
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The performance characteristics of a multi-analyte method for the determination of all 10 carotenoids authorised as feed additives within the EU were assessed via an interlaboratory study. The analytical method is based on reversed phase high performance liquid chromatography (RP-HPLC) coupled to an optical detector set at 410 nm. The analysis is particularly challenging due to the presence of various stereoisomers of each carotenoid, and the use of these compounds via natural or synthetic formulations, requiring a special sample preparation. EU regulations specifying the conditions of use set legal limits for these substances in compound feedingstuffs ranging from 6 mg kg−1 to 138 mg kg−1, depending on the individual carotenoid and the target animal for which the feed is supplemented with this carotenoid. The purpose of the multi-analyte method validated in this paper is to facilitate the monitoring of carotenoids at relevant levels when used as feed additives in compound feedingstuffs and pre-mixtures. The interlaboratory study delivered precision data for 43 different analyte/mass fraction/matrix combinations, covering a mass fraction range of the target analytes from 2.6 mg kg−1 to 3861 mg kg−1. The relative standard deviations for repeatability (RSDr) varied from 2.2 to 16.2 % with a mean value of 6 %, while the relative standard deviations for reproducibility (RSDR) varied from 6.8 to 39 % with a mean value of 21 %. Given the broad scope of the method covering 10 carotenoids added to compound feedingstuffs and pre-mixtures via different formulations, this multi-analyte method is considered fit for the intended purpose.
Introduction
Carotenoids are used as feed additives in animal nutrition and require a premarket authorisation within the European Union (EU) according to a procedure specified by European legislation (European Union Citation2003). This procedure foresees that the applicant asking for authorisation prepares a dossier with technical information related to the feed additives, which is subsequently evaluated by the European Food Safety Authority (EFSA). Moreover, the applicant needs to submit appropriate methods of analysis for the active substance of the product along with results of validation studies. The evaluation of the proposed methods is done by the EU Reference Laboratory for Feed Additives (EURL-FA), formerly called Community Reference Laboratory (European Union Citation2005). The EURL-FA is hosted by the European Commission’s Joint Research Centre and its operation is described in a previous publication (von Holst et al. Citation2016). Based on the risk assessment of EFSA and the report of the EURL-FA, the European Commission decides on the approval of the request for authorisation, specifying by Commission Implementing Regulations the above-mentioned target levels of the active substance and the analytical methods that the Member States’ official laboratories need to apply to enforce these levels.
Currently there are 10 carotenoids authorised as feed additives, namely astaxanthin (AXN), canthaxanthin (CXN), adonirubin (ADR), astaxanthin dimethyldisuccinate (AXN DMDS), capsanthin (CSN), beta-carotene (BCAR), ethyl ester of beta-apo-8ʹ-carotenoic acid (BACARE), citranaxanthin (CIXN), lutein (LUT) and zeaxanthin (ZEA). They are mainly used as colourants under the category ‘sensory additives’ (European Union Citation2003) to adapt the colour of egg yolk and fish flesh to the preferences of the consumer. The details of the authorisation conditions in terms of legal limits and a reference to the analytical methods as established by corresponding Regulations are easily accessible via the EU register of feed additives (European Union Citation2020), abbreviated in this paper as register. Carotenoids are introduced into feedingstuffs via specific preparations, which are either natural products such as the red carotenoid-rich bacterium Paracoccus carotinifaciens or industrially produced preparations containing the carotenoids from synthesis. Because carotenoids are sensitive to heat and light, the latter preparations are encapsulated products to render these substances more stable. Each carotenoid is listed separately in this register, while in the case of the above-mentioned natural product, Paracoccus carotinifaciens, is included in the register (European Union Citation2020) as an authorised feed additive, containing the active substances astaxanthin, canthaxanthin and adonirubin.
Monitoring of the proper use of this group of feed additives is quite important given their widespread application and related safety considerations. Legal limits in complete feedingstuffs have been established for all carotenoids – except for beta-carotene – by EU legislation when used as feed additive, either for individual substances or for a sum of these substances (European Union Citation2020). Moreover, the legal limits for individual carotenoids always refer to the sum of the corresponding cis-trans stereoisomers, which is an important requirement for the quantification of the respective analyte. An overview of the various carotenoids authorised in the European Union, their legal limits and corresponding legal acts is given in a previous publication (Vincent et al. Citation2017).
While this regulatory system ensures that detailed protocols of well-validated methods are available for each carotenoid, the implementation of these methods under routine control conditions turned out to be quite difficult for various reasons. One drawback of this system is that the methods differ depending on the specific analyte and in some cases even on the type of feed additive preparation containing specific carotenoids, while laboratories prefer the use of multi-analyte/multi-product methods that work well for as many as possible of the feed additives and preparations currently used in the European market. Moreover, the majority of these methods require the use of normal phase high performance liquid chromatography (NP-HPLC) to address the correct separation and subsequent quantification of the various stereoisomers of each carotenoid, while nowadays the by far preferred analytical approach is reversed phase HPLC (RP-HPLC). Furthermore, some of the methods require the use of chlorinated solvents, which should be avoided if possible on environmental grounds.
