
Abstract
Herein, graphene oxide (GO)-encapsulated silica (SiO2) hybrids (GO@SiO2) were prepared via electrostatic self-assembly of the 3-aminopropyltriethoxysilane (APS)-modified SiO2 and GO. The as-prepared GO@SiO2 was introduced into polydimethylsiloxane (PDMS) elastomer to simultaneously increase the dielectric constant (k) and mechanical properties of PDMS. Then, the in situ thermal reduction of GO@SiO2/PDMS composites was conducted at 180°C for 2 h to increase the interfacial polarizability of GO@SiO2. As a result, the values of k at 1000 Hz are largely improved from 3.2 for PDMS to 13.3 for the reduced GO@SiO2 (RGO@SiO2)/PDMS elastomer. Meanwhile, the dielectric loss of the composites remains low (<0.2 at 1000 Hz). More importantly, the actuated strain at low electric field (5 kV/mm) obviously increases from 0.3% for pure PDMS to 2.59% for the composites with 60 phr of RGO@SiO2, an eightfold increase in the actuated strain. In addition, both the tensile strength and elastic modulus are obviously improved by adding 60 phr of RGO@SiO2, indicating a good reinforcing effect of RGO@SiO2 on PDMS. Our goal is to develop a simple and effective way to improve the actuated performance and mechanical strength of the PDMS dielectric elastomer for its wider application.
1. Introduction
Dielectric elastomers (DEs), as an attractive branch of electro-active polymers (EAPs), can give rise to surprisingly large deformation when stimulated by an electric field, and work efficiently over a broad frequency range. Thus, they have received much attention in the past two decades [Citation1,Citation2]. A dielectric elastomer actuator (DEA) can shrink in the thickness direction and expand in the plane direction by applying an electric field across the film thickness [Citation3]. By virtue of the excellent properties such as large strain, fast response, lightweight, reliability, high energy density, and high electromechanical coupling efficiency, DEAs find applications in artificial muscles, sensors, micro air vehicles, flat-panel speakers, micro-robotics, and responsive prosthetics [Citation4–Citation8]. A key limitation for the practical application of DEAs is the requirement of high electric field (>100 kV/mm) to drive them [Citation9–Citation11], which could be harmful to humans and can damage equipment, particularly in biological and medical fields [Citation12]. Therefore, getting a large actuated strain at a low electric field is the biggest challenge for DEAs.
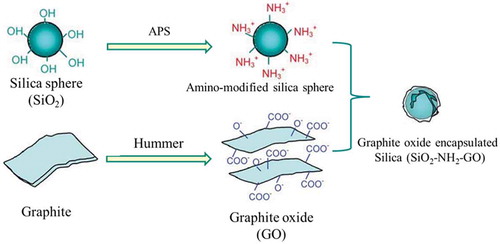
Figure 1. The schematic illustration of the fabrication of SiO2–APS–GO hybrids.
Based on the assumption of constant modulus and free boundary conditions for a DE film, the thickness strain Sz can be approximated by [Citation13].
where P is Maxwell’s stress on the material, εr is the relative permittivity, ε0 is the permittivity of free space, E is the applied electric field, and Y is the elastic modulus of a DE film. According to the law of volume constancy [Citation14], the planar strain Sp can be approximated by:
According to Equations (1) and (2), to obtain a DE with high actuated strain at a low electric field, a high electromechanical sensitivity (β) is required, which is defined as the ratio of the dielectric constant (k) to the elastic modulus (Y)() [Citation1]. However, most DEs such as silicone elastomer and acrylic rubber have quite low values of k, which are 2.8 and 4.8, respectively [Citation15]. Therefore, a key issue is to increase the k of DEs, while retaining other excellent properties such as low dielectric loss, low modulus (Y), and good flexibility. One common method to obtain a DE with high k is to introduce high-k ceramics such as TiO2 into the elastomer matrix [Citation10,Citation15–Citation17]. Usually, a high content (up to 50 vol%) of ceramics is required to obtain high k, leading to low flexibility and poor processability, and thus largely limiting the wide application of DEs [Citation18]. Another method is to add conductive fillers such as carbon nanotubes (CNTs) into the matrix, or blending with chemical modification of silicone with organic dipoles is a promising approach to increase ε′ (dielectric constant) [Citation19]. Because of the curl and entanglement of CNTs with fibrous nanostructure, even quite a low content of CNTs can result in the formation of CNT network, thus leading to the large increase in k [Citation19,Citation20]. However, in this case, a high dielectric loss was also obtained owing to the high direct current (DC) conductance caused by the direct connection of CNTs [Citation21].
Compared with CNTs, graphene sheets are usually synthesized from natural graphite, which is abundant, cheap, and easily available. In addition, graphene sheets have a larger aspect ratio, and thus have been considered as good fillers to improve the dielectric constant of elastomers, as has been reported in previous studies [Citation19,Citation20,Citation22,Citation23]. For instance, Romasanta et al. [Citation24] reported that the value of k of polydimethylsiloxane (PDMS) increased from 2.7 to 23 at low frequency (10 Hz) by addition of 2.0 wt.% of thermally expanded graphene sheets. Obviously, the increase in the value of k of these polymers by addition of graphene oxide (GO) is far less than expected. The main reasons are the poor dispersion of graphene in matrix, the restacking of graphene sheets during preparation caused by π–π stacking and hydrophobic interactions, and/or weak interfacial adhesion between graphene sheets and the polymer matrix [Citation25–Citation27]. A good dispersion of GO in the polymer matrix and a strong interfacial adhesion between GO and the matrix can be obtained via the organic functionalization of GO [Citation28,Citation29]. A disadvantage of this approach is that it is complicated and time-consuming. In addition, the coating of GO on silica (SiO2), metal oxide particles, or carbon nanosphere is an efficient method to prevent GO from self-agglomerating, as reported in previous studies [Citation25,Citation30–Citation32].
