
ABSTRACT
Selective estrogen receptor modulators (SERMs), anti-estrogens and aromatase inhibitors are prohibited in human sports doping. However, they also present a risk of being used illegally in animal husbandry for fattening purposes. A method was developed and validated using UHPLC-MS/MS for the determination and confirmation of SERMs, anti-estrogens and aromatase inhibiters in bovine and porcine urine. This method was used in a survey of more than 200 bovine and porcine urine samples from Dutch farms. In 18 out of 103 porcine urine samples (17%) and two out of 114 bovine samples (2%) formestane, an aromatase inhibitor, was detected. None of the other compounds was detected. From human doping control it is known that formestane can, in some cases, be of natural origin. Analyses of reference samples from untreated bovine and porcine animals demonstrated the presence of formestane in bovine animals, but not yet in porcine animals. Future research will focus on whether the detected formestane in porcine and bovine urine is from endogenous or exogenous origin, using GC-c-IRMS.
Introduction
Substances with anti-estrogenic activity are therapeutically used mainly for the treatment or prevention of breast cancer (Mauras et al. Citation2000; Kang et al. Citation2007; Gerace et al. Citation2012). Anti-estrogenic substances can be divided into different groups according to their pharmacological properties: aromatase inhibitors (AIs), selective estrogen receptor modulators (SERMs) and anti-estrogens (AEs). AEs are compounds that bind to estradiol receptors in the body and thus block estradiol binding. Because of this, AEs reduce the effect of 17ß-estradiol. SERMs are non-steroidal therapeutic compounds that also bind to the estradiol receptor in the body. Depending on the compound and the target organ, this binding can enhance or reduce the effect of 17ß-estradiol (Dutertre & Smith Citation2000). SERMs are also used for the treatment or prevention of breast cancer. AIs inhibit the enzyme aromatase, which is a key enzyme in the production of estradiol in the body. They are used in fertility treatments (Lønning Citation2004).
There are no reference points of action (RPAs) – an analytical concentration for a non-allowed pharmacologically active substance that can be determined by official control laboratories and is low enough to adequately protect the consumers of food commodities that contain that substance (EFSA Citation2013) – for SERMs, AEs and AIs in bovine and porcine urine. On the other hand, the World Anti-Doping Agency (WADA) mentions limits in human urine in Technical Document 2015MRPL (WADA Citation2015). This document sets minimum required performance levels (MRPLs) for the SERMs, AIs and AEs in human urine of 20 ng ml−1, except for formestane, which has an MRPL of 50 ng ml−1. This is the case because formestane can occur endogenously in human urine. According to the document, gas chromatography combustion isotopic ratio mass spectrometry (GC-c-IRMS) should be performed (WADA Citation2016) when formestane is found in a concentration of 50– 150 ng ml−1 to determine whether it is of endogenous or exogenous origin.
WADA has prohibited the use of hormone antagonists and modulators, including SERMs, AEs and AIs, since 2010 because of their potential abuse in human sports (they boost free testosterone) and ability to treat and prevent the side-effects produced from anabolic androgenic steroids (AASs) misuse (Mazzarino et al. Citation2011). Because of the widespread availability of the compounds, also on the black market (Krug et al. Citation2014), and the known anabolic effects in humans, there is also a chance that abuse in livestock production will take place. This phenomenon has been observed in the past many times; examples of this are stanozolol, methyltestosterone and chlortestosterone (Poelmans et al. Citation2002; Regal et al. Citation2010). All these compounds have a history of abuse in both farm animals and sports doping. Therefore, a method is needed for the detection of SERMs, AEs and AIs in bovine and porcine urine because abuse for fattening purposes is most likely in these species. As far as we know, no literature on the detection of these compounds in bovine and porcine urine is available. Furthermore, the combination of this high number of compounds from the three groups mentioned above in urine other than human has not been published before. The detection of the individual compounds or a limited number of compounds in human urine, on the other hand, has been published extensively (Mareck et al. Citation2005; Mazzarino & Botrè Citation2006; Gerace et al. Citation2012).