Consequently, the development of a multi-analyte method suitable for monitoring the correct use of carotenoids as feed additives had to cope with the following major challenges: (1) The extraction conditions have to be applicable to natural and encapsulated preparations; (2) The liquid chromatography has to separate all carotenoids currently authorised within the EU at levels of interest in complete feedingstuffs as defined by EU legislation and in pre-mixtures; (3) The identification of the various stereoisomers individually for each analyte should be achievable by visual inspection of the chromatogram. Moreover, alternatives to chlorinated solvents had to be explored and the chromatographic separation should be accomplishable with RP-HPLC. To respond to the needs for efficient official control, the authors of this study had developed a method based on RP-HPLC coupled to an optical detector in the visible range (VIS) that fulfils the above-mentioned criteria. The experimental details of this method have been reported in previous publications (Mitrowska et al. Citation2012; Vincent et al. Citation2017).
At first, chromatographic conditions were optimised, to achieve sufficient separation of all analytes. In addition, a unique isosbestic wavelength was identified at which the various stereoisomers of each analyte have the same absorption coefficient, thus facilitating the quantification of the sum of all stereoisomers for a specific carotenoid (Mitrowska et al. Citation2012). The equivalent absorption coefficient for all stereoisomers of the same analyte at the isosbestic wavelength is a key prerequisite for using RP-HPLC, which has the intrinsic characteristic that a sufficient chromatographic separation of the various stereoisomers of each analyte cannot be achieved in all cases. However, when measuring at the isosbestic wavelength, all stereoisomers of a specific carotenoid can be quantified against the corresponding all-trans isomer, even if the various stereoisomers are not separated. Moreover, for those analytes for which the stereoisomers are well separated under the conditions of this RP-HPLC procedure, the peaks of all stereoisomers have to be identified and the corresponding areas need to be summed for the correct mass fraction calculation of the analytes.
In a subsequent study, specific extraction conditions for the analytical method were determined that can be applied to the analysis of feedingstuffs containing real-world feed additive/preparations from different origin. The optimised method comprises the following three steps: (1) enzymatic digestion followed by solid-liquid extraction (SLE) or pressurised liquid extraction (PLE); (2) separation of the carotenoids applying RP-HPLC; and (3) quantification with an optical detector using an identical wavelength for all carotenoids. Moreover, the optimised method was subjected to a single-laboratory validation and acceptable results expressed in terms of precision and trueness were obtained (Vincent et al. Citation2017), thus fulfiling the criteria to be selected for a subsequent interlaboratory study.
The purpose of this paper is to present the results from an interlaboratory method performance study of this multi-analyte method (Vincent et al. Citation2017) for the determination of all carotenoids currently authorised within the EU at target levels in complete feedingstuffs and pre-mixtures.
Methods and materials
Study design
Since the participation in this interlaboratory study required that the laboratories had to apply a specific method protocol, a training phase was organised prior to the actual validation study. The main purpose of this training phase was to ensure that the participants were sufficiently knowledgeable about all details of the method and in particular regarding the more critical steps such as the correct identification of the various stereoisomers of each analyte. In addition to the method protocol and templates for reporting the results of analysis, all laboratories taking part in the study obtained test samples and the target analytes, sent out separately for the training and validation study. For the training period, the participants received four compound feed samples with unknown content of the target analytes and pure calibration standards of the all-trans isomer of each analyte in solution at a concentration of 200 μg ml−1. The laboratories also obtained AXN DMDS and BCAR as solid pure standard substances to practice the whole procedure. The participants had to send back to the organiser of the study along with the results of analysis detailed information on the concrete application of the method and chromatograms from the analysis of the test samples. This feedback was closely scrutinised and the organiser of the study contacted the participants to clarify open questions on how correctly to apply the method protocol in their laboratory environment.
After finalising the training period, the laboratories were sent via a second shipment 16 compound feed samples with unknown content of the target analytes, a blank compound feed sample and a fresh set of solid calibration standards. Based on the experiences gained in the training period the laboratories also received 10 solutions in acetone containing individually the various stereoisomers of each analyte and a solution of a mixture of all isomerised standards. The purpose of both types of solutions was to help the laboratories with the correct attribution of the peaks in the chromatogram to the respective stereoisomers of the analytes, in the easier case when the other carotenoids are absent and in the more complex case when the other carotenoids are present. Moreover, the laboratories received a revised version of the method protocol clarifying questions from the participants raised during the training period and containing supplementary information regarding the identification of the stereoisomers.
Statistics
This method performance study was designed according to the corresponding IUPAC harmonised protocol (Horwitz Citation1995). The study presented here is based on the principle that a set of duplicate samples of 8 different feeds are analysed by the participating laboratories and the results are reported back to the organiser of the study for further statistical assessment of the precision of the method.
First, extreme values were excluded from the study, especially when obvious experimental deviations from the protocol were identified or when specific quality criteria of the method were not met. In the second step, following the IUPAC protocol, the remaining valid data were purged of all outliers flagged by the harmonised outlier removal procedure. This procedure consists of the sequential application of the Cochran and Grubbs tests (at 2.5% probability level, one-tail for Cochran and two-tails for Grubbs) until no further outliers are flagged or until a drop of 22.2% (=2/9) in the original number of laboratories providing valid data occurred.
The remaining data were subsequently subjected to analysis of variance (ANOVA) to estimate the values for the relative standard deviation for repeatability (RSDr) and the relative standard deviation for reproducibility (RSDR).