In addition, a disadvantage of GO is that the graphite structure is disrupted by oxidation, resulting in a decrease in the dielectric constant [Citation33,Citation34]. Thus, reduction of GO is required to obtain a DE with both high k and low dielectric loss. In most cases, GO was reduced via chemical reduction using hydrazine hydrate [Citation35], Vitamin C [Citation36], etc., leading to a high reduction degree, and thus a good electric conductivity of the composites. Thus, chemical reduction of GO can result in a high dielectric loss of the composites because of the large DC conductance [Citation10]. In recent years, some studies reported that in situ thermal reduction of graphene oxide nanosheets (GONSs) at a moderate temperature (180–200°C) within a polymer is a simple and effective technique for the moderate reduction of graphitic structure [Citation26,Citation30].
PDMS, as one of the most important DEs, has a rather low modulus, a good thermal stability over a wide temperature range, a fast speed of response, high efficiency, and excellent biocompatibility for artificial muscle. However, its dielectric constant and mechanical strength are too low for practical application. Racles et al. [Citation37] developed where cyanopropyl-groups were distributed along the backbone of PDMS chains. Good overall results were obtained for blends of the cyanopropyl-functional PDMS and PDMS.
Herein, GO-encapsulated silica (SiO2) (GO@SiO2) hybrids were prepared via electrostatic self-assembly in which the 3-aminopropyltriethoxysilane (APS) acts as a ‘glue’ molecule to connect GO and SiO2.Therefore, in this study, GO@SiO2 is introduced in the PDMS matrix to simultaneously increase the dielectric constant and mechanical properties of PDMS. Then, the reduced GO@SiO2 (RGO@SiO2)/PDMS composites were obtained by in situ thermal reduction of GO@SiO2/PDMS composites to increase the interfacial polarizability, and thus increase the k of the composites. Our goal is to develop a simple and effective way to improve the actuated performance and mechanical strength of PDMS DE for its wider application.
2. Experimental
2.1. Materials
Natural graphite flakes with a mean size of 18 μm were supplied by Huadong Graphite Factory (China). Submicron-sized silica (average diameter: 3 00 nm) was supplied by Guangdong Shengyi Technology Co., LTD (China). APS was supplied by Momentive Performance Materials (Shanghai). A commercial grade of siloxane (PDMS) (DC3481 184) and the curing agent (81-F) were purchased from Dow Corning Co., Ltd. (America). The viscosity and density of PDMS material used are 22.1 Pa s and 1.213 g/cm3, respectively. Potassium permanganate (KMnO4, 99.5%), sulfuric acid (H2SO4, 98%), sodium nitrate (NaNO3, 99.0%), hydrogen peroxide (H2O2, 30%), hydrochloric acid (HCl, 37%), and methanol (CH3OH, 99.5%), which were all used for the preparation of graphite oxide, were supplied by Beijing Chemical Reagents Co., Ltd. (China). Tetrahydrofuran (THF) was used as a solvent to dissolve PDMS and was purchased from Beijing Chemical Reagents Co., Ltd. (Beijing, China).
2.2. Preparation of graphene oxide-encapsulated silica (GO@SiO2) hybrids
The graphene oxide-encapsulated silica (SiO2–NH2–GO) hybrids were prepared in two steps: the modification of silica particles with APS coupling agent and the surface assembly of GONSs with the modified silica microspheres.
Graphite oxide was prepared from natural graphite by Hummers method. The as-prepared graphite oxide was then dispersed in an aqueous solution (1 mg/mL) and exfoliated via sonication for 1 h using a GA 98-IIIultrasonic cell disruptor (Shangjia Biological Technology, Shanghai, China) with a power of 1000 W to form a uniform colloidal suspension of GO. The nonexfoliated GO sheets were removed by centrifugation at 8000 rpm for 10 min.
The surface modification of SiO2 with APS coupling agent was carried out in a mixture of ethyl alcohol and water. In a typical process, SiO2 powder (1 g) was first dispersed well in 400 mL of ethyl alcohol by sonication for 1 h, followed by the addition of 40 mL water. Subsequently, 1 mL of APS coupling agent dissolved in a small amount of ethyl alcohol was added to the above solution, and the mixture was stirred at 60°C for 8 h until the grafting reaction was realized. The obtained particles (SiO2−NH2) were centrifuged from the mixture, washed with ethyl alcohol and deionized water thrice.
The GO@SiO2 hybrids were simply fabricated by mixing the neutral aqueous suspension of SiO2−NH2 and GO solution. A total volume of 400 mL of SiO2−NH2 suspension (2.5 mg/mL) was added to 200 mL of aqueous GO solution (0.1 mg/mL) under mild stirring for 4 h. When stirring stopped, the GO precipitated with SiO2−NH2 at the bottom of the beaker, leaving a transparent aqueous solution. The sediment solid (GO@SiO2) was collected and washed with deionized water several times to remove the unbound GO and then freeze-dried under vacuum.
2.3. Preparation of PDMS-based composites with solution compounding
The preparation of GO@SiO2/PDMS composites is schematically illustrated in . First, a certain weight of GO@SiO2 hybrids was dispersed in THF by ultrasonic treatment and mild magnetic stirring. Meanwhile, 3 g of PDMS was dispersed in 20 ml THF by mild magnetic stirring for 1 h to form a stable solution. Then, the uniform GO@SiO2/THF suspension was mixed with PDMS/THF solution under stirring for 2 h, followed by the addition of a curing agent. After mechanical stirring for 10 min, the GO@SiO2/PDMS/THF solution was poured into a mold to remove the solvent and cure at room temperature in a fume cupboard with a thickness of 0.5 mm. The GO@SiO2/PDMS film was obtained after the removal of solvent, and the RGO@SiO2/PDMS composites were obtained by in situ thermal reduction of GO@SiO2/PDMS composites at 180°C for 2 h at a pressure of 4 MPa. The composites with different contents of GO@SiO2 (30 wt%, 60 wt%) were achieved by changing the content of GO@SiO2. For the purpose of comparison, a pure PDMS film was also prepared by the same method. Here, it should be noted that the GO content is only 0.6 and 1.2 wt% for the composites with 30 and 60 wt% of GO@SiO2, respectively.
2.4. Characterization
The morphology and microstructure of SiO2 and GO@SiO2 were investigated by scanning electron microscopy (SEM, S-4800, Hitachi Co. Japan), transmission electron microscopy (TEM, Tecnai G2 20, FEI Co. Hong Kong), and high-resolution transmission electron microscopy (HRTEM, JEM-3010, Hitachi, Japan). Samples for TEM and HRTEM observation were prepared by suspending GO@SiO2 in water at a concentration of 0.005 mg/mL under ultrasonication for 30 min, dropping the suspension on micro grids, and then drying in a vacuum oven.
