the presence of matrix on the calibration function was calculated by comparing the slopes of matrix-matched and matrix-free curves. ME was calculated as:
where a negative ME indicates ion suppression; and a positive ME means ion enhancement. To study the effect of differences in the composition of samples of a given matrix on ME, blank tomato samples, which originated from three different sources, were subjected to the whole sample preparation. The purified sample residues were spiked at 10× the estimated LOQ level with the standard mixture, and samples were reconstituted and measured. The relative standard deviation (RSD%) of the respective mycotoxin peak areas obtained for the three different tomato samples was taken as a measure of the relative ME (Van Eeckhaut et al. Citation2009; Tölgyesi et al. Citation2012). The LOD was estimated as the lowest concentration of a compound in a sample that could be detected and identified with an acceptable SNR, as well as quantifier to qualifier ion ratio. The SNRs that are higher than 3 on each ion transition were used as acceptance criteria. Two concentration levels were used to evaluate the repeatability: intermediate precision and recovery. These were 3 and 10× the estimated LOQ (). Four replicates at each level were prepared and analysed; the whole analytical procedure was repeated on five different days. During the 5 days, two operators prepared the samples, different lots of SPE cartridges, tomato samples and instruments (Ultima PT and TSQ Quantum Ultra) were used. All 20 results at each level were used to calculate the performance characteristics applying the single-factor analysis of variance (ANOVA) test. A 12 run Plackett–Burman design was utilised for the robustness experiment. Seven factors, expected to have an impact on the reliability of results, were tested at two levels. The chosen factors were: extraction solvent volume (4.5 or 5.5 ml), extraction time (36 or 44 min), derivatisation reagent volume (90 or 110 µl), derivatisation time (54 or 66 min), sample volume loaded onto the SPE cartridge (36 or 44 ml), elution solvent volume (4.5 or 5.5 ml) and evaporation temperature (40 or 50°C). Levels corresponded to the nominal settings of ±10%. Samples were fortified at 100 µg kg−1 level for each analyte.
Results and discussion
Optimisation of MS/MS detection
The development of the method started on the Ultima PT instrument, and then the optimised parameters were later adapted to the TSQ LC-MS/MS system. MS/MS settings were optimised on the Ultima PT and TSQ instruments using a direct infusion of individual standard solutions and flow injection analysis, respectively (Tölgyesi et al. Citation2012). Ion transitions were tuned with 10 µg ml−1 methanolic standard solutions and 1.91 µg ml−1 derivatised TEA solution in methanol. As the derivatisation step was carried out before the clean-up step in the sample preparation, some polar matrix components, which also reacted with DNPH, had a significantly reduced polarity after the derivatisation and they concentrated together with the target compounds during the SPE clean-up. This led to an increased noise level and a lower selectivity of ion transitions. Therefore, the selection of daughter ions was mainly aimed at ensuring selectivity and intensity was taken into account in second place. For example, the most intense ion transition of AOH (257.1 > 213.0 m/z) and TEA (376.4 > 182.1 m/z), which were already used in the existing methods (Magnani et al. Citation2007; Asam et al. Citation2013), were not selective enough during real sample analysis. A change in ion trace selection resulted in decreased noise levels and enhanced selectivity for appropriate identification. Ion source parameters were optimised after setting the final mobile phase composition. The highest responses could be achieved at those temperatures and voltages described in . The APCI source with the tuned ion transitions was also tested in an additional experiment on the Ultima PT system. Both ion sources (ESI and APCI) resulted in high ME, which led to the selection of ESI as preferred interface.