Test material and reference standards
Test materials
The composition of the test materials was designed in order to ensure a minimum of four different matrix/analyte/concentration combinations for each of the 10 target analytes across the range of mass fractions tested at and around the additive levels, except for adonirubin, where the number of different matrix/analyte/concentration combinations was two. The composition of the test materials is given in . The ideal scenario of at least 5 analyte/mass fraction/matrix combinations for all 10 carotenoids could not be implemented due to various external restrictions, namely (1) legal provisions of using only specific carotenoids for poultry and fish feedingstuffs; (2) the need of including these analytes both from natural and synthetic origin in the study; (3) the unique characteristics of the natural product Paracoccus carotinifaciens containing three carotenoids (astaxanthin, canthaxanthin and adonirubin); and (4) the need to limit the overall number of samples of the study considering the available resources of the participating laboratories.
Table 1. Composition of the nine test materials (MATs) of the study, classified in terms of the target species, i.e. fish versus poultry and type of additive, i.e. by synthesis versus natural: The target mass fractions of the carotenoids in the different test materials are shown in , and . AXN DMDS: Astaxanthin dimethyldisuccinate; BACARE: Ethyl ester of beta-apo-8ʹ-carotenoic acid. Materials used in the study: Complete feedingstuffs: For fish: MAT 1–3; For poultry: Grower feed: MAT 5, Laying hens: MAT 6 and 7. Premixtures: For fish: MAT 8, For poultry: MAT 9
Table 2. Results from the collaborative trial: Performance of the method for the determination of carotenoids exclusively used in fish feedingstuffs. AXN DMDS: Astaxanthin dimethyldisuccinate; Calculated value: Mass fraction of the carotenoids in the test material based on the certificate of analysis of the specific feed additive used; Average: Average obtained from the statistical assessment of the results from the laboratories. RSDr: relative standard deviation of repeatability; RSDR: relative standard deviation of reproducibility. No. of laboratories indicates the number of laboratories retained after removal of outliers identified by Cochran and Grubbs tests. The number in brackets is the number of laboratories identified as outliers
Table 3. Results from the collaborative trial: Performance of the method for the determination of carotenoids exclusively used in poultry feedingstuffs. BACARE: Ethyl ester of beta-apo-8ʹ-carotenoic acid; Calculated value: Mass fraction of the carotenoids in the test material based on certificate of analysis of the specific feed additive used. Average: Average obtained from the statistical assessment of the results from the laboratories. RSDr: relative standard deviation of repeatability; RSDR: relative standard deviation of reproducibility. No. of laboratories indicates the number of laboratories retained after removal of outliers identified by Cochran and Grubbs tests. The number in brackets indicates the number of laboratories identified as outliers
Table 4. Results from the collaborative trial: Performance of the method for the determination of carotenoids used in fish feedingstuffs (MAT 1-MAT 3, MAT 8) and poultry feedingstuffs (MAT 5-MAT 7, MAT 9). Calculated value: Mass fraction of the carotenoids in the test material based on certificate of analysis of the specific feed additive used, *: Average from the homogeneity study due to a strong discrepancy with the calculated value probably caused by a problem with the preparation of the test material. Average: Average obtained from the statistical assessment of the results from the laboratories. RSDr: relative standard deviation of repeatability; RSDR: relative standard deviation of reproducibility. No. of laboratories indicates the number of laboratories retained after removal of outliers identified by Cochran and Grubbs tests. The number in brackets is the number of laboratories identified as outliers
Two different complete compound feed for poultry (for laying hens and grower feed), one complete compound feed for fish and two pre-mixtures – one for poultry and one for fish – were used for the preparation of the test materials. For the target carotenoids commercial feed additive preparations were employed, which were either natural or synthetic products. The commercial feed additives of the latter type were encapsulated preparations containing various excipients and the respective carotenoid at mass fractions in the range of 10 % as specified for instance in the Scientific Opinion of the European Food Safety Authority (EFSA) for astaxanthin (EFSA Citation2014). Three different natural feed additives were utilised in the study, namely for fish feedingstuffs a product from Paracoccus carotinifaciens containing astaxanthin, adonirubin and canthaxanthin, while for poultry feedingstuffs saponified paprika extract containing capsanthin and processed leaves from plants containing lutein and zeaxanthin. An overview of the content of the target carotenoids in all the feed additives utilised in this study is provided in a previous publication (Vincent et al. Citation2017). The corresponding mass fractions of the analytes in the test materials are given in to 4.
First, the commercial feeds were tested in the laboratory to establish that they did not contain the target carotenoids. Then the feeds were ground and fortified with the above-mentioned feed additive preparations. The expected mass fractions of the respective carotenoids in the test samples were taken from the certificates of analysis of the producer of the feed additives. Finally, the materials were homogenised and distributed in bottles containing approximately 15 g of material. Prior to shipment of the test materials, 10 samples were randomly taken from each type of test material and analysed in duplicate. Results were evaluated according to ISO 13528:2015 (ISO Citation2015), showing that the test materials were sufficiently homogenous for the purpose of the study.
Given the light and temperature sensitivity of carotenoids, a proven stability of the final test material during the validation study was considered a crucial element. Therefore, the stability of the samples was tested according to the following design. Eight samples from all materials were split into three groups. Four samples were analysed, in duplicate, at the beginning of the study. The second group comprised of two samples were then kept for the whole period of the validation study at −20°C and the remaining two samples were kept for the same period at 4°C, reflecting the target transport and storage conditions of the test samples. The samples stored at −20°C and 4°C were simultaneously analysed, in duplicate, for the target analytes, at the end of the study. The obtained mass fractions from all three groups were then subjected to statistical assessment. First, a one-tailed F-test showed that there was no significant difference of the variances between the three groups. Then, the subsequent t-test (α = 0.05) showed that no significant difference of the mean values of the carotenoid mass fractions from the three groups could be observed, thus confirming sufficient stability of the test material.