This paper describes the development and validation of an ultra-high performance liquid chromatography triple quadrupole mass spectrometry (UHPLC-MS/MS) method for the determination and confirmation of SERMs, AEs and AIs in bovine and porcine urine. Furthermore, a survey was performed of more than 200 bovine and porcine urine samples from Dutch farms. shows the investigated compounds, structures, chemical names and groups (SERM, AE or AI) in which the compounds are classified.
Table 1. Compounds, structures, chemical names, compound group (SERM, AE or AI) and MS parameters.
Materials and methods
Materials
Methanol (MeOH) and acetonitrile (ACN) were obtained from Actu-All (Oss, the Netherlands). Sodium hydroxide, acetic acid, formic acid, sodium hydrogen carbonate, sodium dihydrogen phosphate, disodium hydrogen phosphate and tert-butyl methyl ether (TBME) were obtained from Merck (Darmstadt, Germany) and ß-Glucuronidase E Coli K12 from Roche (Indianapolis, IA, USA). Potassium carbonate was obtained from Boom B.V. (Meppel, the Netherlands).
Milli-Q water was prepared using a Milli-Q system with a resistivity of at least 18.2 MΩ cm–1 (Millipore, Billerica, MA, USA). The reference standards anastrozole, exemestane, letrozole, tamoxifene, toremifene, fulvestrant, DL-aminoglutethimide, clomiphene, 4-hydroxytamoxifen, raloxifene, formestane, mesterolone and tamoxifen-13C2-15N were obtained from Sigma-Aldrich (St. Louis, MO, USA). Dromostanolone was obtained from Steraloids (Newport, RI, USA) and androsta-1,4,6-triene-3,17-dione from Carbosynth (Compton, UK). Testolactone and 4-hydroxycyclofenil were obtained from LGC (Teddington, UK); and 1alpha-methyl-5alpha-androstan-3alpha-ol-17-one and 2alpha-methyl-5alpha-androstan-3alpha-ol-17-one were obtained from NMIA (West Lindfield, NSW, Australia). The AASs 1-alpha-methyl-5-alpha-androstan-3-alpha-ol-17-one and 2-alpha-methyl-5-alpha-androstan-3-alpha-ol-17-one are included in this study because they form the main active metabolites of mesterolone and dromostanolone respectively (Ho et al. Citation2007). 17α-Methyltestosteron-d3 was obtained from EURL (Wageningen, the Netherlands). Stock solutions were prepared in MeOH at 1000 mg l−1. Dilutions were prepared in MeOH. Tamoxifen-13C2-15N was used as an internal standard for tamoxifen, toremifene and clomiphene. 17α-Methyltestosteron-d3 was used as internal standard for all other compounds.
Sample preparation
Sample preparation was based on a method of De Rijke et al. (Citation2013) for the detection of selective androgen receptor modulators (SARMs). To 2 ml of urine internal standard solution was added (40 µl of tamoxifen-13C2-15N and 20 µl of 17α-methyltestosteron-d3). Additionally, 750 µl 0.2 M Na2HPO4/NaH2PO4 (1:2, v/v) buffer was added and the pH was checked with pH indicator strips and, if necessary, adjusted to pH 6.5–7.5 with acetic acid or sodium hydroxide. After addition of 20 µl ß-glucuronidase E Coli K12 the sample was incubated for 1 h at 50°C. After the sample was cooled to RT, 100 mg of a mixture of K2CO3/NaHCO3 (2/1) was added. After mixing the sample with a vortex, 4 ml TBME were added as an extraction solvent, the sample was mixed for 15 min by means of head-over-head and centrifuged for 10 min at 4000g. A total of 2 ml of the upper layer was evaporated under a gentle stream of nitrogen at 40°C until just dry. The residue was dissolved in 300 µl H2O/ACN (70:30, v/v) and transferred into an LC-MS sample vial for analysis.
