
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
It remains a challenge to achieve rapidly recoverable hydrogels by molecular hydrogen-bonding interaction because of its slow interaction kinetics. This work for the first time reports a trehalose (Tre)-based molecular movement mechanism inside a single network of polyacrylamide (PAM) that accelerates the kinetics of hydrogen-bonding interaction, and thereby endows the hydrogel with high toughness and rapid shape and mechanical recoverability. The resultant PAM@Tre hydrogel is capable of full shape recovery after 10,000 loading/unloading cycles at a strain of 500%. Even after being stretched at a strain of 2500%, it can recover to its original shape within 10 seconds. Moreover, the molecular movement of trehalose also endows the PAM@Tre hydrogel with fracture energy and toughness as high as ~9000 J m–2 and ~1600 kJ m–3, respectively, leading to strong resistance to both static and dynamic piercing. The PAM@Tre hydrogel is thus believed to have enormous potentials in protection devices, bionic skin, soft actuator, and stretchable electronics.
Graphical abstract
Introduction
Double-network hydrogels are composed of two types of polymers. One with high stiffness is to achieve elevated strength while the other is soft and ductile to restrain the rupture [Citation1,Citation2]. Crosslinked polyacrylamide (PAM) is a typical soft polymer, which is often selected and hybridized with stiffer polymers or inorganic materials to build double-network hydrogel systems [Citation3,Citation4]. Compared to the single-network counterparts, the double-network hydrogels are mechanically tough, owing to the toughening mechanism as revealed by Gong’s research group[Citation5]. Further chemical or physical treatments on the single/double networks endowed hydrogels with more-prominent mechanical behaviors. For example, the crosslinked PAM hydrogel exhibited higher strength when graphene oxide nanosheets were used to substitute the cross-linker N,N’-methylenebisacrylamide[Citation6]. After being treated with freeze-casting and salting out, poly(vinyl alcohol) (PVA) hydrogel exhibited an ultimate stress of 23.5 MPa, strain levels of 2900%, toughness of 210 MJ m‒3, and fracture energy of 170 kJ m‒2, which are comparable to natural tendons[Citation7]. PAM-alginate double-network hydrogels equilibrated with CaCl2 aqueous solution were found able to sustain strain of 400% and exhibited a fracture toughness of 5000 J m−2 even at the temperature of −57°C[Citation8]. A co-crosslinkling network of acrylic acid chitosan and acrylamide, after being treated with FeCl3 solution, showed improved strength of 2.75 MPa and recovery efficiency of 96%[Citation9]. Moreover, with the elaboratively designed molecular structures, the hydrogel was substantially strengthened by mechanical muscle-training[Citation10]. These practices demonstrated the effectiveness of using covalent/non-covalent intermolecular interaction in producing stretchable and tough hydrogels. However, once the bonding interactions are broken under large deformation, it is hard to re-build them because of the intrinsical rigidity and lower motility of these materials inside the network[Citation11]. As a result, the hydrogels can barely maintain their shape recoverability after a large stretching or many cycles of cyclic loading.
To increase the mechanical recoverability of hydrogels, the elements in chemical frameworks should be able to maintain the mechanical stability or to rapidly reconstruct the broken bonding interaction induced by external loading. Suo et al. reported a hydrogel containing both covalently cross-linked PAM and un-cross-linked PVA, which demonstrated the particular importance of non-covalent bonding interaction for maintaining the high structure stability of hydrogel[Citation12]. A type of protein was chemically crosslinked into PAM network, resulting in the hydrogel with low hysteresis (<5%) in loading/unloading cycles, where the physical folding of proteins improved the recoverability[Citation13]. Alternatively, dynamic bonds could also boost the mechanical recovery of hydrogels owing to the high dissociation and association rates, which have been achieved by a peptide-metal complex as the physical cross-linker in hydrogel networks[Citation14].