In total, the laboratories received 16 samples supplemented with the carotenoids (MAT 1 – MAT 3, MAT 5 – MAT 9) and a blank compound feed sample (MAT 4) in order to assess the probability of false positive results of the method.
Reference standards
The organiser of the study provided all participants with reference standards in powder form; the analytes were the all-trans-isomers. Adonirubin, ethyl ester of beta-apo-8ʹ-carotenoic acid, capsanthin, citranaxanthin, lutein and zeaxanthin were purchased at CaroteNature GmbH (Lupsingen, Switzerland). Astaxanthin and canthaxanthin were obtained from Dr. Ehrenstorfer GmbH (Augsburg, Germany) and beta-carotene from Sigma–Aldrich (St. Louis, MO, USA). Astaxanthin dimethyldisuccinate was kindly provided by DSM Nutritional Products Ltd (Kaiseraugst, Switzerland).
Method
The laboratories were asked to take two sub-samples from each sample jar in order to carry out two independent experiments for the determination of the target analytes. The difference between the results served as additional criterion for proper implementation of the method and must not be above 15 % relative to the higher result. This procedure has been adopted from the routine control methods developed by industry. When passing this test, the average value of the two measurements was used for subsequent statistical assessment.
The analytical procedure for the compound feed and pre-mixture samples consisted of the following three steps: (1) enzymatic release of the target analytes from the feed additive preparations used to prepare the test samples; (2) extraction of these analytes using PLE or SLE; and (3) determination of the carotenoids with RP-HPLC/VIS. Details of the original method protocol applying PLE are given in a previous publication on the single-laboratory validation of the method (Vincent et al. Citation2017). The use of SLE was included as alternative technique for this study to allow the participation of laboratories that did not have the required PLE instrumentation. The laboratories participating in the study were free to opt for PLE or SLE.
Scope of the method
The purpose of the method presented in this study was to determine 10 carotenoids (astaxanthin, canthaxanthin, adonirubin, astaxanthin dimethyldisuccinate, capsanthin, beta-carotene, ethyl ester of beta-apo-8ʹ-carotenoic acid, citranaxanthin, lutein and zeaxanthin) used as feed additives in complete compound feed at mass fraction levels specified by European legislation and in pre-mixtures containing the analytes at higher mass fractions.
Regulation (EC) No 767/2009 (European Union Citation2009) requires that for feed additives such as carotenoids with legal limits in compound feed, commercial materials containing these substances have to be labelled with the specific feed additive and the mass fraction of the corresponding carotenoid. A main objective of official control is therefore to check samples for compliance with these levels indicated on the label and with legal limits, if applicable.
Enzymatic release
A 5-g aliquot of the test material added to an aqueous solution containing the enzyme was kept at 50 °C for 20 min as further specified in the previous publication (Vincent et al. Citation2017).
Extraction of the carotenoids
The mixture from the previous step was further processed using PLE or SLE. In the latter case, 50 ml of acetone was added to this mixture, shaken for 1 min and centrifuged at 1800–1900 g for 5 min. The supernatant was separated and transferred to a 150 ml volumetric flask; 50 ml of acetone was added to the remaining material, the previous procedure was repeated and the supernatant solution transferred to the 150 ml volumetric flask. An ultra-Turrax mixer was used to facilitate dissolution of the sample if needed. In the third and last extraction step, 40 ml of acetone was added to the remaining material, the previous procedure repeated and the supernatant solution transferred to the 150 ml volumetric flask and filled up to the mark with acetone. The solution was vigorously shaken and a 1.8 ml aliquot taken, centrifuged for 1 min at 13000 g and the supernatant transferred to a HPLC amber vial for subsequent HPLC analysis.
Determination with RP-HPLC/VIS
The carotenoids were measured with a RP-HPLC instrument equipped with a spectrophotometric detector adjusted at the isosbestic wavelength of 410 nm. A specific eluent gradient protocol (Vincent et al. Citation2017) with two different solutions, namely phase A (acetonitrile: methyl tert-butyl ether: water mixture (70:20:10; v:v:v)) and phase B (acetonitrile: methyl tert-butyl ether mixture (70:30; v:v)) was used to achieve a sufficient separation of the target analytes. Since individual carotenoids may be present in the sample with a number of different cis-trans isomers, the chromatographic conditions have scrupulously to be applied to ensure that the groups of peaks belonging to a specific carotenoid are well separated in the chromatogram from the group of peaks belonging to other carotenoids. The procedure also requires that a quality control solution containing astaxanthin and canthaxanthin at a defined concentration is included in the sample sequence. The signal area of the response of both analytes and the corresponding retention times of the quality control sample are then evaluated to confirm sufficient suitability of the HPLC system. Finally, the mass fraction of each carotenoid is determined from the corresponding summed area of all stereoisomers of the specific analyte, using an external calibration curve established with the all-trans standards of this carotenoid.