UHPLC-MS/MS analysis
The samples were analysed using UHPLC-MS/MS. The UHPLC system consisted of a Waters Acquity Ultra Performance LC equipped with Waters Acquity UPLC BEH analytical column (100 × 2.1 mm, 1.7 µm). Column temperature was set to 60°C, flow rate at 0.6 ml min−1 and injection volume was 5 µl. Gradient elution was used with mobile phase 5 mM formic acid in water (A) and 5 mM formic acid in ACN (B). After injection the mobile phase was held at 100% A during 0.5 min followed by a gradient in 7.5 min to 100% B and back to 100% A after a 1 min hold at 100% B. The total run time of the analysis was 12 min.
Detection was carried out using a Waters Xevo TQ-S mass spectrometer in positive electrospray ionisation (ESI) ion mode using SRM. Capillary voltage was set at 3.0 kV, source temperature at 150°C, desolvation temperature at 600°C, cone gas flow at 150 l h−1 and desolvation gas flow at 600 l h−1. Argon (p = 2.2 · 10–3 mbar; purity > 99.998%) was used as the collision-induced dissociation (CID) gas. The SRM transitions are given in with the cone voltage used and collision energy settings.
Confirmatory analysis of formestane was carried out using GC-MS/MS. This was performed using an internal sample pre-treatment and detection procedure for hormone analysis (RIKILT Citation2013).
Method validation
A full validation was carried out according to Commission Decision 2002/657/EC (EC Citation2002). According to this procedure, the following parameters had to be determined: linearity, trueness, repeatability, within-laboratory reproducibility, decision limit (CCα), detection capability (CCß), selectivity, robustness and stability. According Commission Decision 2002/657/EC, trueness should be determined using certified reference material (CRM). For the compounds and materials used in this study no CRM is available. In such a case the document states trueness can be determined using additions of known amounts of the compounds to blank matrix. Based on both WADA MRPL levels and pre-validation studies, validation target levels were set for the compounds, which can be found in section S1 in the supplemental data online. The validation was carried out on 3 different days with 21 different bovine urine samples and additionally 1 day with seven different porcine urines.
Quantification was performed using a matrix-matched standard (MMS) calibration curve. Commission Decision 2002/657/EC states that a compound is confirmed when the relative retention time of the chromatographic peak and the relative ion ratio of the two product ions fulfil the criteria specified. Linearity was calculated with linear regression on a MMS curve for a concentration range of 0.25–3.0 times the target level for validation. The calculated correlation coefficient (R2) should be > 0.990. For repeatability, within-laboratory reproducibility and accuracy, seven different blank urine samples were fortified with 0.5, 1.0 and 1.5 times the target level for validation. The same procedure was repeated on 2 additional days with seven different fortified blank urine samples on each day. Repeatability, within-laboratory reproducibility and accuracy were calculated from these data and had to fulfil the criteria described in tables 2 and 3 of Commission Decision 2002/657/EC. CCα and CCβ were determined by analysing 21 blank urines and 21 urines fortified at 0.5 times the target level for validation. CCα was calculated according ISO-11843 using the equation:
Table 2. CCα and CCβ (ng ml–1) in bovine and porcine urine.
where SD is the standard deviation of within-laboratory reproducibility. CCβ was calculated by using:
Robustness was tested by analysing four urine samples in duplicate. The first sample was analysed using the developed method; the three other samples were analysed with a slightly different sample pre-treatment procedure. The differences in sample pre-treatment were: prolonging of the incubation time, reduction of the addition of K2CO3/NaHCO3 and prolonging of the evaporation time after the sample extract is already dry. The method can be considered as robust in case the accuracy of a specific compound lies within the repeatability limit of the method. Specificity was checked by the analysis of 20 blank samples without the addition of the compounds. The chromatograms were monitored for peaks interfering with those of the compounds of interest. When no interference on a relevant level is observed, the method can be considered specific for this compound. Stability of the compounds in solution and matrix was determined. Stability in solution was determined by comparing −80°C stored standard solutions with standard solution stored in the freezer for 1, 3, 6, 9 and 12 months. Stability in extracts was determined by comparing the results of sample extracts of validation day 1 which were stored in the freezer for 1 week after analyses with the initial results.