Optimisation of LC-MS/MS separation
The chromatographic separation was developed on HPLC columns packed with different core–shell particles. Investigated stationary phases were as follows: cyano (ES-CN), phenyl-hexyl and C-18. In addition, 2.7 and 5 µm particle sizes were also tried. High resolution was important in order to use time segments during the MS detection and hence maximise the dwell time of ion transitions for the Ultima PT detector. Since a pronounced ME during real sample analysis was observed, it was necessary to avoid the co-elution of analytes, because target compounds can also suppress each other’s ionisation. An acidic eluent improved the peak shapes due to weak acidic property of several Alternaria toxins and CIT. Baseline separation for all compounds could be achieved using the phenyl-hexyl or ES-CN columns. The separation on the phenyl-hexyl column required a high concentration of formic acid (0.2%, v/v) in the eluent, which reduced the sensitivity of [M – H]– precursor ions, mainly for ALT. The separation of toxins on ES-CN or C-18 phases could be carried out with lesser formic acid in the mobile phase. All compounds with the exception of CIT had improved peak shapes on the ES-CN column, while the obtained plate numbers for CIT was the lowest. A separation attempt on a C-18 column gave satisfactory resolution for most of the analytes, but AOH and TEN nearly co-eluted (). The resolution between these two compounds could be improved using a column packed with 2.7 µm particles, but baseline separation could not be achieved. Methanol as an organic modifier in the mobile phase composition allowed for better sensitivity compared with acetonitrile.
Figure 2. (colour online) (a) Quantifier MRM chromatograms of a spiked tomato sample. Spiking levels are 50 µg kg–1 for ALT, 5 µg kg–1 for CIT, 5 µg kg–1 for AOH, 5 µg kg–1 for TEN, 10 µg kg–1 for TEA and 2 µg kg–1 for AME; (b) quantifier MRM chromatograms of a blank tomato sample. All analyses were performed on an Ultima PT instrument.
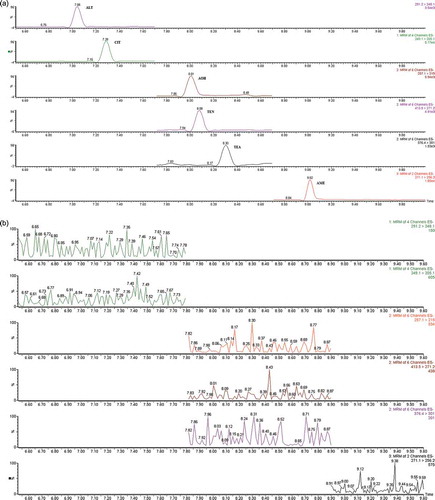
The determination of mycotoxins was influenced by considerably high ME on both HPLC columns (ES-CN and C-18). ME was evaluated in tomato juice samples. ME on ES-CN columns were −50%, 98%, −86%, −72%, −35% and −78%, for ALT, CIT, AOH, TEN, TEA and AME, respectively. The relative ME for different tomato juice samples varied from 8.7% to 13.0% for the individual toxins. When the measurement was carried out on the C-18 phase, MEs were −64%, 71%, −83%, −74%, −73%, −69%, for ALT, CIT, AOH, TEN, TEA and AME, respectively. The inter-sample ME was between 3.3% and 12.6%. During the method adaption to the TSQ system, the same ME influenced the analysis. Therefore, before the derivatisation, an additional SPE clean-up was tried to reduce the number and concentration of polar matrix compounds. However, such a two-step SPE procedure did not decrease the ME. Considerable ME for ALT, AOH and AME in orange flavedo and albedo tissue and food supplements was reported by Magnani et al. (Citation2007) and Di Mavungu et al. (Citation2009). In a recent paper, aflatoxin M1 was used as an ISTD to compensate ME for the determination of Alternaria toxins in tomato products (Prelle et al. Citation2013); however, due to lack of chemical and physical similarity with the target mycotoxins, it was not used in our study. Finally, the C-18 LC column with 2.7 µm packing materials was used for the separation because it provided acceptable peak shapes and better column stability.
The HPLC separation, optimised on the Ultima PT, could be transferred to the TSQ system equipped with a Shimadzu binary UFLC pump after slight modification in the gradient programme. This was done by extending the washing step with 100% methanol in the gradient to 12 min to obtain reproducible signals for CIT.