Prior to their use, all laboratories had to determine the correct concentration of the analytes in the calibration solutions by conducting an absolute spectrophotometric measurement separately for each carotenoid. For this purpose, solutions of the analytes are prepared in analyte specific solvents and spectra covering a range from 300–500 nm are measured against the pure solvent to determine the analyte specific wavelength at which the absorbance shows a maximum. The measured absorbances at this wavelength are then used in conjunction with the analyte specific absorption coefficients () to calculate the correct mass fraction of the analytes in the solutions. The carotenoids are dissolved in the following solvents and the respective absorption coefficients (
), as specified in the literature (Britton et al. Citation2004; US-Pharmacopeia Citation2020; FAO Online Edition Citation2020) are written in brackets next to each analyte: (1) AXN (2100) and AXN DMDS (1519) in n-hexane; (2) CXN (2200), ADR (2150), BACARE (2550), BCAR (2500) and CIXN (2680) in cyclohexane; (3) LUT (2550) and ZEA (2480) in ethanol; and (4) CSN (2157) in acetone.
Method for the isomerisation of the individual carotenoids
The purpose of the following protocol is to form separately for each carotenoid the various cis-trans isomers with their corresponding equilibrium concentrations by subjecting the respective all-trans isomer to specific experimental conditions. The protocol is that 1 ml of the standard solutions are individually filled into LC vials, tightly closed and kept at 80 °C for 2 hours. The obtained solutions can be subsequently measured with the RP-HPLC system to obtain a carotenoid specific profile of the isomers, which facilitates the correct chromatographic identification of the various cis-trans isomers of each carotenoid in the test sample. The correct identification of the cis-trans isomers belonging to the same carotenoid is crucial for a correct quantification of these substances.
Results and discussion
Training phase
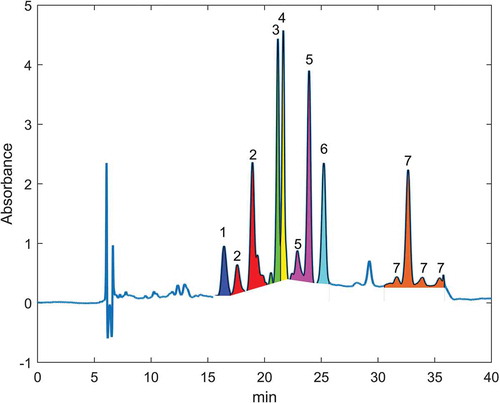
The results of the training phase and the evaluation of the supplementary information from the participants showed that the correct identification of the various stereoisomers of some analytes turned out to be one of the major problems. The specific challenge for the laboratories is exemplified by the following chromatograms. In two chromatograms of a standard solution containing exclusively astaxanthin dimethyldisuccinate are shown, namely the blue curve from the analysis of the standard solution, which contains only the all-trans isomer, and the red curve from the analysis of the standard solution after subjecting it to the isomerisation experiment. While the blue curve includes just one peak labelled as “1” corresponding to the all-trans stereoisomer, two additional peaks labelled as “2” appeared in the red chromatogram that belong to two cis stereoisomers formed in the isomerisation experiment. This is quite important, because isomerisation of the carotenoids is permanently occurring within the test samples. In consequence, it may happen when comparing the chromatogram of a sample against the chromatogram of the standards, which exclusively show the all-trans stereoisomer, that one of the two peaks of the cis stereoisomer from the chromatogram of the sample is omitted, leading to a significantly lower signal area for this analyte. This is even more complicated when evaluating the peaks from a chromatogram of MAT 6 (), which contains 7 carotenoids with their various stereoisomers as specified in the caption of this figure. It is obvious that in some cases, such as for capsanthin, the various stereoisomers almost co-elute, thus forming peak shoulders close to the all-trans isomer signal. In contrast to that, for instance the stereoisomers of lutein and BACARE are completely separated. In any case, the corresponding peaks need to be identified and the corresponding signal areas have to be summed. In order to address this critical aspect of the correct interpretation of the chromatograms, the organiser adapted the protocol by including examples of the chromatograms showing the peaks of the isomerised analytes. Moreover, the participants of the study obtained for the validation study isomerised analytes in solution, ready for injection as described in the section above on the study design and intended for easier peak identification.
Figure 1. Two chromatograms of a solution containing exclusively the astaxanthin dimethyldisuccinate standard substance, namely the chromatograms prior to the isomerisation experiment (blue line) and after this experiment (red line). While the chromatogram of the standard prior to the experiment exclusively shows a single peak labelled “1” corresponding to the all-trans isomer, the chromatogram shows after the experiment two additional peaks labelled “2”, which are two main cis isomers
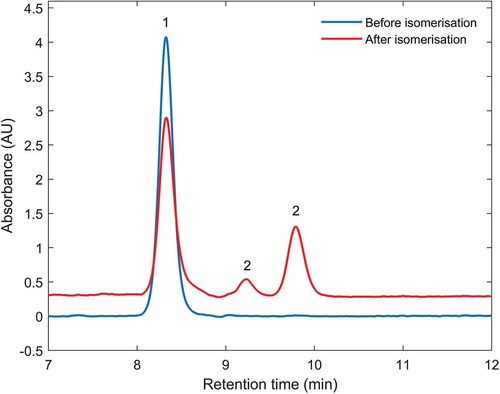
Figure 2. Chromatographic profile for some isomerised carotenoids obtained by analysis of Mat 6. The peaks of the same colour are different cis-trans isomers of the same anlayte labelled with the following numbers: (1) canthaxanthin, (2) capsanthin, (3) citranaxanthin, (4) ethyl ester of beta-apo-8ʹ-carotenoic acid, (5) lutein (6) zeaxanthin and (7) beta-carotene
Validation phase
After completion of the training period, all 17 laboratories were invited to take part in the subsequent validation study. Finally, 16 laboratories covering official control laboratories, private laboratories and laboratories from feed additive producers performed the required analysis of the test materials and provided results of analysis.