Survey
To gain insight into the current situation of the possible abuse of SERMs, AEs and AIs in the Netherlands, a survey was performed. For this survey 114 bovine urines (both calf and adult) and 103 porcine urines from Dutch farms were analysed using the method described in this paper. The urines used were sampled by the Dutch Food Safety Authority (NVWA) for use in the regulatory control programme of the Netherlands and stored in the freezer before analysis. No additional information on gender and age of the animals was available to the researchers for this study. The samples were analysed using the described method.
Results and discussion
Method validation
A summary of the validation results (CCα and CCβ values) for bovine and porcine urine is given in . Complete validation results can be found in section S2 in the supplemental data online. As can be seen, the method is not suitable for the quantification of all compounds. This is the case if one or more of the parameters linearity, trueness, repeatability or within-laboratory reproducibility does not fulfil the criteria stated in Commission Decision 2002/657/EC. If quantification of a compound is necessary, it should be analysed using a multilevel standard addition method or a method dedicated to this compound. In the case of 4-hydroxycyclofenil confirmation is not possible. In this case the ion ratio does not fulfil the criteria for confirmation stated in Commission Decision 2002/657/EC. A reason for not fulfilling the criteria for some compounds is most likely ion suppression in the MS and the lack of isotope-labelled internal standards. 4-Hydroxytamoxifen has two isomers that are chromatographically separated. The Z-isomer is the most active form. However, no enantiomeric pure standards were available so no decisive answer can be given at this time as to which peak belongs to which isomer. Clomiphene also has two isomers; both the Z and E isomers are active forms, but they cannot be separated chromatographically with this method. Due to this, the validation parameters are calculated for the sum of the isomers. In porcine urine 4-hydroxycyclofenil cannot be analysed due to interfering peaks around the retention time of the chromatographic peak of 4-hydroxycyclofenil. The peak interferences are not present in bovine urine chromatograms. For compounds that can be measured quantitatively, CCα and CCß are calculated as described above. For compounds that can be analysed qualitatively, CCα and CCß are set to the lowest validation level. Linearity of the MMS calibration lines of all compounds fulfils the set criteria. The robustness experiments all fulfilled the set criteria, so for the tested changes the method can be considered robust. With the specificity measurements no peaks at 0.03× the target level for validation were found. Low abundant peaks (< 0.03× the target level for validation), when present, were probably due to cross-contamination from the UHPLC system. Because the peak areas are more than 10 times lower than the lowest point in the MMS, this is not considered to be a problem. The stability results show that the extracts could not be stored in the freezer for 1 week after analysis, so sample extracts should be analysed directly after sample pre-treatment. The results of the stability of the standard solutions show that the compounds are stable for 1 year in the freezer, with the exception of raloxifene, fulvestrant and 4-hydroxytamoxifen. These standard solutions are stable for 3 months in the freezer.
Survey
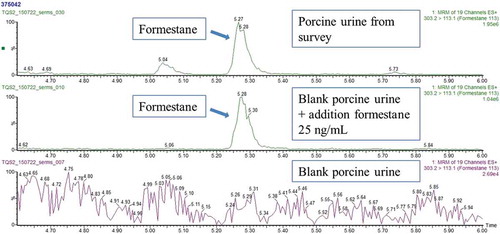
In the majority of the bovine urine samples no SERMs, AEs and AIs were detected. In 18 of the porcine urine samples and two of the bovine samples, formestane, an AI, was detected with indicative levels of 15–72 ng ml−1. shows three chromatograms: a blank porcine urine sample, a blank porcine urine sample with the addition of 25 ng ml−1 and a porcine urine sample from the survey suspected of containing formestane. Because no confirmation and quantification of formestane was possible with the UHPLC-MS/MS method (see Table S1 in the supplemental data online), a selection of the samples was analysed using a quantitative and confirmative in-house GC-MS/MS method dedicated for formestane (RIKILT Citation2013). In the selected samples (porcine urine, n = 3) formestane was confirmed according Commission Decision 2002/657/EC and quantification was performed using an external calibration curve of formestane. Concentrations in the samples were 9, 24 and 33 ng ml−1. Based on these positive confirmation results, we assume all 20 samples contained formestane. This assumption is valid in this project because the main goal of the survey was to gain insight into possible misuse of the compounds in the Netherlands, and no enforcement based on these results was performed.