Extraction and derivatisation
The method development was carried out with tomato juice samples that are considered to be a more complex matrix compared with tomato, because of the additives and preservatives. Since the tomato juice samples contain a high level of water (approximately 90%), an extraction with a water-immiscible solvent such as ethyl acetate is unfavourable unless a repetitive extraction is planned. Therefore, extraction was tested with water miscible organic solvents such as methanol or acetonitrile with and without additives (formic acid or ammonium hydroxide). AOH and AME require the highest level of organic solvent for extraction due to their lipophilic character. Extraction with methanol or acetonitrile enabled similarly good recovery (> 70%) for all compounds at 20 and 40 µg kg−1. There was no difference between the pure and acidic (2% formic acid in the extraction medium) extraction solvent concerning recovery and ME. Extraction with methanol was preferable to acetonitrile since the derivatisation of TEA was faster in methanol by a factor of four compared with acetonitrile.
Effective derivatisation for TEA was obtained immediately after extraction in the methanolic extract. It was achieved by adding 100 µl derivatisation reagent to the sample extract (approximately 6 ml). The reaction was stopped after 1 h by adding 500 µl stop solution. The effect of derivatisation conditions on the other mycotoxins was tested. Blank samples (n = 3) were extracted with pure methanol and two of them were spiked with standard solution resulting in a level of 40 µg kg−1 and derivatisation reagent was added to the three extracts (one blank and two spiked samples). Samples were allowed to react overnight (approximately for 16 h) at RT (22°C). ALT, AOH, TEN and AME in the derivatised samples were quantified after a reaction time of 16 h by spiking (at 40 µg kg−1 level) the blank sample and using it as a reference for calibration. No degradation of the Alternaria toxins was detected. Recovered concentrations were between 90% and 100%, confirming their stability during the derivatisation reaction. Derivatisation time was tested for 20, 30 and 60 min. The response of derivatised TEA improved by a factor of 1.5 following an increase in the time from 20 to 30 min. However, only a slight enhancement could be observed between 30 and 60 min, suggesting that the reaction rate decreased considerably after 30 min. Prolonged derivatisation time (16 h) yielded slightly higher responses, but were considered impractical and not suitable to meet the goal, given that a 60 min derivatisation period allowed sufficiently precise quantification of TEA at 10 µg kg−1 level. Other authors used 30 min for derivatisation, but applied higher concentrations of DNPH and lower pHs (Siegel et al. Citation2010; Asam et al. Citation2011). Five % (v/v) undecanal solution has been used previously (Siegel et al. Citation2010; Asam et al. Citation2011) and were confirmed to be suitable for stopping the reaction under the conditions of the here described method as well.
SPE clean-up
For sample purification mixed-mode and polymeric reversed-phase (RP) SPE cartridges were tested. Normal-phase SPE on silica sorbent (Strata Si-1) would require dichloromethane and was not considered in the presence of other alternatives. Selective clean-up with mixed-mode anion exchange and mixed-mode cation exchange cartridges were tried under basic and acidic conditions, respectively. Mixed-mode cartridges possess both RP and strong ion exchange phases. Strata-XL-A, mixed-mode anion exchange cartridge was tested to purify sample extracts under basic condition (pH 10). This allowed ionic interaction between the acidic toxins and the SPE sorbent. However, TEN remained trapped on the RP part of the Strata-XL-A column under this condition; hence a selective extraction of all mycotoxins could not be achieved. Strata-XL-C, mixed-mode cation exchange SPE column showed sufficient retention for all analytes on the RP of the cartridge under acidic condition (pH 3). The advantage of a mixed-mode cation exchange cartridge is the selective adsorption of basic matrix components at acidic pH via ionic interaction (Tölgyesi et al. Citation2012). Therefore, the target acidic and neutral toxins can be separated from the basic compounds. However, this did not lessen the influence of co-extracted matrix compounds on quantification by LC-MS/MS. Moreover, lower recoveries were observed for AOH and AME compared with results obtained with polymeric RP columns. The best recoveries could be achieved with polymeric RP columns (Strata-XL). This cartridge enabled good retention for all compounds at pH > 3. Lower pH values resulted in a low recovery for CIT. The washing and elution conditions were carefully studied with Strata-XL columns. A total of 15% (v/v) methanol in water, followed by n-hexane allowed for the removal of a substantial amount of matrix compounds and to wash out the non-reacted quenching agent (undecanal) from the cartridges. Sample elution was tested with methanol, acetonitrile and ethyl acetate with and without additives (2% formic acid or 2% ammonium hydroxide). Elution with pure methanol resulted in clean eluates and acceptable recoveries for all compounds while acetonitrile failed to give acceptable recoveries for AOH.
