Abstract
Twin formation energy is an intrinsic quantity for bulk crystals. At the nanoscale, the twin formation energy of covalent SiC nanowires goes up with decreasing dimension. In contrast, this article reports that the twin formation energy of metallic nanowires goes down with decreasing dimension. This result is based on classical molecular statics simulations of four representative metals. Cu and Al represent face-centered cubic (FCC) metals of low and high twin formation energies. Ta represents a body-centered cubic (BCC) metal, and Mg represents a hexagonal close-packed (HCP) metal. For all the four metals, the dependence of twin formation energy on size correlates with lower twin formation energy near surfaces, according to atomic-level analysis. Based on this atomic-level insight, the authors propose a core–shell model that reveals the twin formation energy as inversely proportional to the diameter of nanowires. This dependence is in agreement with the results of molecular statics simulations.
1. Introduction
Twin boundaries in metallic nanostructures can increase strength without causing loss of ductility or electrical conductivity [Citation1]. In covalent nanostructures, they can change the crystal phase, and thereby the band gaps of semiconductor nanowires, such as SiC [Citation2]. In addition, with twin boundaries, nanowires can exhibit superelasticity or shape memory effects [Citation3,Citation4]. With so much benefit, it is desirable to introduce twin boundaries and necessary to understand their introduction [Citation5,Citation6]. In general, the introduction of twin boundaries into nanostructures is feasible either by mechanical deformation [Citation7] or during growth [Citation8,Citation9]. In either case, the nucleus of a twin boundary has to have an origin, either near a surface or interface, or in the interior of a crystalline segment. If the twin formation energy is smaller near a surface or interface, nucleation of twin boundaries may start there. Otherwise, the nucleation will more likely start at the interior. Does a surface or interface, near which the bonding nature is usually different from that in bulk, increase or decrease the twin formation energy in a nanostructure such as a nanowire?
Aiming to answer this question, we first briefly summarize two seemingly contradicting trends. In one direction, our earlier study [Citation10,Citation11] shows that for SiC nanowires a surface may increase the twin formation energy. This trend is consistent with the experimental observation that twinning of PbS nanocrystals is more difficult as the size goes down [Citation12], and the observation that twin spacing of SiC nanowires is finitely large even though the twin formation energy of SiC in bulk is essentially zero [Citation2,Citation13]. In the opposite direction, twin nucleation in Cu nanowires starts near surfaces [Citation5,Citation14], indicating that the twin formation energy there is lower.
To understand how a surface affects the twin formation energy in metals, we determine the twin formation energy of four representative metallic nanowires and examine the atomic-level effects of surfaces. The first metal is Cu, which has a face-centered cubic (FCC) structure and low twin formation energy. The second metal is Al, which also has an FCC structure but high twin formation energy. Due to the more prominent role of twin boundaries in FCC structures, relative to metals of other structures, we will consider only one body-centered cubic (BCC) metal and one hexagonal close-packed (HCP) metal; they are Ta and Mg, respectively.
2. Methods
Aiming to understand the atomic-level effects of surfaces on twin boundary formation energy, we use classical molecular statics simulations in this study. In contrast to experimental investigations, this method provides direct atomic-level insights. Further, due to the computational efficiency, this method allows the investigation of a range of nanowire diameters, up to about 18 nm in this work; density functional theory (DFT)-based ab initio calculations are more computationally demanding and will not allow for the investigation of nanowires of this large diameter.
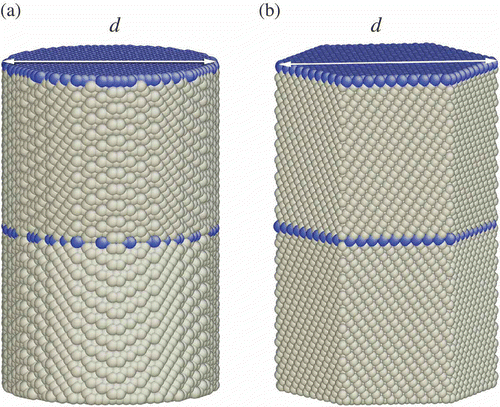
The nanowires used in the simulations are of circular cylindrical shape, as shown in . In order to show whether surface faceting changes our conclusion, we also consider a hexagonal cylinder with side faces being faceted, as shown in . This test is done only for FCC metals to confirm that surface faceting does not change the conclusion. The diameter d, measured as the farthest separation of two atoms on a cross section, ranges from 1 to 18 nm in our studies. The length of a nanowire is 32 nm in the simulation cell. By using a periodic boundary condition along the axial direction of the nanowire, it is effectively infinitely long with equally spaced twin boundaries. The dimension along the axial direction is sufficiently large, so the neighboring twin boundaries do not interact with one another. The twin boundaries are of {111} for FCC, {112} for BCC, and for HCP. Such twin boundaries are commonly seen in these metals.