Figure 1. (colour online) Chromatograms of a blank porcine urine (bottom), a blank porcine urine with addition of 25 ng ml−1 formestane (middle) and a porcine urine from the survey samples that contain formestane. The areas on the y-axes of the chromatograms are not normalised.
From studies on human urine it is known that formestane can be of endogenous origin (Piper et al. Citation2012; Polet et al. Citation2013; De la Torre et al. Citation2014) and thus can be found at low levels in human urine. Limited studies show exogenous formestane detection in sports horse urine (Leung et al. Citation2013), but no literature is published of the presence of formestane in bovine or porcine urine. Therefore, additionally 10 porcine, 20 bovine and 20 ovine urine samples of known untreated animals (Sterk et al. Citation1998) were analysed for formestane using the GC-MS/MS confirmatory method (RIKILT Citation2013). In the porcine and ovine urine samples no formestane was detected. However, in nine of the 20 bovine samples formestane was detected at levels around 1 ng ml−1. Therefore, formestane can be considered to be of endogenic origin in this species. The relative large number of findings in the survey samples of formestane in porcine animals needs further studies. In order to assess whether formestane found in the bovine or porcine urine samples is endogenous or exogenous, the urine samples should be analysed using a suitable GC-c-IRMS method. With GC-c-IRMS it should be possible to make a distinction between endogenous and exogenous formestane. This technique has already proved its value for hormones like testosterone and estradiol in bovine urine (Piper et al. Citation2012), but it remains to be developed for formestane in bovine and porcine urine.
Conclusions
In order to detect possible abuse of SERMs, AEs and AIs in animal husbandry, a UHPLC-MS/MS method was developed for the determination and confirmation of SERMs, AEs and AIs in bovine and porcine urine. Additionally, the method was validated according to Commission Decision 2002/657/EC. A survey on more than 200 Dutch bovine and porcine urine samples was performed to gain insight into the current situation of possible misuse of the compounds in Dutch animal husbandry. In 18 of 103 porcine urine samples (17%) and two of 114 bovine urine samples (2%) formestane (AI) was detected and additionally confirmed (a selective number of cases) using a dedicated GC-MS/MS method. From the literature it is known that formestane can be of endogenous origin in humans, but no data for bovine and porcine animals are available. Analyses of reference samples obtained from untreated animals demonstrated the endogenous origin of formestane in bovine animals. However, until now in porcine animals no findings of endogenous formestane have been reported in the literature. Additional information of the natural occurrence and metabolic pathways of formestane in porcine and bovine animals is needed. Therefore, the development of a GC-c-IRMS method for formestane needs to be undertaken.
Acknowledgements
The authors thank the Dutch Food Safety Authority (NVWA) for providing sample materials.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
The Supplemental data for this article can be accessed here.
Additional information
Funding
References
- De la Torre X, Colamonici C, Curcio D, Jardines D, Molaioni F, Parr MK, Botrè F. 2014. Detection of formestane abuse by mass spectrometric techniques. Drug Test Anal. 6:1133–1140.
- De Rijke E, Essers ML, Rijk JCW, Thevis M, Bovee TFH, van Ginkel LA, Sterk SS. 2013. Selective androgen receptor modulators: in vitro and in vivo metabolism and analysis. Food Addit Contam Part A. 30:1517–1526.
- Dutertre M, Smith CL. 2000. Molecular mechanisms of selective estrogen receptor modulator (SERM) action. J Pharmacol Exp Ther. 295:431–437. Epub 2000 Oct 25.
- EFSA Panel on Contaminants in the Food Chain. 2013. Guidance on methodological principles and scientific methods to be taken into account when establishing Reference Points for Action (RPAs) for non-allowed pharmacologically active substances present in food of animal origin. EFSA J. 11:3195.
- [EC] European Commission. 2002. Commission decision 2002/657/EC implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Commun L. 221:8–36.