The reconstitution of evaporated samples required neat methanol as the sample residues could not be fully re-dissolved in aqueous solutions due to the lipophilic character of AOH and AME. This limited the injection volume to 5 µl to avoid peak distortion of CIT.
Validation
The comparison of MRM chromatograms of blank and spiked samples showed that no interfering peaks co-eluted with any of the target compounds (,)), hence selectivity was proven on both instruments. The average of ion ratios was calculated in the calibration samples (n = 5). Maximum permitted tolerances for ion ratios were evaluated according to Commission Decision 2002/657/EC (European Commission Citation2002). The ion ratios obtained in spiked and naturally contaminated samples were within this tolerance interval (). Determination coefficients (r2) in the linearity investigation were 0.9635–0.9982 and 0.9557–0.9994 for matrix-free and matrix-matched calibrations respectively. However, due to strong MEs (see below) matrix-matched calibration was used throughout the validation experiment.
Overall, it seems that observed MEs (ranging from −67% to 144%) showed strong variation not only between different instruments as it is likely to be expected, but also between rather similar matrices (tomato and tomato juice). Therefore, care was taken to obtain consistency, which was achieved with the here described protocol. The resulting repeatability of ME% was 3% for TEA despite a relatively high ME of 144% in tomato. The LOQ levels estimated at the beginning of the validation could be confirmed for all toxins except TEA. For TEA, LOQs were twice as high but still fulfilled the criteria ().
Recoveries within the entire validation range, evaluated from 20 results at each level ranged from 89% to 94%. Repeatability (RSDr,%) varied from 5.4% to 14.3% at the lower validation level and improved (3.5–5.9%) with the increase of concentration level. Intermediate precision (RSDwR,%) was between 9.1% and 15.9% at the lower spiking level and RSDwR,% varied from 5.2% to 7.0% at the higher fortification level (). At the lower spiking level, repeatability was approximately half of intermediate precision except for TEA and AME. No difference was observed between repeatability and intermediate precision at the higher spiking level. Acceptance criteria for recovery ranged from 70% to 110% at these levels; RSDr,% and RSDwR,% had to be lower than 20% (Practical Guide to ISO 5725-2 Citation1994). The validation results fulfilled the acceptance criteria. Relative expanded uncertainty was calculated at both spiking levels ().
Table 4. Recovery, repeatability, intermediate precision, and relative expanded uncertainty.
During the investigation of method robustness, seven factors were studied. Since no reference material is available for these toxins in tomato, the robustness testing was done with fortified samples. Two factors influenced the recovery, namely the derivatisation reagent volume and the elution solvent volume. This also confirmed that the 1 h-long derivatisation time chosen in the final method was enough for the analysis of TEA at the levels of interest. The higher the volume of derivatisation reagent used, the better was the recovery for TEA. The elution solvent volume could affect the recovery of AOH and AME. The more solvent used, the higher the recoveries that were to be achieved due to their non-polar character. Generally, there was no specific factor that influenced all mycotoxins. The method showed acceptable robustness.