Zeta potential measurements were performed using a Zetasizer 90 (Malvern Instruments, Malvern, England). The aqueous suspensions of silica, SiO2––NH2, and GO were diluted to 0.05 mg/mL and sonicated for 30 min to form a homogeneous solution before conducting the measurements.
Raman spectra of GO and GO@SiO2 dried powder samples were obtained by using a multichannel confocal micro spectrometer (Renishaw in Via, England) with a laser wavelength of 633 nm.
The chemical compositions of GO, SiO2–NH2, and GO@SiO2 were obtained using an ESCALAB 250 X-ray photoelectron spectrometer (XPS) purchased from Thermo Fisher Scientific Company (American).
The volume resistivity (ρv) of GO@SiO2/PDMS composites was measured using a high-resistance meter (EST121, Beijing Huajinghui Technology Co., Ltd, China). ρv is calculated as follows: ρv = (RV·S)/L, where RV is the volume resistance of the specimen, S is the contact area between the composite and the electrode, which is 2122.64 mm2 for this instrument, and L is the average thickness. The conductivity is the reciprocal of volume resistivity.
Dielectric properties were measured using an HP4294A impedance analyzer (Agilent, USA) in the frequency range of 102–106 Hz at room temperature. The volume resistivity of the composites was tested using a high-resistance meter (EST 121, Beijing HuaJingHui Scientific and Technical Co., Ltd., China) at room temperature. Mechanical properties were obtained using a tensile apparatus (CMT4104, Shenzhen SANS Testing Machine Co., Ltd., China); 5–10 samples were tested in the tensile test for each material at a strain rate of 50 mm/min, and the Y of pure PDMS and GO@SiO2/PDMS composites was obtained by calculating the slopes of the stress–strain curves at the strain of 5%, which was obtained using a tensile apparatus.
The actuated strain was measured using a circular strain test and the diameter of the active area is 1 cm. The samples for actuated strain tests had a thickness of about 0.5 mm and a diameter of 5 cm, and compliant electrodes with a diameter of 11 mm were applied to each side of the DE. Before the test, two main surfaces of the films were sprayed with graphite suspension using an airbrush to fabricate compliant electrodes. The film was first fixed on two circular frames without a pre-strain, and electric field was applied on both sides of the film. The electric field needed was equipped with a high-voltage DC generator (DTZG-60, Wuhan Dotek Electric Co., Ltd.). The strain was calculated as the change of the pixel of the electrode area divided by the original pixel. A video camera was fixed at the same focal length to capture the actuator plain before and after voltage exertion. Three actuators were studied three times for each composition, and the average values are reported. The thickness of pure PDMS and GO@SiO2/PDMS composites was 0.5 mm. The electric breakdown filed was obtained by testing the actuated stains of composites until electric breakdown occurred.
3. Results and discussion
3.1. Self-assembly of GO and APS-modified silica
The overall synthetic procedure of GO coated on silica is demonstrated in . GONSs are negatively charged in aqueous solution because of the ionization of the carboxylic acid groups and phenolic hydroxy groups on their surface. When GO is mixed with other positively charged particles, the assembly can be realized by the electrostatic force. However, a silica (SiO2) particle in an aqueous solution is also negatively charged because of the ionization of H2SiO3 on its surface. So the surface modification of SiO2 is necessary realize the assembly of GO on silica. APS molecules with amino groups can ionize to form a positively charged surface and APS can also undergo condensation reaction with materials that have hydroxyl groups.
Here, SiO2 was first modified by APS to form a positively charged surface. Therefore, it is easy to assume that electrostatic driving force can take place between SiO2−NH2 and GO. To clarify this process, zeta potential was tested to examine the surface charges of GO, SiO2, and SiO2–NH2 aqueous suspensions, as shown in .
Table 1. The zeta value of SiO2, SiO2–NH2, and GO.
With a pH value of 7.0, GO had a highly negative surface charge with zeta value of −42.5 mV. Meanwhile, the surface charges of the SiO2 switched from negative (zeta = − 26.8 mV) to positive (zeta = 39.1 mV) after the modification, suggesting the successful graft of APS on SiO2. When the oppositely charged GONSs and SiO2−NH2 particles collided through simple solution mixing, the electrostatic assembly might have been triggered, forming the core−shell structure of GO@SiO2 hybrid afterward.
The morphology and microstructure of the GO@SiO2 hybrids and the pristine SiO2 were observed by SEM, TEM, and HRTEM, as shown in . Compared with the SiO2 particles (specific area ~ 120 m2/g) that have a microsphere morphology and a smooth surface (), GO@SiO2 hybrids show crinkled and rough textures that are associated with the presence of flexible GO sheets. In most cases, the edges of individual as well as overlapping GO layers can be observed, particularly at the interface between aggregated particles, where the graphene layers appeared to link the neighboring spheres together ()). The typical TEM and HRTEM images of GO@SiO2 hybrid () also confirm that the flexible GO sheets are successfully wrapped around the SiO2−NH2 microspheres, with the GO shell thicknesses of about 3 nm. The core–shell structure of the GO@SiO2 hybrids can prevent the restacking of GO and the self-aggregation of SiO2. Considering the large specific surface area gap between GO and silica (the specific surface area of an individual GO sheet is 2600 m2/g), it is easy to understand why only 2 wt% GO is needed to assemble on the SiO2 when the equilibrium coverage is reached.
Figure 2. The SEM images of (a) pristine SiO2 (b) GO@SiO2 hybrid; (c)TEM; and (d) HRTEM of GO@SiO2 hybrid.