Figure 1. Schematic of the simulation cell of a <111> nanowire with a circular cross section (a) and a hexagonal cross section (b). Blue (dark) spheres represent atoms at twin boundaries and gray spheres represent atoms in an FCC structure.
The key to the reliability of molecular statics simulation results is the choice of interatomic potentials. For this study, the guiding principle is that the potential must well describe (1) either the twin or the stacking fault formation energies and (2) surface formation energies. The twin and the stacking fault formation energies are generally correlated. For instance, in FCC metals, the twin formation energy can be roughly estimated as half of the stacking fault formation energy since the former consists of one layer of atoms with HCP stacking and the latter of two layers of such atoms. Based on this principle, we choose the embedded atom method (EAM) potentials from Mishin et al. [Citation15,Citation16] for Cu and Al, from Li et al. [Citation17] for Ta, and from Sun et al. [Citation18] for Mg. For Cu and Al, the potentials give the twin formation energy of 22.2 and 76 mJ/m2, which compare well with the ab initio result of 24 [Citation15] and 75 mJ/m2 [Citation16], respectively. The potentials also reproduce the surface formation energies of {100}, {110}, and {111} from the ab initio calculations. The potentials for Ta [Citation17] and Mg [Citation18] are calibrated with first-principles data including a variety of structures such as clusters, surfaces, interstitials, vacancies, and stacking faults, which make them good fits for this work.
For each nanowire, we minimize the total energy using the molecular statics method and relax the axial dimension according to the Parrinello–Rahman algorithm [Citation19]. The twin formation energy in the nanowire derives from two configuration energies. The configuration energy E
ref corresponds to a reference configuration with no twin boundary, and the configuration energy E
twin corresponds to a configuration after a twin boundary is introduced with the same number of atoms and the same cross-sectional area A. The area A is πd2/4 for circular cross section and /8 for hexagonal cross section. The twin formation energy is defined as
The factor of 2 is due to the fact that there are two twin boundaries in the simulation cell setup based on the periodic boundary conditions.
3. Results
In this section, we first focus on FCC metals to report the size dependence, the atomic-level insights, and a proposed model. Following that, we use the results of BCC and HCP metals for generalization.
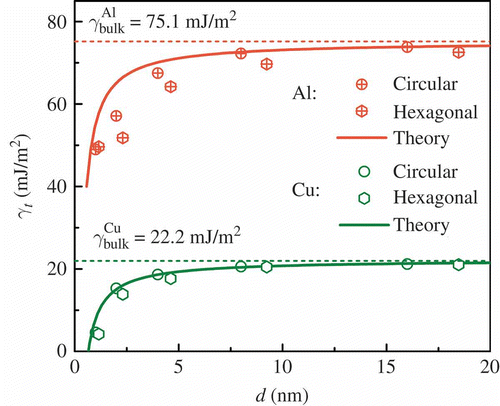
shows the twin formation energy of a nanowire as a function of nanowire diameter for both FCC metals Al and Cu. First, the twin formation energy goes down with decreasing nanowire diameter, regardless of whether the twin formation energy (in bulk) is large or small. Second, this dependence remains the same regardless of whether the cross section is circular or hexagonal. In nanowires, atoms near free surfaces are associated with imperfect bonds, and the fraction of them over the entire volume increases as the diameter decreases. With that in mind, the size dependence in suggests that the presence of surfaces decreases the twin formation energy. Considering the nondirectional nature of metallic bonding, we hypothesize that the bonding near surface is less perturbed than that in the bulk when a twin boundary forms.
Figure 2. Twin formation energy γ t as a function of nanowire diameter d for both circular and hexagonal cross sections. The dashed lines show the bulk values γbulk and the solid lines represent theory (discussed in Section 3).