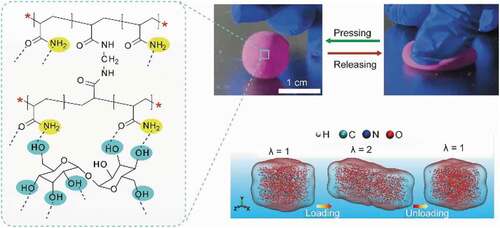
Despite these progresses, it remains a challenge to achieve rapidly recoverable hydrogels through molecular hydrogen bonding interaction. No effective protocol to improve the molecular hydrogen bonding interaction kinetics has been reported so far. Even though the molecular hydrogen bonding interactions could endow hydrogel with extremely high stretchability (stain reaching 10,000%), its hysteresis ratio was still high, reaching 93%; it required more than 1 min to recover its original shape from the large deformation[Citation15]. Such high hysteresis ratio and low recoverability were attributed to the slow hydrogen-bonding interaction kinetics that was caused by the immobility of polymer chains, in which the broken hydrogel bonds could not be rapidly rebuilt (). The covalent-bonds-constructed hydrogel also showed low recoverability at a large strain because the broken covalent bonds could not be rapidly rebuilt ().
Figure 1. Schematic illustration on molecular covalent/non-covalent interactions. (a) Strong hydrogen bonding interaction between two polymers enables the hydrogel with high stretchability yet low recoverability. (b) Hydrogel is of low recoverability as its network is built with irreversible covalent bonds. (c) Reversible non-covalent bonding interaction achieved by calcium hydroxide nanospheres inside a single network, which endows the hydrogel with high recoverability. (d) Conceptual illustration on construction of reversible non-covalent bonding interaction between polyhydroxy trehalose molecules and PAM network.
We thus conjectured if hydrogen bonds-contributing elements (electron donors or acceptors) were capable of moving inside a hydrogel network, they might be able to improve the kinetics of hydrogen-bonding interaction, and thereby accelerate the mechanical recovery of hydrogel from the large deformation. For example, the hydrogen-bond-cross-linked hydrophobic domains (diameter: ∼13 nm) were found to endow the hydrogel with high recoverability[Citation16]. The underlying mechanism might be the hydrophobic domains that enhanced the kinetics of hydrogen-bonding interaction by the rapid local motion. Typically, calcium hydroxide nanospheres with the size <5 nm were hybridized with PAM network, giving rise to a highly stretchable hydrogel with rapid shape recovery. 200-p.p.m. nanospheres could enable hydrogel to restore 90% of its original length upon releasing from 100-MPa compressive stress[Citation17]. We speculated that the hydrogen-bonding interaction between calcium hydroxide nanospheres and PAM network played a key role in such high stretchability and recoverability. One calcium hydroxide molecule contained two – OH groups. After aggregating into nanospheres, there were lots of – OH groups on the surface of the calcium hydroxide nanospheres, which acted as physical cross-linkers to improve the mechanical properties of the hydrogel by forming hydrogen bonds between the nanospheres and the PAM network. Upon the hydrogen bonds being broken by external force, the free movements of the calcium hydroxide nanospheres were capable of accelerating the association of hydrogen-bonding elements, and improving the kinetics of hydrogen-bonding interaction, so that enabling the rapid recovery of the hydrogel (). Indeed, element motion inside networks has been found to enable the functionalization of hydrogels. As demonstrated by Zhang’s research group, electrochemistry-induced movements of carrageenan inside PAM network could improve the mechanical strength of hydrogel by 7.37 times[Citation18].
Above analysis indicates that introducing movable components into PAM network might be a feasible strategy for developing highly recoverable hydrogels. In this work, we demonstrated this conception by applying trehalose (Tre) as a movable physical cross-linker to improve the reversible kinetics of hydrogen-bonding interaction inside a single network. Trehalose is a type of non-reducing disaccharide, composed of two glucose molecules. One trehalose molecule contains eight hydroxyl groups, offering many functional sites for non-covalent bonding interaction (e.g. hydrogen bond). In the meantime, it is a low-molecular-weight molecule with the size <1 nm[Citation19], which, when hybridized into PAM network, is expected to move inside the PAM network and facilitates the reconstruction of the broken hydrogen bonds ().