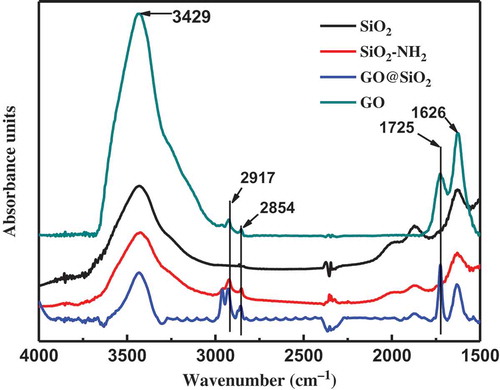
The encapsulation of GO on SiO2 can be confirmed by XPS, as shown in . The nitrogen content of 3.39% in SiO2–NH2 suggests a successful introduction of amino groups on SiO2. The increase in carbon content from 25.12% of SiO2–NH2 to 33.79% of GO@SiO2 also indicates the immobilization of GO on SiO2. Fourier transform infrared spectroscopy (FT-IR) was also used to confirm the covalently attached APS coupling agent and the GO wrapping on the SiO2, as shown in . The main peaks of SiO2 spectra curve at 1621 and 3430 cm−1 are designed as stretching vibration of Si−O−Si, blending vibration of O–H, and stretching vibration of −OH, respectively. Several minor bands at around 2800–3000 cm−1 were detected in the spectra of SiO2−NH2, which were attributed to the C−H stretching vibration of the hydrocarbon chains of the grafting APS. A weak band at 1725 cm−1 appeared in the SiO2@GO spectra curve, which is the characteristic of the C=O stretching vibration band of GO. As GO has a large amount of –OH groups, the peak at 3429 cm−1 of SiO2−GO is higher than that of SiO2−NH2.
Table 2. The element analysis of SiO2, SiO2–NH2, GO@SiO2, and GO.
Figure 3. The FT-IR spectra curves of pristine SiO2, SiO2–NH2, GO@SiO2, and GO.
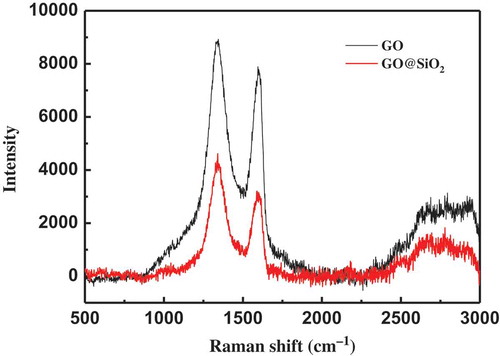
Raman spectroscopy has been used as a powerful tool for the characterization of graphene and its derivatives. Therefore, it was also employed to further confirm the GO shells on SiO2 hybrids. As shown in the , there are two peaks at about 1337 and 1595 cm−1 in the Raman spectroscopy of graphite oxide, corresponding to the D and G bands. The existence of the two bands was also observed in the spectroscopy of GO@SiO2, which demonstrates the successful assembly of GO on SiO2. Meanwhile, it was reported that both G and 2D bands can be used to monitor the number of layers [Citation38–Citation40]. The G peak of the single-layer graphene shifts to lower wavenumbers, and the 2D band decreases in intensity and lower frequency peaks after stacking more GO layers. In this study, the peak positions of the G bands of the GO and GO@SiO2 were centered at about 1595 and 1602 cm−1, respectively. This indicates that GO@SiO2 contains less layers compared to original GO. However, the 2D peaks of the two materials are not obvious in this study and cannot demonstrate the change in number of GO layers.
Figure 4. The Raman spectrum of GO and GO@SiO2.
3.2. In situ thermal reduction of GO@SiO2/PDMS
Many researchers have demonstrated that the increase in k is far less than that expected using GO as the dielectric filler because of the severe disruption of the graphite structure of GO [Citation41]. Therefore, the reduction of GO@SiO2/PDMS composites is required to increase the interfacial polarization ability of GO@SiO2. So, the in situ thermal reduction of GO@SiO2/PDMS composites was conducted at 180°C for 2 h because of the following advantages. First, the directional distribution of GO@SiO2 around PDMS latex particles and network structure can be retained during in situ thermal reduction at 180°C. Second, the method is simple and efficient for the partial reduction of GO shells of GO@SiO2
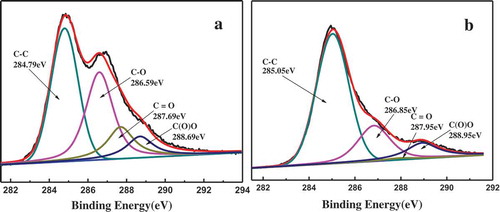
Thermal reduction of pure GO was conducted under the same conditions to evaluate the degree of reduction of GO shells. The degree of reduction was characterized using XPS and TGA, and the results are shown in and . The oxygen content obviously decreases from 28.88% for GO to 21.38% for RGO, as shown in , and the carbon oxygen ratio increases from 2.46% for GO to 3.73% for RGO, suggesting partial reduction of GO. shows the XPS C1s core-level spectra of pure GO. We can observe four characteristic peaks corresponding to carbon atoms in different functional groups: C–C (unoxidized graphite carbon skeleton) at 284.79 eV, C–O (hydroxyl and epoxy groups) at 286.59 eV, C=O (carbonyl group) at 287.69 eV and COOH (carboxyl and ester groups) at 288.69 eV. The presence of the same peak in ) suggests that there are still some oxygen-containing groups in RGO. But the peaks corresponding to the oxygen-containing groups in the spectrum of RGO are weakened, especially that of C=O (carbonyl), indicating that GOs have been partially reduced during thermal reduction at 180°C.
Table 3. The element analysis of GO and in situ thermal reduction of graphene.
Figure 5. The C1s X-ray photoelectron spectroscopy of GO and RGO.
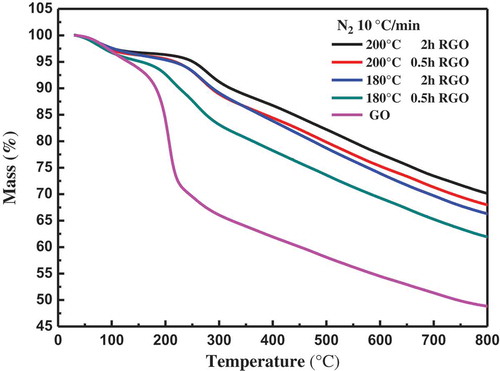
The mass loss of pure GO, RGO was measured at a heating rate of 10°C min−1 in nitrogen atmosphere, as shown in . The mass loss below 100°C of GO is due to the removal of absorbed water. The maximum rate of mass loss, corresponding to the loss of most of the oxygen-containing groups, occurs at 150–200°C for both GO and RGO. The mass loss of RGO continues with further increase in temperature because of the remaining oxygen-containing groups caused by the partial reduction of GO during thermal reduction at 180°C
Figure 6. The TGA curves of GO and RGO.