- Gerace E, Salomone A, Abbadessa G, Racca S, Vincenti M. 2012. Rapid determination of anti-estrogens by gas chromatography/mass spectrometry in urine: method validation and application to real samples. J Pharm Anal. 2:1–11.
- Ho EN, Leung DK, Leung GN, Wan TS, Wong HN, Xu X, Yeung JH. 2007. Metabolic studies of mesterolone in horses. Anal Chim Acta. 596:149–155. Epub 2007 Jul 10.
- Kang M-J, Hwang YH, Lee W, Kim D-H. 2007. Validation and application of a screening method for β2-agonists, anti-estrogenic substances and mesocarb in human urine using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 21:252–264.
- Krug O, Thomas A, Walpurgis K, Piper T, Sigmund G, Schanzer W, Laussmann T, Thevis M. 2014. Identification of black market products and potential doping agents in Germany 2010-2013. Eur J Clin Pharmacol. 70:1303–1311.
- Leung GNW, Kwok WH, Wan TSM, Lam KKH, Schiff PJ. 2013. Metabolic studies of formestane in horses. Drug Test Anal. 5:412–419.
- Lønning PE. 2004. Aromatase inhibitors in breast cancer. Endocr Relat Cancer. 11:179–189.
- Mareck U, Sigmund G, Opfermann G, Geyer H, Thevis M, Schänzer W. 2005. Identification of the aromatase inhibitor letrozole in urine by gas chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 19:3689–3693.
- Mauras N, O’Brien KO, Klein KO, Hayes V. 2000. Estrogen suppression in males: metabolic effects. J Clin Endocrinol Metabolism. 85:2370–2377.
- Mazzarino M, Botrè F. 2006. A fast liquid chromatographic/mass spectrometric screening method for the simultaneous detection of synthetic glucocorticoids, some stimulants, anti-oestrogen drugs and synthetic anabolic steroids. Rapid Commun Mass Spectrom. 20:3465–3476.
- Mazzarino M, Braganò MC, de la Torre X, Molaioni F, Botrè F. 2011. Relevance of the selective oestrogen receptor modulators tamoxifen, toremifene and clomiphene in doping field: endogenous steroids urinary profile after multiple oral doses. Steroids. 76:1400–1406.
- Piper T, Fusshöller G, Emery C, Schänzer W, Saugy M. 2012. Investigations on carbon isotope ratios and concentrations of urinary formestane. Drug Test Anal. 4:942–950.
- Poelmans S, De Wasch K, De Brabander HF, Van De Wiele M, Courtheyn D, van Ginkel LA, Sterk SS, Delahaut P, Dubois M, Schilt R, et al. 2002. Analytical possibilities for the detection of stanozolol and its metabolites. Anal Chim Acta. 473:39–47.
- Polet M, Van Renterghem P, Van Gansbeke W, Van Eenoo P. 2013. Profiling of urinary formestane and confirmation by isotope ratio mass spectrometry. Steroids. 78:1103–1109.
- Regal P, Nebot C, Vázquez BI, Cepeda A, Fente CA. 2010. Determination of the hormonal growth promoter 17α-methyltestosterone in food-producing animals: bovine hair analysis by HPLC–MS/MS. Meat Sci. 84:196–201.
- RIKILT Wageningen University & Research. 2013. SOP 1160 – bovine and porcine urine, meat fish and liver. The analysis of a large number of hormones. Wageningen: GC-MS/MS.
- Sterk SS, van Tricht EF, Stephany RW. 1998. Lyophilised samples of blank urine for the validation and quality control of residue analysis for growth promoting compounds. Bilthoven: RIVM. Monograph Wageningen UR Library C1 -NN31014, 1998-45.
- World Anti Doping Agency. 2015. WADA technical document – TD21015MRPL – minimum required performance levels for detection and identification of non-threshold substances. Montreal: World Anti Doping Agency.
- World Anti Doping Agency. 2016. WADA technical document – TD2016IRMS. Detection of synthetic forms of endogenous anabolic androgenic steroids by GC/C/IRMS. Montreal: World Anti Doping Agency.