Our results show that the high motility of trehalose molecules enabled recovery of 94% of broken hydrogen bonds in a few seconds, endowing the single-network PAM@Tre hydrogel with prominently improved mechanical properties (e.g. fracture energy, toughness, and recoverability). Specifically, the as-prepared hydrogel reaches fracture energy as high as ~9000 J m–2, toughness of ~1600 kJ m–3, and tensile strength of 0.12 MPa at a fracture strain of 3000%. These properties are maintained after 10,000 loading/unloading cycles at a strain of 500%. Even after being stretched by a strain of 2500%, the properties of the hydrogel can be fully recovered in 10 seconds. Such high recoverability in mechanical properties can be attributed to the highly reversible kinetics of non-covalent bonding interaction induced by the trehalose molecules, which are demonstrated by the diffusion ordered spectroscopy, molecular dynamics simulation, fluorine element tracking, and infrared spectroscopy. The highly recoverable tough hydrogel is demonstrated to have enormous application potential in resisting both static piercing and dynamic impact.
Results and discussion
Synthesis of hydrogel and recoverability demonstration
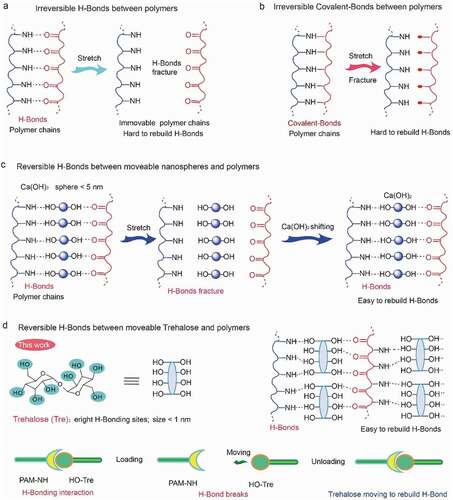
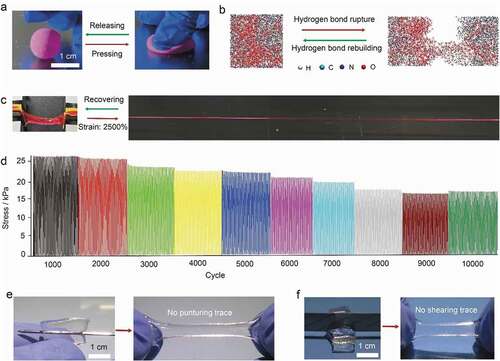
As shown in (see the details in Experimental Methods), we prepared the hydrogel by free radical polymerization of acrylamide (AM) and N,N’-methylenebisacrylamide (MBA) in trehalose aqueous solution with ammonium persulfate as an initiator (unless otherwise stated, the water content in the composite hydrogel was fixed at 78 wt%). In contrast to the traditional double-network hydrogel systems [Citation20–22], our PAM@Tre hydrogel contains only a single network; however, it exhibited higher recoverability. For example, as a solid ball was produced with the PAM@Tre hydrogel (PAM: Tre = 10: 7) by photo-polymerization in a ball-shaped mold and stained with red dye (Rhodamine B: 0.01 wt.% of PAM) for better visualization, it could sustain severe compression (, and Movie S1, ESI†) and instantly jump up when the compression was released. Molecular dynamics simulation showed that the PAM network with a homogeneous distribution of polyhydroxy small molecules (trehalose) constructed a trehalose-PAM non-covalent (hydrogen bond) interaction (). Upon stretching, the dissociation of the hydrogen bonds dissipated energy while the integrity of the covalent PAM network remained intact. In the meantime, the movable trehalose molecules allowed the reconstruction of the broken hydrogel bonds between the trehalose molecules and PAM network, leading to the rapid recovery of the mechanical properties of the hydrogel. As a result, a 2-cm-long PAM@Tre hydrogel strip could be stretched to the strain of 2500%, and contracted to 2.1 cm within 10 seconds upon the release of the loading (, and Movie S2, ESI†), reaching a length-recovering ratio of ≈95%. The evolution of stress during a loading/unloading cyclic test under a given strain load of 500% showed a slight softening behavior of the PAM@Tre hydrogel after 2000 cycles; nevertheless, its recoverability was still kept within the 10,000 cycles (). The stiffening behavior in the later stage (after 9000 cycles) was attributed to the dehydration of the hydrogel strip. The molecular movements of trehalose also enabled the PAM@Tre hydrogel to withstand extreme mechanical attack. Puncturing with a needle did not result in any apparent trace inside the hydrogel ( and Movie S3, ESI†). Cutting with a razor blade only caused a large deformation of the PAM@Tre hydrogel, without leaving any damaged trace as well ( and Movie S4, ESI†).