3.3. Microstructure of RGO@SiO2/PDMS composites
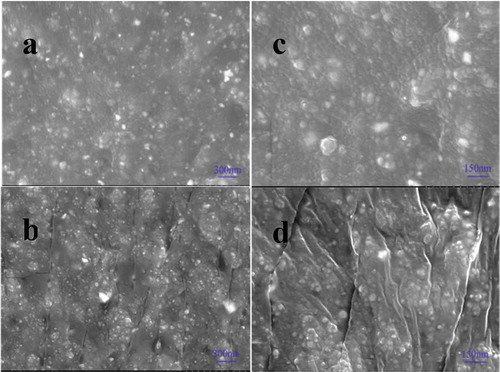
The fracture surfaces of RGO@SiO2/PDMS composites with different filler content were observed by SEM, and the results are shown in the . A uniform distribution of RGO@SiO2 in PDMS matrix is observed in both composites filled with 30 phr and 60 phr of RGO@SiO2, as shown in (a) and (b), respectively. To clearly observe the interspace between the fillers, the SEM images of the composites with larger magnification were obtained, as shown in (c) and (d), respectively. Obviously, although some agglomeration of RGO@SiO2 in the matrix is observed with the increase in the content of RGO@SiO2 to 60 phr, the interspace between the filler particles largely decreases. As a result, a strong filler network is observed in the composite with 60 phr of RGO@SiO2, facilitating the increase in conductivity and dielectric constant.
Figure 7. The SEM figures of fracture surfaces of RGO@SiO2/PDMS composites with different filler content (a, b) 30 phr (c, d) 60 phr.
3.4. Electromechanical properties of GO@SiO2/PDMS composites
The stress–strain curves of pure PDMS and the RGO@SiO2/PDMS composites with 30 phr and 60 phr of RGO@SiO2 are shown in and the corresponding mechanical properties are summarized in . We can observe that both the tensile strength and elastic modulus of PDMS obviously increase with the increase in the content of RGO@SiO2, indicating the good reinforcing effect of RGO@SiO2 on the PDMS matrix. For example, the tensile strength increases from 1.0 MPa for pure PDMS to 1.5 MPa for the composite with 60 phr of RGO@SiO2, and the corresponding elastic modulus increases from 0.27 to 0.9 MPa. Although the elongation at break decreases with increasing the content of RGO@SiO2, the elongation at break of the composites is larger than 200%, facilitating the practical application of DE. (The standard deviation is ±0.3.)
Table 4. The conductivity of RGO@SiO2/PDMS composites.
Table 5. The electromechanical properties of RGO@SiO2/PDMS composites.
Figure 8. Strain–stress curve of PDMS and PDMS with 30 and 60 phr of RGO@SiO2.
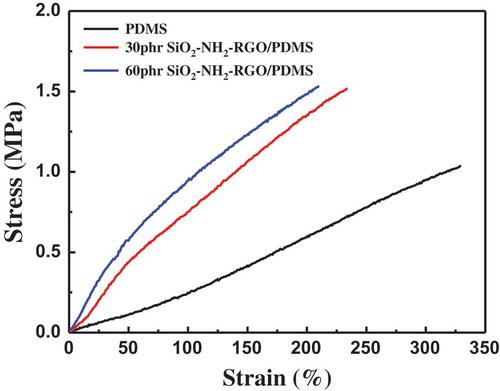
Figure 9. Frequency response of dielectric constant (a) and dielectric loss (b).
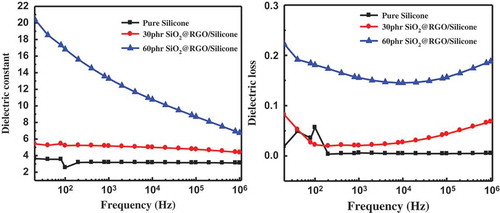
The dielectric properties of pure PDMS and RGO@SiO2/PDMS composites as a function of frequency at room temperature are shown in . As expected, the k of all the RGO@SiO2/PDMS composites is largely decreased with an increase in frequency, suggesting a strong frequency dependence of k of all the composites. The strong dependence of k is ascribed to interfacial polarization effect (also named as the Maxwell–Wagner–Sillars (MWS) effect) of RGO@SiO2 on PDMS molecules caused by the accumulation of many free charges at the internal interfaces between RGO@SiO2 and PDMS [Citation42]. Is has enough time to accumulate charge at the interfaces between RGO@SiO2 and the rubber matrix at low frequency, resulting in the high k of the composites. However, the interfacial polarization cannot catch up with the change in electrical field frequency at high frequency, leading to the large decrease in k at high frequency [Citation43].
Interestingly, a significant increase in k is observed with the increase in the content of RGO@SiO2 at the same frequency (see (a)) in the whole frequency range. The dielectric loss slightly increases with the increase of filler content, especially in the high frequency range of 103–106 Hz. For example, the k of the RGO@SiO2/silicone rubber (SR) composite with 60 wt% of RGO@SiO2 at 100 and 1000 Hz is 16.8 and 13, respectively, which is much higher than that of pure SR (3.6). The increase in k is ascribed to the large increase in the interfacial polarizability of RGO@SiO2 caused by the in situ thermal reduction of GO. The dielectric loss of RGO@SiO2/SR composite (see ) at 103 Hz with 30 wt% and 60 wt% GO@SiO2 remains low (<0.2), much lower than that of many other conductive filler/polymer dielectric composites. The relatively low loss is mainly attributed to the low DC conductance of RGO@SiO2 caused by the partial reduction of GO shells by in situ thermal reduction at 180°C, as demonstrated by the relatively low electrical conductivity of all the composites, as shown in .
The elastic moduli of pure PDMS and RGO@SiO2/PDMS composites are summarized in . We can observe that the elastic modulus increases from 0.27 MPa for pure PDMS to 0.6 and 0.9 MPa for the composites with 30 and 60 phr of RGO@SiO2, respectively. There is a large increase in k and a mild increase in the elastic modulus of PDMS composites by addition of RGO@SiO2.