Application of the method
Proficiency test
The accuracy of the developed method for the analysis of naturally contaminated samples was tested by participating in a proficiency test organised by BfR with tomato juice samples containing Alternaria toxins at levels from 1.56 to 53.0 µg kg−1. Employing the here described method z-scores for test samples, as well as a standard solution, ranged from −0.1 to 0.8 and from −1.3 to 0.1 respectively (). Results are successful if the z-scores are between −2 and 2. Therefore, these satisfactory scores highlight the applicability of the proposed method.
Table 5. Detected concentrations in the proficiency test samples and in the standard solution (Z-scores).
External laboratory test
After the validation and proficiency test, our method was tested by BfR using a third type of LC-MS/MS instrument (AB Sciex 4000). Recovery (n = 3) in tomato samples at 10 and 20 µg kg−1 levels (100 and 200 µg kg−1 for ALT) ranged from 76% to 99% and from 80% to 98%, respectively, and the RSD% was less than 13%. These data are comparable with those obtained in our laboratory ().
Conclusions
An improved method for the simultaneous analysis of Alternaria toxins and CIT in tomato-based samples was developed and single-laboratory validated. The employed derivatisation for TEA allowed the simultaneous determination of TEA with other Alternaria toxins and CIT. Matrix-matched calibration compensated MEs. The method was successfully tested on three different types of LC-MS/MS systems and the results proved the transferability of the method. This aspect is relevant since the method will be subjected to inter-laboratory validation using the ISO 5725:1994 protocol.
Disclosure statement
No potential conflict of interest was reported by the authors.
Acknowledgement
The authors thank Andreas Breidbach for assistance.
References
- Asam S, Konitzer K, Schieberle P, Rychlik M. 2009. Stable isotope dilution assays of alternariol and alternariol monomethyl ether in beverages. J Agr Food Chem. 57:5152–5160.
- Asam S, Lichtenegger M, Muzik K, Liu Y, Frank O, Hofmann T, Rychlik M. 2013. Development of analytical methods for the determination of tenuazonic acid analogues in food commodities. J Chromatogr A. 1289:27–36.
- Asam S, Liu Y, Konitzer K, Rychlik M. 2011. Development of a stable isotope dilution assay for tenuazonic acid. J Agr Food Chem. 59:2980–2987.
- Devari S, Jaglan S, Kumar M, Deshidi R, Guru S, Bhushan S, Kushwaha MP, Gupta AG, Gandhi SP, Sharma J, et al. 2014. Capsaicin production by Alternaria alternata, an endophytic fungus from Capsicum annum; LC–ESI–MS/MS analysis. Phytochemistry. 98:183–189.
- Di Mavungu JD, Monbaliu S, Scippo M-L, Maghuin-Rogister G, Schneider Y-J, Larondelle Y, Callebaut A, Robbens J, Van Peteghem C, De Saeger S. 2009. LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food Addit Contam. 26:885–895.
- European Commission. 2002. COMMISSION DECISION of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. 2002/657/EC. Off J Eur Union. L 221:8.
- European Commission. 2006. COMMISSION REGULATION (EC), No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off J Eur Union. L 364:5.
- [EFSA] European Food Safety Authority. 2011. Scientific Opinion on the risks for public and animal health related to the presence of Alternaria toxins in food and feed. EFSA J. 9:1–97.
- [EFSA] European Food Safety Authority. 2012. Scientific Opinion on the risks for public and animal health related to the presence of citrinin in food and feed. EFSA J. 10:1–82.
- Lau BP-Y, Scott PM, Lewis DA, Kanhere SR, Cleroux C, Roscoe VA. 2003. Liquid chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry of the Alternaria mycotoxins alternariol and alternariol monomethyl ether in fruit juices and beverages. J Chromatogr A. 998:119–131.