Figure 2. Scheme showing the preparation of PAM@Tre hydrogel by hybridization of low-molecular-weight trehalose into PAM matrix. This structural design concept is different from the traditional double-network, where the movability of water-soluble trehalose molecules improved the shape recoverability and other physical properties of the hydrogel.
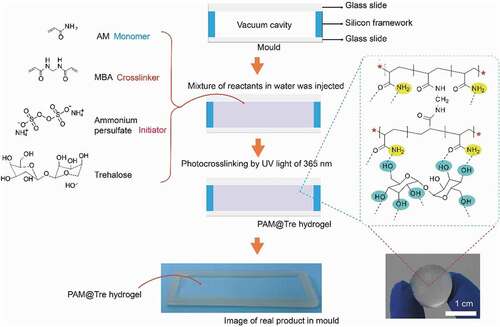
Figure 3. Mechanical analysis of highly recoverable hydrogel. (a) Demonstration on the shape recoverability by manual press. The PAM@Tre hydrogel was shaped into a hydrogel ball. As being pressed by a finger, it deformed greatly yet still maintaining its integrity. (b) Molecular dynamics simulation showing that the non-covalent bonds broke, leading to energy dissipation in the loading process. Upon unloading, the movable trehalose molecules could promote the reconstruction of the hydrogen bonds, leading to the recovery of the hydrogel’s original state. (c) Snapshots showing the rapid shape recovery of a PAM@Tre hydrogel strip upon being released from a stretched state. (d) Cyclic loading/unloading test. The PAM@Tre hydrogel was loaded and unloaded for 10,000 cycles with a strain of 500% and a loading rate of 200 mm/min. The relative humidity surrounding the hydrogel strip was kept at 95–100% by a humidifier to reduce the dehydration effects. Note that we stopped the loading/unloading and saved the data every 1000 cycles owing to the limits of device. Each pause time was less than 1 min until totally reaching 10,000 cycles. This operation caused the stress that did not decrease gradually but step by step with the cycles. (e,f) Snapshots showing the PAM@Tre hydrogel that could withstand external puncturing and cutting damages without causing visible traces.
To further demonstrate its high recoverability, a PAM@Tre hydrogel ball (2 g) was dropped from a height of 31 cm and hit a plastic ground. It could bounce back to a height of 16 cm, implying a 50% transformation rate from the gravitational potential energy (6.3 mJ) to elastic potential energy (, and Movie S5, ESI†). This rate could be increased further to 70% if the water content in the hydrogel was reduced to 60 wt.%. Moreover, the hydrogel ball was also good at absorbing the kinetic energy and converting it to elastic potential energy. One strike by a wood hammer (200 g) was able to trigger a hydrogel ball (2 g) to jump up to a height of 5 cm, implying ≈0.98 mJ elastic potential energy was stored during the hammering (Fig. S1a, ESI†). In the hammering process, the hydrogel ball highly deformed, yet its spherical shape got quickly recovered after jumping up. This feature was unusual since many conventional hydrogels fractured under hammering [Citation23–25]. A strip of the PAM@Tre hydrogel (10 cm × 2 cm × 2 mm) could survive the wheel rolling by a car weighted 2 tons, showing a prominent recoverability (Fig. S1b, ESI†).