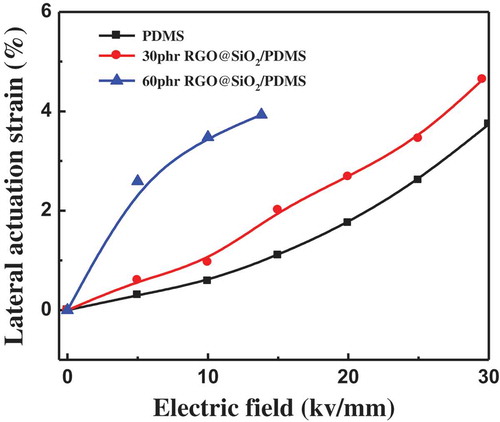
shows the actuated strains of pure PDMS and RGO@SiO2/PDMS composites as a function of the electric field. The actuated strain of all the samples obviously increases with the increase in electric field, because it has a quadratic relationship with the applied electric field. Although the maximum actuated strain of the composites is lower than that of pure PDMS and the breakdown strength of PDMS obviously decreases with the increase in RGO@SiO2 content, the actuated strain of the composites at low electric field is much higher than that of pure PDMS, as summarized in . For example, the actuated strain at 10 kV/mm obviously increases from 0.58% for pure PDMS to 3.48% for the composites with 60 phr of RGO@SiO2. The k of the RGO@SiO2/SR composite with 60 wt% of RGO@SiO2 at 100 and 1000 Hz is 16.8 and 13, respectively, which is much higher than that of pure SR (3.6). According to the equation Sz = –εrε0E2/Y, the Y of composites with 60 wt% of RGO@SiO2 is close to the Y of composites with 30 wt% of RGO@SiO2. The β of 60 wt% RGO@SiO2 has been increased due to the significantly improved dielectric constant of composites. So the actuator strain of 60 wt% RGO@SiO2 composites has been enhanced, as the electric field strength increased, the composites with 60 wt% of RGO@SiO2 possess high dielectric loss. So the electrical energy conversion to heat energy leads to the actuator strain non-linear increase. More interestingly, the actuated strain at 5 kV/mm obviously increases from 0.3% for pure PDMS to 2.59% for the composites with 60 phr of RGO@SiO2, an eightfold increase in the actuated strain, much higher than that of other previously reported DEs [Citation13,Citation43–Citation47], as summarized in . The improvement in the actuated strain at low electric fields is in favor of the application of DE in the biological and medical fields (such as synthesis of artificial skin), tactile displays, and braille displays, etc. [Citation47].
Table 6. Comparison of actuated performances of advanced DE composites.
Figure 10. Lateral actuation strain of PDMS and RGO@SiO2/PDMS composites as a function of electric field.
3.5. Conclusions
We developed a simple and efficient method to simultaneously improve the actuated performance and mechanical strength of a silicone elastomer. GO@SiO2 were first prepared via electrostatic self-assembly of APS-modified SiO2 and GO, and then introduced into PDMS matrix to prepare GO@SiO2/PDMS dielectric composites. RGO@SiO2/PDMS composites are then prepared by in situ thermal reduction of GO@SiO2/PDMS composites at 180°C for 2 h to increase the interfacial polarizability. The results show that the k of PDMS largely increases by adding 60 phr of RGO@SiO2, whereas the dielectric loss of the composites remains low (<0.2 at 1000 Hz). The actuated strain at low electric field (5 kV/mm) obviously increases from 0.3% for pure PDMS to 2.59% for the composites with 60 phr of RGO@SiO2, an eightfold increase in the actuated strain. On the other hand, both the tensile strength and elastic modulus are obviously improved by adding 60 phr of RGO@SiO2 due to the good reinforcing effect of RGO@SiO2 on PDMS.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- A. O’Halloran, F. O’Malley, and P. McHugh, A review on dielectric elastomer actuators, technology, applications, and challenges, J. Appl. Phys. 104 (7) (2008), pp. 071101. doi:10.1063/1.2981642
- S. Bauer, S. Bauer-Gogonea, I. Graz, M. Kaltenbrunner, C. Keplinger, and R. Schwödiauer, 25th Anniversary Article: A Soft Future: From Robots and Sensor Skin to Energy Harvesters, Adv. Mater. 26 (1) (2014), pp. 149–162. doi:10.1002/adma.201303349
- K. Wongtimnoi, B. Guiffard, A. Bogner-Van De Moortèle, L. Seveyrat, C. Gauthier, and J.-Y. Cavaillé, Improvement of electrostrictive properties of a polyether-based polyurethane elastomer filled with conductive carbon black, Compos. Sci. Technol. 71 (6) (2011), pp. 885–892. doi:10.1016/j.compscitech.2011.02.003
- L.Z. Chen, C.H. Liu, C.H. Hu, and S.S. Fan, Electrothermal actuation based on carbon nanotube network in silicone elastomer, Appl. Phys. Lett. 92 (26) (2008), pp. 263104. doi:10.1063/1.2955513
- A. Tiwari, A.P. Mishra, S.R. Dhakate, R. Khan, and S.K. Shukla, Synthesis of electrically active biopolymer–SiO2 nanocomposite aerogel, Mater. Lett. 61 (23–24) (2007), pp. 4587–4590. doi:10.1016/j.matlet.2007.02.076
- R. Palakodeti and M.R. Kessler, Influence of frequency and prestrain on the mechanical efficiency of dielectric electroactive polymer actuators, Mater. Lett. 60 (29–30) (2006), pp. 3437–3440. doi:10.1016/j.matlet.2006.03.053
- S.M. Ha, W. Yuan, Q. Pei, R. Pelrine, and S. Stanford, Interpenetrating Polymer Networks for High-Performance Electroelastomer Artificial Muscles, Adv. Mater. 18 (7) (2006), pp. 887–891. doi:10.1002/(ISSN)1521-4095
- R. Pelrine, High-Speed Electrically Actuated Elastomers with Strain Greater Than 100%, Science 287 (5454) (2000), pp. 836–839. doi:10.1126/science.287.5454.836
- H. Stoyanov, M. Kollosche, S. Risse, D.N. McCarthy, and G. Kofod, Elastic block copolymer nanocomposites with controlled interfacial interactions for artificial muscles with direct voltage control, Soft Matter 7 (1) (2011), pp. 194–202. doi:10.1039/C0SM00715C