Fifteen laboratories had utilised SLE and one laboratory of this group used in addition PLE, thus reporting two sets of results. One laboratory applied exclusively PLE.
First, the results of the laboratories were individually screened to establish whether the difference of the duplicate samples obtained from the analysis of each sample jar was below 15 %. In cases where this quality criterion was exceeded, the corresponding analytical results for a specific analyte/mass fraction/matrix combinations and from the laboratory concerned was removed from the data set. The remaining data were subsequently subjected to the statistical treatment as described before.
Extraction method: influence of PLE
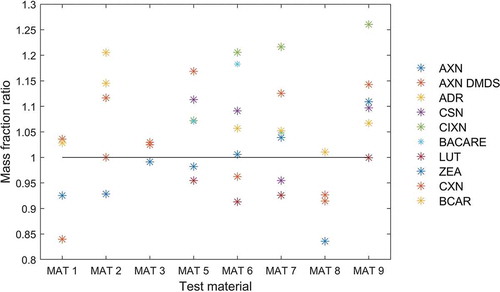
The impact of SLE and PLE on the results of analysis had been assessed and the evaluation did not indicate significant difference of the results (data not shown). Therefore, all data of the current study have been pooled regardless of the extraction method used. Prior to this step, the results of the laboratories were evaluated to establish whether PLE showed also a similar performance in the current study. For this purpose, a mass fraction ratio has been calculated by dividing separately for each analyte/matrix combination the average value for PLE reported by two laboratories by the corresponding average value of the results of all laboratories. The obtained mass fraction ratios for all 43 statistical assessments is given in indicating that they are well distributed above and below an ideal ratio of 1, with slight preference towards values higher than 1. This is confirmed by calculating the average value of all mass fraction ratios, which was 1.04 with a relative standard deviation of 10 %. This indicates a small positive method bias of PLE. The distribution of the mass fraction ratios amongst the various materials may also hint at a systematic effect, e.g. that the extraction efficiency for MAT 8 is systematically lower, whilst the opposite applies for MAT 9. However, all analytes are determined from the same extraction solution and therefore random effects that occurred in a single extraction step could influence simultaneously the results of all analytes. Moreover, the PLE results are based only on the experiments performed in two laboratories. Therefore, we assume that the scattering of the mass fraction ratios within and between the test materials reflects the overall measurement uncertainty of the carotenoid determination. Considering that the relative standard deviation of the mass ratio factor and the potential method bias (4 %) are low, we concluded that the results obtained with SLE and PLE could be pooled for the subsequent statistical assessment of the method performance characteristics.
Figure 3. Comparison of the results from two laboratories having applied PLE with the results of all laboratories shown by the mass fraction ratio. This parameter is obtained by dividing separately for each analyte/matrix combination the average mass fraction reported by two laboratories having applied PLE by the corresponding average value of the mass fractions of all laboratories. AXN: astaxanthin. AXN DMDS: astaxanthin dimethyldisuccinate. ADR: adonirubin. CSN: capsanthin. CIXN: citranaxanthin. BACARE: ethyl ester of beta-apo-8ʹ-carotenoic acid. LUT: lutein. ZEA: zeaxanthin. CXN: canthaxanthin. BCAR: beta-carotene
Precision profile of the method
The laboratories participating in the study performed duplicate analyses from each of the 16 sample jars, thus delivering analytical results from 32 independent measurements covering the whole analytical procedure. This allows for estimating the precision of the method separately for 43 analyte/mass fraction/matrix combinations. In addition, the laboratories performed a single analysis of the blank sample. An overview of the results is presented in for carotenoids exclusively used in fish feedingstuffs, in for carotenoids exclusively used in poultry feedingstuffs and in for carotenoids used in both types of feedingstuffs. The relative standard deviations for repeatability (RSDr) varied from 2.2 to 16.2 % with a mean value of 6 %, while the relative standard deviations for reproducibility (RSDR) varied from 6.8 to 39 % with a mean of 21 %. One may compare the obtained precision against the Horwitz ratio (Horwitz and Albert Citation2006), taken as a normalised performance parameter and a benchmark for the fitness for purpose of an analytical method. This approach is frequently applied for evaluating methods targeting a single analyte. However, a corresponding criterion has not been established yet for assessing the performance of methods measuring a group of analytes. Indeed, during the development of the method discussed here some compromises were accepted in terms of the chromatographic conditions and the selected detection wavelength to accommodate all 10 carotenoids in one method protocol. For instance, an isosbestic wavelength was selected to facilitate the quantification of 10 analytes, but at the expense of a decreased sensitivity for some of these analytes. Moreover, the results of the single-laboratory validation of the method indicated already that there was no relationship between the precision of the method and the mass fraction of the corresponding analyte as foreseen by the Horwitz equation. Given the high number of available precision data obtained from the 43 statistical assessments in this study, we looked at the applicability of the Horwitz equation by plotting the relative standard deviations for reproducibility against the corresponding mass fractions of the analytes. The results presented in did not show any influence of the level of the mass fraction on the precision of the method, even though the mass fractions covered a range of three orders of magnitude. Therefore, we decided not to apply the Horwitz equation for the evaluation of the method performance profile of the multi-analyte method of this study.