Figure 4. Qualitative analysis on the recoverability. (a) Sketch showing the free-falling and rebounding of a PAM@Tre hydrogel ball. (b) Stress-strain profiles of a continuous loading/unloading test on a PAM@Tre hydrogel strip under a strain load of 1500%. (c) Stress-strain profiles showing the effects of stationary time on the recovery of the mechanical behavior of a PAM@Tre hydrogel strip. (d) Effect of the stationary time on the dissipated energy showing the time-dependent reconstruction of the hydrogen bonds in a stretched PAM@Tre hydrogel strip. After the first loading/unloading cycle, the hydrogel strip was kept in a stationary state for 5, 10, and 15 min, separately, before the next loading/unloading cycle. The s.d. from three measurements are shown as error bars. (e) Stress-strain profiles of a loading/unloading test on a PAM@Tre hydrogel strip under a lower stain load of 30%. (f) Stress-strain behavior of PAM@Tre hydrogel strips with different trehalose contents.
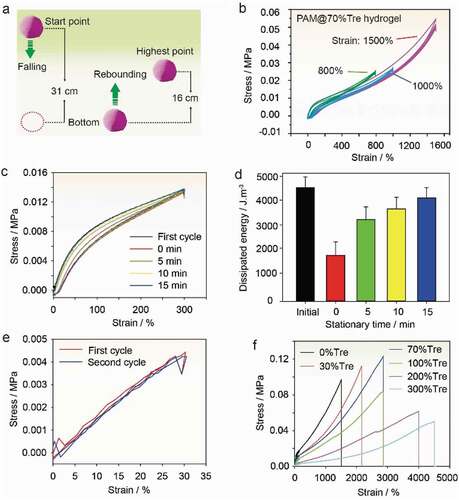
To quantitatively testify the recoverability, three PAM@Tre hydrogel strips containing 70 wt.% trehalose were, respectively, tested at different strain levels (800%, 1000%, and 1500%) on a tensile tester in a cyclic loading/unloading mode; all of them showed less hysteresis as compared to the conventional double-network hydrogels (). To measure the recovery efficiency of the dissipated energy, we carried out a loading/unloading test on a hydrogel strip with a strain load of 300% (). The PAM@Tre hydrogel strip dissipated 4300 ± 435 J m‒3 energy in the first loading/unloading cycle. If the second cycle was carried out immediately after the first cycle, the dissipated energy was reduced to 1700 ± 411 J m‒3 (). However, if the hydrogel strip was kept in a stationary state for 5 min prior to the second cycle, the dissipated energy increased to 3100 ± 421 J m‒3. If the stationary time was extended to 15 min, the dissipated energy could recover to 3900 ± 369 J m‒3 in the next cycle, which was already 97% of the original value. This result indicated that the broken hydrogen bonds during the deformation could be re-built by keeping the PAM@Tre hydrogel in a stationary state for some time, leading to the recovery of mechanical properties. At an even smaller strain of 30%, the energy dissipation of the hydrogel was negligible (), indicating that few hydrogen bonds were broken under such a small strain load. The mechanical comparison indicated that trehalose was capable of improving the mechanical recovery of PAM@Tre hydrogel (Fig. S2, ESI†).
The elastic stretchability of PAM@Tre hydrogel could be improved by increasing the trehalose content (). The maximum strain of a neat PAM hydrogel reached 1500% at the rupture stress (tensile strength) of 0.10 MPa. After the hybridization with 70 wt.% trehalose (70 wt.% of the amount of PAM), its rupture strain rose to 2800%, and its rupture strength reached 0.12 MPa. Increasing the trehalose content to 300 wt.% further enhanced the rupture strain to 4500%, and the hydrogel still exhibited high recoverability. The high content of trehalose in the network would also limit the application of PAM@Tre hydrogel in water environment since trehalose is water-soluble and may release from the network. However, there are also many application fields that require hydrogels yet not in water environments, where our PAM@Tre hydrogel might be applicable. The content of crosslinker (MBA) affected the elastic stretchability of PAM@Tre hydrogel as well. By keeping the trehalose content at 70 wt.%, the optimal MBA content was 0.05 wt.% of the amount of AM monomer (Fig. S3, ESI†). In comparison with the reported works, our PAM@Tre hydrogel demonstrated optimal stretchability and recoverability (Fig. S4, ESI†), even though it did not show the highest strength.