- F. Carpi, P. Chiarelli, A. Mazzoldi, and D. De Rossi, Electromechanical characterisation of dielectric elastomer planar actuators: Comparative evaluation of different electrode materials and different counterloads, Sensors and Actuators A: Physical 107 (1) (2003), pp. 85–95. doi:10.1016/S0924-4247(03)00257-7
- M. Molberg, D. Crespy, P. Rupper, F. Nüesch, J.-A.E. Månson, C. Löwe, and D.M. Opris, High Breakdown Field Dielectric Elastomer Actuators Using Encapsulated Polyaniline as High Dielectric Constant Filler, Adv. Funct. Mater. 20 (19) (2010), pp. 3280–3291. doi:10.1002/adfm.201000486
- S. Liu, M. Tian, B. Yan, Y. Yao, L. Zhang, T. Nishi, and N. Ning, High performance dielectric elastomers by partially reduced graphene oxide and disruption of hydrogen bonding of polyurethanes, Polymer 56 (2015), pp. 375–384. doi:10.1016/j.polymer.2014.11.012
- D.M. Opris, M. Molberg, C. Walder, Y.S. Ko, B. Fischer, and F.A. Nüesch, New Silicone Composites for Dielectric Elastomer Actuator Applications In Competition with Acrylic Foil, Adv. Funct. Mater. 21 (18) (2011), pp. 3531–3539. doi:10.1002/adfm.201101039
- G. Kofod, H. Kollosche, M. Kollosche, S. Risse, H. Ragusch, D. Rychkov, M. Dansachmuller, and D.N. McCarthy, Nano-scale materials science for soft dielectrics:Composites for dielectric elastomer actuators. IEEE International Conference on Solid Dielectrics (ICSD 2010) 10th, 2010.
- F. Carpi and D. De Rossi, Dielectric elastomer cylindrical actuators: Electromechanical modelling and experimental evaluation, Mater. Sci. Engineering: C 24 (4) (2004), pp. 555–562. doi:10.1016/j.msec.2004.02.005
- P. Brochu and Q. Pei, Advances in Dielectric Elastomers for Actuators and Artificial Muscles, Macromol. Rapid Commun. 31 (1) (2010), pp. 10–36. doi:10.1002/marc.v31:1
- S. Vudayagiri, S. Zakaria, L. Yu, S.S. Hassouneh, M. Benslimane, and A.L. Skov, High breakdown-strength composites from liquid silicone rubbers, Smart Mater. Struct. 23 (10) (2014), pp. 105017. doi:10.1088/0964-1726/23/10/105017
- B. Li, H. Chen, and J. Zhou, Electromechanical stability of dielectric elastomer composites with enhanced permittivity, Composites Part A: Applied Science Manufacturing 52 (2013), pp. 55–61. doi:10.1016/j.compositesa.2012.11.013
- K. Goswami, A.E. Daugaard, and A.L. Skov, Dielectric properties of ultraviolet cured poly(dimethyl siloxane) sub-percolative composites containing percolative amounts of multi-walled carbon nanotubes, RSC Adv 5 (17) (2015), pp. 12792–12799. doi:10.1039/C4RA14637A
- H. Liu, Y. Shen, Y. Song, C.-W. Nan, Y. Lin, and X. Yang, Carbon Nanotube Array/Polymer Core/Shell Structured Composites with High Dielectric Permittivity, Low Dielectric Loss, and Large Energy Density, Adv. Mater. 23 (43) (2011), pp. 5104–5108. doi:10.1002/adma.201102079
- N. Ning, X. Bai, D. Yang, L. Zhang, Y. Lu, T. Nishi, and M. Tian, Dramatically improved dielectric properties of polymer composites by controlling the alignment of carbon nanotubes in matrix, RSC Adv 4 (9) (2014), pp. 4543–4551. doi:10.1039/C3RA45769A
- J.-K. Yuan, S.-H. Yao, A. Sylvestre, and J. Bai, Biphasic Polymer Blends Containing Carbon Nanotubes: Heterogeneous Nanotube Distribution and Its Influence on the Dielectric Properties, J. Phys. Chem. C 116 (2) (2012), pp. 2051–2058. doi:10.1021/jp210872w
- I. Alig, D. Lellinger, S.M. Dudkin, and P. Pötschke, Conductivity spectroscopy on melt processed polypropylene–multiwalled carbon nanotube composites: Recovery after shear and crystallization, Polymer 48 (4) (2007), pp. 1020–1029. doi:10.1016/j.polymer.2006.12.035
- L.J. Romasanta, M. Hernández, M.A. López-Manchado, and R. Verdejo, Functionalised graphene sheets as effective high dielectric constant fillers, Nanoscale Res. Lett. 6 (2011), pp. 508. doi:10.1186/1556-276X-6-508
- S.K. Kumar, M. Castro, A. Saiter, L. Delbreilh, J.F. Feller, S. Thomas, and Y. Grohens, Development of poly(isobutylene-co-isoprene)/reduced graphene oxide nanocomposites for barrier, dielectric and sensingapplications, Mater. Lett. 96 (2013), pp. 109–112. doi:10.1016/j.matlet.2013.01.036
- M. Tian, J. Zhang, L. Zhang, S. Liu, X. Zan, T. Nishi, and N. Ning, Graphene encapsulated rubber latex composites with high dielectric constant, low dielectric loss and low percolation threshold, J. Colloid Interface Sci. 430 (2014), pp. 249–256. doi:10.1016/j.jcis.2014.05.034
- J. Oh, J.-H. Lee, J.C. Koo, H.R. Choi, Y. Lee, T. Kim, N.D. Luong, and J.-D. Nam, Graphene oxide porous paper from amine-functionalized poly(glycidyl methacrylate)/graphene oxide core-shell microspheres, J. Mater. Chem. 20 (41) (2010), pp. 9200–9204. doi:10.1039/c0jm00107d
- D. Yang, M. Tian, D. Li, W. Wang, F. Ge, and L. Zhang, Enhanced dielectric properties and actuated strain of elastomer composites with dopamine-induced surface functionalization, J. Mater. Chem. A 1 (39) (2013), pp. 12276. doi:10.1039/c3ta12090b
- F. B. Madsen, A. E. Daugaard, S. Hvilsted, and A. L. Skov, Novel silicone compatible cross-linkers for controlled functionalization of PDMS networks, Electroactive Polymer Actuators and Devices (EAPAD). 8687 (2013), pp. 86871H.