To prove this hypothesis, we plot the change of atomic potential energy when a twin boundary is introduced. For this purpose, we partition the cross section of a nanowire 16 nm in diameter into a series of circular layers (for circular cylinders) and determine the twin formation in each layer γ
t
layer following Equation Equation(1)(1). The thickness of each layer is one first nearest neighbor distance, which is 0.28 nm for Al and 0.26 nm for Cu. The cross-sectional area of each layer equals the total atomic volume divided by the axial dimension of the simulation cell. It is recognized that the energy of an individual atom is not well defined in a many-body system. Nevertheless, the partition of energy into shells provides insight into how energy changes with twin boundary formation. shows the variation of the twin formation energy of each shell with the radial coordinate r, which corresponds to the average radius of each shell. Indeed, for both Al and Cu, the twin formation energy of the outermost shell is about 20 mJ/m2 lower than that of the interior shells.
Figure 3. Twin formation energy of individual shells as a function of radial coordinate r. The dashed lines represent the twin formation energies in the bulk.
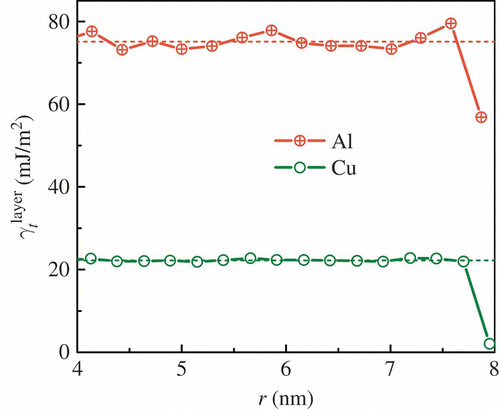
Combining results in and , we have shown that the surfaces of FCC metallic nanowires decrease the twin formation energy. Based on this atomistic result, we propose a core–shell model of nanowires. The shell region is a thin layer of atoms that are identical to atoms in the interior in chemistry but are different in coordination. The thickness of the shell region is the same as that of the outermost shell of , and its thickness Δd is 0.28 nm for Al and 0.26 nm for Cu; the core region is the collection of all of the inner shells. The twin formation energy of the shell γshell is the value shown on the right end of , and it is 56.82 mJ/m2 for Al and 2.04 mJ/m2 for Cu. The twin formation energy of the core region γcore is that of the bulk γbulk, and it is 75.1 mJ/m2 for Al and 22.2 mJ/m2 for Cu; this is also close to the average of those of the inner shells in We propose that the effective twin formation energy γ t is the area weighted average:
The corresponding areas A core and A shell are
Therefore, by omitting the second and higher order terms of Δd,
As shown by the solid lines in , this theory is in agreement with the dependence of atomistic results of twin formation energy as a function of diameter. Such agreement indicates that the proposed core–shell model is a valid theory for the twin formation energy of nanowires or surface effects on the twin formation energy of metals in more generic sense.
Due to the generic nature of metallic bonds, we expect that the dependence in and is generic for all metals. To confirm this expectation, we next examine the molecular statics simulation results of Ta and Mg.
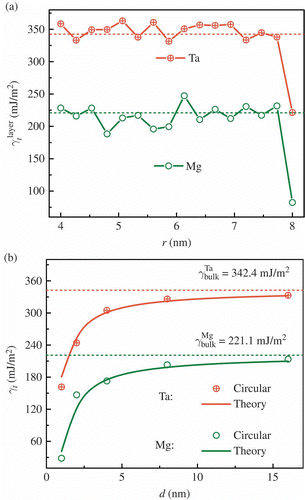
shows that indeed the dependence of twin formation energy of each shell is similar to that for FCC metals (). Here, the cross sections are also circular like those of FCC metallic nanowires, and the trend is the same as in The change of twin formation energy from bulk to surface is on the same order of magnitude as the formation energy itself; it is ∼100 mJ/m2 for BCC and HCP and ∼10 mJ/m2 for FCC. Using the expression of Equation Equation(4)(4), our theory agrees with the atomistic simulation results for both Ta and Mg, as shown in . These results indicate that indeed the size dependence of twin formation energy, the atomic insights on surface effects, and the theory apply to all the three categories of metals: FCC, BCC, and HCP.
Figure 4. (a) Twin formation energy of individual shells as a function of radial coordinate r and (b) comparison of theory and simulation results. The dashed lines represent the twin formation energies in the bulk.
4. Conclusions
In summary, we use molecular statics simulations to show that the twin formation energy of a metallic nanowire decreases as the diameter goes down. That is, surfaces decrease the twin formation energy. Further, we have proposed a core–shell model for nanowires, with the core having twin formation energy identical to its bulk counterpart and the shell having smaller twin formation energy. This model is in agreement with the molecular statics simulation results and the agreement indirectly shows the validity of the core-shell model.