Figure 4. Precision expressed in terms of relative standard deviation for reproducibility RSD (%) of the method obtained from the 43 statistical assessments versus the mass fraction of the 10 carotenoids. AXN: astaxanthin. DMDS: astaxanthin dimethyldisuccinate. ADR: adonirubin. CSN: capsanthin. CIXN: citranaxanthin. BACARE: ethyl ester of beta-apo-8ʹ-carotenoic acid. LUT: lutein. ZEA: zeaxanthin. CXN: canthaxanthin. BCAR: beta-carotene
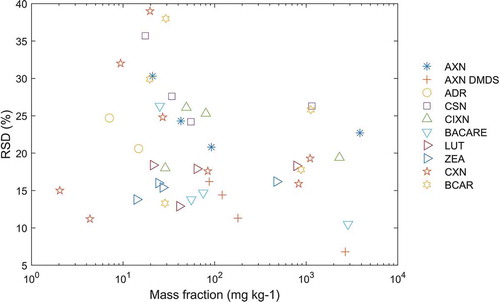
While the majority of the relative standard deviations for reproducibility covered a range of about 10 to 30 %, which are typical values found in feed analysis, there were four cases with elevated values for the RSDR above 30 %. Looking carefully at the corresponding chromatograms received from the laboratories revealed that the determination of the analytes especially in MAT 6 and MAT 7 posed the biggest problem. This is most likely related to the fact that these materials contain more analytes compared to other materials, thus leading to a higher number of peaks in the chromatogram, which in turn complicates the correct identification of all peaks that belong to the same analyte. For instance, the various peaks of canthaxanthin, capsanthin and citranaxanthin appear in the chromatogram very close to each other as shown in . Moreover, if the specific chromatographic conditions implemented in some laboratories did not allow for a sufficient separation of the stereoisomers or if less peaks per analyte appeared due to a somewhat lower sensitivity of the detector, the integration of the peaks related to these analytes was not performed in the optimal manner. In consequence, these findings led to larger deviations of the calculated mass fractions from the target values. It is important to highlight in this context that up to seven analytes were added to MAT 6 and MAT 7 to accommodate all carotenoids in the study, while the number of these substances added to compound feeds is much lower under real-world conditions. This means that under realistic conditions fewer peaks appear in the chromatogram, which facilitates the correct integration of the signals for the target analytes. Therefore, we concluded that the values of the RSDR above 30 % partly reflect the difficulty for the laboratories to identify the various stereoisomers of the analytes in case of the high number of peaks that appear in the chromatograms of these materials. Given the fact that the majority of the obtained values of the RSDR from the 43 statistical assessments were below 30 %, the method and taking into account the specific challenge of the correct identification of the various stereoisomers the method is considered fit for the intended purpose.
When implementing this method under routine conditions for official control, it is important to recall that in the case of a dispute on the found content of a specific carotenoid in the feed samples, the specific single-analyte method from the corresponding regulation authorising the carotenoid has to be applied. While this would often require the additional use of the less preferred normal phase liquid chromatography method, it can be expected that such a scenario will only take place in a minor number of cases.
The study also confirmed a sufficient specificity of the method, because no false positive results were reported on the analysis of the blank samples (MAT 4).
Trueness of the method
While the main objective of this study was to assess the precision profile of the method when applied in different laboratories, the calculated mass fractions of the target analytes in the test materials and the corresponding average values obtained from the statistical evaluation could also be used to estimate the trueness of the method. The trueness of the method expressed in terms of the recovery rate has been extensively investigated in the single-laboratory validation (Vincent et al. Citation2017) confirming quite acceptable values for the recovery rates. The average value of the recovery rate reported in the former study was 96 % with 3 values between 120 and 130 %. In the current study, an average recovery rate of 89 % was observed with 6 analyte/mass fraction/matrix combinations, in which the recovery rates were below 70 %. Given the thermal instability of the target analytes we could not exclude that these apparently low values for the recovery rate are partly due to adverse effects of the preparation procedure of the test material on the final mass fractions of the carotenoids. While we consider the average recovery of 89 % acceptable, it was not possible to identify the actual reasons for the recovery rates below 70 % given the above-mentioned restrictions of the preparation of test material. Therefore, further investigations of the apparently low recovery rates obtained in this study were not pursued.
Standardisation of the method
This interlaboratory study was organised within the work programme of the European Committee for Standardisation (CEN), Technical Committee 327, which has the scope to standardise methods of analysis for animal feedingstuffs (CEN Citation2020). The method for the determination of 10 carotenoids in compound feedingstuffs and pre-mixtures is referenced as CEN prEN 17550 and its status is currently ‘Under Enquiry’ with a presumed recommendation for adoption.
Conclusions
The availability of the analytical method validated here further via an interlaboratory study should facilitate the efforts of control laboratories to monitor the correct use of an important class of feed additives in animal nutrition. One should keep in mind that legal limits have been established for nine of these carotenoids.
Declaration of interest statement
No potential conflict of interest was reported by the authors.
Acknowledgments
The authors are grateful to Joseph Schierle and Andre Duesterloh (DSM Nutritional Products, Kaiseraugst, Switzerland) and to the members of CEN/TC 327/WG 3 (Animal feedingstuffs - Methods of sampling and analysis, Feed additives and drugs) for their continuous scientific support during the discussion of the results of the study.