- M. Tian, Q. Ma, X. Li, L. Zhang, T. Nishi, and N. Ning, High performance dielectric composites by latex compounding of graphene oxide-encapsulated carbon nanosphere hybrids with XNBR, J. Mater. Chem. A 2 (29) (2014), pp. 11144. doi:10.1039/c4ta01600a
- L. Chu, Q. Xue, J. Sun, F. Xia, W. Xing, D. Xia, and M. Dong, Porous graphene sandwich/poly(vinylidene fluoride) composites with high dielectric properties, Compos. Sci. Technol. 86 (2013), pp. 70–75. doi:10.1016/j.compscitech.2013.07.001
- L. Seveyrat, A. Chalkha, D. Guyomar, and L. Lebrun, Preparation of graphene nanoflakes/polymer composites and their performances for actuation and energy harvesting applications, J. Appl. Phys. 111 (10) (2012), pp. 104904. doi:10.1063/1.4718577
- F.R. Carpi, D.D., Improvement of electromechanical actuating performances of a silicone dielectric elastomer by dispersion of titanium dioxide powder. Dielectrics and Electrical Insulation, IEEE Trans. 12 (2005), pp. 835–843.
- L. Gu, T. Wang, W. Zhang, G. Liang, A. Gu, and L. Yuan, Low-cost and facile fabrication of titanium dioxide coated oxidized titanium diboride–epoxy resin composites with high dielectric constant and extremely low dielectric loss, RSC Adv. 3 (19) (2013), pp. 7071. doi:10.1039/c3ra23239e
- X. Li, A. Umar, Z. Chen, T. Tian, S. Wang, and Y. Wang, Supramolecular fabrication of polyelectrolyte-modified reduced graphene oxide for NO2 sensing applications, Ceram. Int. 41 (9) (2015), pp. 12130–12136. doi:10.1016/j.ceramint.2015.06.030
- M. Tian, B. Yan, Y. Yao, L. Zhang, T. Nishi, and N. Ning, Largely improved actuation strain at low electric field of dielectric elastomer by combining disrupting hydrogen bonds with ionic conductivity, J. Mater. Chem. C 2 (39) (2014), pp. 8388–8397. doi:10.1039/C4TC01140F
- C. Racles, M. Cazacu, B. Fischer, and D.M. Opris, Synthesis and characterization of silicones containing cyanopropyl groups and their use in dielectric elastomer actuators, Smart Mater. Struct. 22 (10) (2013), pp. 104004. doi:10.1088/0964-1726/22/10/104004
- I. Calizo, A.A. Balandin, W. Bao, F. Miao, and C.N. Lau, Temperature Dependence of the Raman Spectra of Graphene and Graphene Multilayers, Nano Lett. 7 (9) (2007), pp. 2645–2649. doi:10.1021/nl071033g
- S. Coh, L.Z. Tan, S.G. Louie, and M.L. Cohen, Theory of the Raman spectrum of rotated double-layer graphene, Phys. Rev. B. 88 (16) (2013). doi:10.1103/PhysRevB.88.165431
- K.S. Kim, Y. Zhao, H. Jang, S.Y. Lee, J.M. Kim, K.S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi, and B.H. Hong, Large-scale pattern growth of graphene films for stretchable transparent electrodes, Nature 457 (7230) (2009), pp. 706–710. doi:10.1038/nature07719
- C. Wu, X. Huang, G. Wang, X. Wu, K. Yang, S. Li, and P. Jiang, Hyperbranched-polymer functionalization of graphene sheets for enhanced mechanical and dielectric properties of polyurethane composites, J. Mater. Chem. 22 (14) (2012), pp. 7010–7019. doi:10.1039/c2jm16901k
- X. Huang, P. Jiang, and L. Xie, Ferroelectric polymer/silver nanocomposites with high dielectric constant and high thermal conductivity, Appl. Phys. Lett. 95 (24) (2009), pp. 242901. doi:10.1063/1.3273368
- Z.-M. Dang, L. Wang, Y. Yin, Q. Zhang, and -Q.-Q. Lei, Giant Dielectric Permittivities in Functionalized Carbon-Nanotube/ Electroactive-Polymer Nanocomposites, Adv. Mater. 19 (6) (2007), pp. 852–857. doi:10.1002/(ISSN)1521-4095
- F. Carpi, G. Gallone, F. Galantini, and D. De Rossi, Silicone–Poly(hexylthiophene) Blends as Elastomers with Enhanced Electromechanical Transduction Properties, Adv. Funct. Mater. 18 (2) (2008), pp. 235–241. doi:10.1002/(ISSN)1616-3028
- G. Gallone, F. Carpi, D. De Rossi, G. Levita, and A. Marchetti, Dielectric constant enhancement in a silicone elastomer filled with lead magnesium niobate–lead titanate, Mater. Sci. Engineering: C 27 (1) (2007), pp. 110–116. doi:10.1016/j.msec.2006.03.003
- B. Kussmaul, S. Risse, G. Kofod, R. Waché, M. Wegener, D.N. McCarthy, H. Krüger, and R. Gerhard, Enhancement Of Dielectric Permittivity And Electromechanical Response In Silicone Elastomers: Molecular Grafting Of Organic Dipoles To The Macromolecular Network, Adv. Funct. Mater. 21 (23) (2011), pp. 4589–4594. doi:10.1002/adfm.v21.23
- L. Jiang, A. Betts, D. Kennedy, and S. Jerrams, The fabrication of dielectric elastomers from silicone rubber and barium titanate: Employing equi-biaxial pre-stretch to achieve large deformations, J. Mater. Sci. 50 (24) (2015), pp. 7930–7938. doi:10.1007/s10853-015-9357-6