- Magnani R, De Souza GD, Rodrigues-Filho E. 2007. Analysis of alternariol and alternariol monomethyl ether on flavedo and albedo tissues of tangerines (Citrus reticulata) with symptoms of alternaria brown spot. J Agr Food Chem. 55:4980–4986.
- Mikula H, Horkel E, Hans P, Hametner C, Fröhlich J. 2013. Structure and tautomerism of tenuazonic acid – a synergetic computational and spectroscopic approach. J Hazard Mater. 250–251:308–317.
- Ostry V. 2008. Alternaria mycotoxins: an overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 1:175–188.
- Paterson RRM, Lima N. 2014. Self mutagens affect detrimentally PCR analysis of food fungi by creating potential mutants. Food Control. 35:329–337.
- Practical guide to ISO 5725-2. 1994. Accuracy (trueness and precision) of measurement methods and results – Part 2: basic method for the determination of repeatability and reproducibility of a standard measurement method [Internet]. [cited 1994 Dec 22]. http://www.iso.org/iso/catalogue_detail.htm?csnumber=11834
- Prelle A, Spadaro D, Garibaldi A, Gullino ML. 2013. A new method for detection of five alternaria toxins in food matrices based on LC–APCI-MS. Food Chem. 140:161–167.
- Qi B-L, Liu P, Wang Q-Y, Cai W-J, Yuan B-F, Feng Y-Q. 2014. Derivatization for liquid chromatography-mass spectrometry. TrAC-Trend Anal Chem. 59:121–132.
- Siegel D, Merkel S, Koch M, Nehls I. 2010. Quantification of the Alternaria mycotoxin tenuazonic acid in beer. Food Chem. 120:902–906.
- Siegel D, Rasenko T, Koch M, Nehls I. 2009. Determination of the Alternaria mycotoxin tenuazonic acid in cereals by high-performance liquid chromatography–electrospray ionization ion-trap multistage mass spectrometry after derivatization with 2,4-dinitrophenylhydrazine. J Chromatogr A. 1216:4582–4588.
- Tölgyesi Á, Sharma VK, Fekete S, Lukonics D, Fekete J. 2012. Simultaneous determination of eight corticosteroids in bovine tissues using liquid chromatography–tandem mass spectrometry. J Chromatogr B. 906:75–84.
- Van de Perre E, Deschuyffeleer N, Jacxsens L, Vekeman F, Van Der Hauwaert W, Asam S, Rychlik M, Devlieghere F, De Meulenaer B. 2014. Screening of moulds and mycotoxins in tomatoes, bell peppers, onions, soft red fruits and derived tomato products. Food Control. 37:165–170.
- Van Eeckhaut A, Lanckmans K, Sarre S, Smolders I, Michotte Y. 2009. Validation of bioanalytical LC–MS/MS assays: evaluation of matrix effects. J Chromatogr B. 877:2198–2207.
- Varga E, Glauner T, Berthiller F, Krska R, Schuhmacher R, Sulyok M. 2013. Development and validation of a (semi-)quantitative UHPLC-MS/MS method for the determination of 191 mycotoxins and other fungal metabolites in almonds, hazelnuts, peanuts and pistachios. Anal Bioanal Chem. 405:5087–5104.
- Walravens J, Mikula H, Rychlik M, Asam S, Ediage EN, Di Mavungu JD, Van Landschoot A, Vanhaecke L, De Saeger S. 2014. Development and validation of a ultra-high-performance liquid chromatography tandem mass spectrometric method for the simultaneous determination of free and conjugated Alternaria toxins in cereal-based foodstuffs. J Chromatogr A. 1372:91–101.
- Xu BJ, Jia XQ, Gu LJ, Sung CK. 2006. Review on the qualitative and quantitative analysis of the mycotoxin citrinin. Food Control. 17:271–285.
- Zöllner P, Mayer-Helm B. 2006. Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography–atmospheric pressure ionisation mass spectrometry. J Chromatogr A. 1136:123–169.