Acknowledgments
The authors acknowledge the financial support of Defense Threat Reduction Agency (HDTRA1-09-1-0027). H.H. also acknowledges the financial support of National Science Foundation (DMR-0906349 and CMMI-0856426).
Notes
Present address of Yongfeng Zhang: Fuels Modeling and Simulation, Idaho National Laboratory, Idaho Falls, ID 83415, USA
References
- Lu , L. , Shen , Y.F. , Chen , X.H. , Qian , L. and Lu , K. 2004 . Ultrahigh strength and high electrical conductivity in copper . Science , 304 : 422 – 426 .
- Shim , H.W. , Zhang , Y.F. and Huang , H.C. 2008 . Twin formation during SiC nanowire synthesis . J. Appl. Phys , 104 : 063511
- Park , H.S. , Gall , K. and Zimmerman , J.A. 2005 . Shape memory and pseudoelasticity in metal nanowires . Phys. Rev. Lett , 95 : 255504
- Liang , W. and Zhou , M. 2005 . Shape memory effect in Cu nanowire . Nano Lett , 5 : 2039 – 2043 .
- Zhang , Y.F. and Huang , H.C. 2010 . Controllable introduction of twin boundaries into nanowires . J. Appl. Phys , 108 : 103507
- Huang , H.C. 2011 . Twin boundaries in nanowires – Controllable introduction . JOM , 63 : 58 – 61 .
- Park , H.S. , Gall , K. and Zimmerman , J.A. 2006 . Deformation of FCC nanowires by twinning and slip . J. Mech. Phys. Solids , 54 : 1862 – 1881 .
- Zhong , S. , Koch , T. , Wang , M. , Scherer , T. , Walheim , S. , Hahn , H. and Schimmel , T. 2009 . Nanoscale twinned copper nanowire formation by direct electrodeposition . Small , 5 : 2265 – 2270 .
- Tian , M. , Wang , J. , Kurtz , J. , Mallouk , T.E. and Chan , M.H.W. 2003 . Electrochemical growth of single-crystal metal nanowires via a two-dimensional nucleation and growth mechanism . Nano Lett , 3 : 919 – 923 .
- Zhang , Y.F. , Shim , H.W. and Huang , H.C. 2008 . Size dependence of twin formation energy in cubic SiC at the nanoscale . Appl. Phys. Lett , 92 : 261908
- Rupich , S.M. , Shevchenko , E.V. , Bodnarchuk , M.I. , Lee , B. and Talapin , D.V. 2010 . Size-dependent multiple twinning in nanocrystal superlattices . J. Am. Chem. Soc , 132 : 289
- Wang , D.H. , Xu , D. , Wang , Q. , Hao , Y.J. , Jin , G.Q. , Guo , X.Y. and Tu , K.N. 2008 . Periodically twinned SiC nanowires . Nanotechnology , 19 : 215602
- Zhang , Y.F. and Huang , H.C. 2009 . Twin Cu nanowires using energetic beams . Appl. Phys. Letts , 95 : 111914
- Mishin , Y. , Mehl , M.J. , Papaconstantopoulos , D.A. , Voter , A.F. and Kress , J.D. 2001 . Structural stability and lattice defects in copper: Ab initio, tight-binding, and embedded-atom calculations . Phys. Rev. B , 63 : 224106
- Mishin , Y. , Farkas , D. , Mehl , M.J. and Papaconstantopoulos , D.A. 1999 . Interatomic potentials for monoatomic metals from experimental data and ab initio calculations . Phys. Rev. B , 59 : 3393 – 3407 .
- Li , Y.H. , Siegel , D.J. , Adams , J.B. and Liu , X.Y. 2003 . Embedded-atom-method tantalum potential developed by the force-matching method . Phys. Rev. B , 67 : 125101
- Sun , D.Y. , Mendelev , M.I. , Becker , C.A. , Kudin , K. , Haxhimali , T. , Asta , M. , Hoyt , J.J. , Karma , A. and Srolovitz , D.J. 2006 . Crystal-melt interfacial free energies in hcp metals: A molecular dynamics study of Mg . Phys. Rev. B , 73 : 024116
- Parrinello , M. and Rahman , A. 1980 . Crystal structure and pair potentials: A molecular-dynamics study . Phys. Rev. Lett , 45 : 1196 – 1199 .
- Shim , H.W. and Huang , H.C. 2007 . Three-stage transition during silicon carbide nanowire growth . Appl. Phys. Letts , 90 : 083106