The authors would like to thank the laboratories for their participation in this interlaboratory study: Österreichische Agentur für Gesundheit und Ernährungssicherheit GmbH, Inst. f. Tierernährung und Futtermittel/Abt. Zusatzstoffanalytik und Prüfung – AGES (Vienna, Austria), National Reference Laboratory of the Central Institute for Supervising and Testing in Agriculture, Ústřední kontrolní a zkušební ústav zemědělský,– ÚKZÚZ (Prague, Czech Republic), Finnish Food Safety Authority Evira Research and Laboratory Department, Chemistry and Toxicology Unit (Helsinki, Finland), BASF (Ludwigshafen, Germany), LUFA-ITL (Kiel, Germany), SGS Institut Fresenius GmbH (Germany), Federal Institute for Risk Assessment (BfR), National Reference Laboratory for Feed Additives, Unit Contaminants, Department Safety in the Food Chain (Berlin, Germany), Landesbetrieb Hessisches Landeslabor (Kassel, Germany), National Food Chain Safety Office, Food and Feed Safety Directorate, Feed Investigation Reference Laboratory (Budapest, Hungary), Ministero politiche agricole alimentari e forestali (Modena, Italy), National Institute of Nutrition and Seafood Research (Bergen, Norway), Nofima AS (Bergen, Norway), Laboratori Agroalimentari (DAAM) de Cabrils –Generalitat de Catalunya (Cabrils, Spain), DSM Nutritional Products (Kaiseraugst, Switzerland), Nutrition Analytical Service (Stirling, United Kingdom), Novus International (St. Charles MO, USA).
References
- Britton G, Liaaen-Jensen S, Pfander H. 2004. Carotenoids handbook. Basel, Switzerland: Birkhaeuser Verlag.
- [CEN] European Committee for Standardisation. 2020. [accessed 2020 Aug 25] https://standards.cen.eu/dyn/www/f?p=204:110:0::::FSP_PROJECT,FSP_LANG_ID:66542,25&cs=1C3EBD2ACB6D80E28100584E11BF8BBA5
- [EFSA] European Food Safety Authority. 2014. Scientific opinion on the safety and efficacy of synthetic astaxanthin as feed additive for salmon and trout, other fish, ornamental fish, crustaceans and ornamental birds. Efsa J. 12(6):3724, 1:35. doi:10.2903/j.efsa.2014.3877.
- European Union. 2003. Regulation (EC) No. 1831/2003 of the European Parliament and the council of 22 September 2003 on additives for use in animal nutrition. Off J Eur Union. L 268:29. as last amended by Commission Regulation (EU) 2015/2294.
- European Union. 2005. Commission Regulation (EC) No. 378/2005 of 4 March 2005 on detailed rules for the implementation of Regulation (EC) No. 1831/2003 of the European Parliament and of the Council as regards the duties and tasks of the community reference laboratory concerning applications for authorisations of feed additives. Off J Eur Union. L 59:8–15. last amended by Commission Implementing Regulation (EU) 2015/1761.
- European Union. 2009. Committee Regulation (EC) No 767/2009 of the European Parliament and of the council of 13 July 2009 on the placing on the market and use of feed, amending European parliament and council regulation (EC) No 1831/2003 and repealing council directive 79/373/EEC, Commission directive 80/511/EEC, Council directives 82/471/EEC, 83/228/EEC, 93/74/EEC, 93/113/EC and 96/25/EC and commission decision 2004/217/EC. Off J Eur Union. L 229:1–48.
- European Union. 2020. European Union register of feed additives. [accessed 2020 Aug 25]. http://ec.europa.eu/food/safety/docs/animal-feed-eu-reg-comm_register_feed_additives_1831-03.pdf
- Food and Agriculture Organization (FAO) Online Edition. 2020. Combined compendium of food additive. [accessed 2020 Aug 25]. http://www.fao.org/food/food-safety-quality/scientific-advice/jecfa/jecfa-additives/en/
- Horwitz W. 1995. Protocol for the design, conduct and interpretation of method-performance studies. Pure Appl Chem. 67:331–343. doi:10.1351/pac199567020331.
- Horwitz W, Albert R. 2006. The Horwitz ratio (HorRat): A useful index of method performance with respect to precision. J AOAC Int. 89:1095–1109. doi:10.1093/jaoac/89.4.1095.
- [ISO] International Organization for Standardization. 2015. ISO 13528:2015. Statistical methods for use in proficiency testing by interlaboratory comparison. Geneva (Switzerland):ISO copyright office.
- Mitrowska K, Vincent U, von Holst C. 2012. Separation and quantification of 15 carotenoids by reversed phase high performance liquid chromatography coupled to diode array detection with isosbestic wavelength approach. J Chromatogr A. 1233:44–53. doi:10.1016/j.chroma.2012.01.089.
- U.S. Pharmacopeia. 2020. [accessed 2020 Aug 25]. https://www.uspnf.com/
- Vincent U, Serano F, von Holst C. 2017. Development and validation of a multi-analyte method for the regulatory control of carotenoids used as feed additives in fish and poultry feed. Food Addit Contam. 34:1285–1297. doi:10.1080/19440049.2017.1315651.
- von Holst C, Robouch P, Bellorini S, González de la Huebra MJ, Ezerskis Z. 2016. A review of the work of the EU reference laboratory supporting the authorisation process of feed additives in the EU. Food Addit Contam. 33:66–77.