To demonstrate temperature effects on the stretchability, we placed a piece of PAM@Tre hydrogel in a cooling chamber with temperature of ‒31°C for 5 min. The hydrogel strip was still stretchable (Fig. S5a, ESI†). Upon longer cooling time, the hydrogel whitened and became stiff. To quantitatively measure the mechanical variation at subzero temperature, a mechanical testing at ‒31°C was carried out on a tensile tester. The neat PAM hydrogel was fragile at ‒31°C and rapidly ruptured under tension. With the increase of trehalose content to 70 wt.%, the PAM hydrogel showed stretchability and therefore anti-freezing capability. Such anti-freezing capability was enhanced prominently as the content of trehalose was increased to 100 wt.%, where the hydrogel represented higher stretchability (Fig. S5b, ESI†). Apparently, the anti-freezing capability was attributed to the trehalose molecules.
Notch-insensitivity and fracture energy
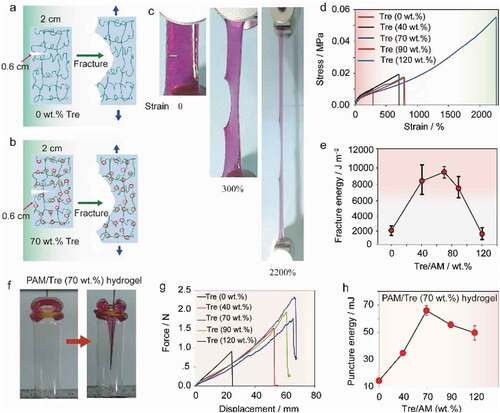
The molecular movements of trehalose endowed the PAM@Tre hydrogel with notch-insensitivity under stretching. For a comparison, we first tested a neat PAM hydrogel strip with a 0.6 cm notch. When it was stretched, a crack propagated along the notch direction. The strip finally ruptured at the critical strain of 260% (). In contrast, for a PAM@Tre hydrogel strip with 40 wt.% trehalose and a notch of the same-size, the stretching load made the notch expand with a much-blunted tip, the strip finally ruptured at a strain of 720%. As the trehalose content was increased to 70 wt.%, the PAM@Tre hydrogel strip exhibited a flaw-tolerance state, when no clear crack propagation was observed before the catastrophic rupture at the strain of 2200% () and Movie S6, ESI†). Its fracture energy reached ~9000 J m–2 (), which is 8.0-fold the fracture energy of cartilage [Citation26–28]. Further increasing the trehalose content to 90 and 120 wt.% would weaken the fracture resistance of the hydrogel, and the rupture strain dropped to 770% under the same tensile condition (). The highest toughness (the area beneath the stress–strain curve) reached ~1600 kJ m–3, when the trehalose content was 70 wt.%, and the weight ratio of MBA to acrylamide was 0.05 (Fig. S6, ESI†).
Figure 5. Qualitative analysis on fracture and puncture. (a, b) Sketches showing the stretchability of (a) a notched neat PAM strip and (b) a notched PAM@Tre hydrogel strip. and (c) Snapshots revealing that the PAM@Tre hydrogel containing 70% trehalose was notch-insensitive. (d) Stress-strain profiles indicating the trehalose weight-ratio effects on the mechanical stretchability of notched PAM@Tre hydrogel strips. (e) Trehalose-to-AM weight-ratio effects on the fracture energy of notched PAM@Tre hydrogel strips. The s.d. from three measurements are shown as error bars. (f) Snapshots showing the qualitative anti-puncturing performance of PAM@Tre hydrogel. (g) Quantitative measurements of trehalose-weight-ratio effects on the anti-puncturing performance. (h) Comparison of puncture energies of PAM@Tre hydrogel containing different trehalose contents. The s.d. from three measurements are shown as error bars.
Resistance to puncturing
The elastic PAM@Tre hydrogel exhibited a prominent puncture-resisting performance. As shown in , a metallic needle with a tip diameter of 1.0 mm was applied to stab at the hydrogels at a rate of 50 mm min‒1. During this process, the energy absorbed by the hydrogel was calculated from the force–displacement curve to be ~15 mJ. The neat PAM hydrogel could only sustain a penetration of 24 mm by the needle before being punctured. For the PAM@Tre hydrogel containing 70 wt.% trehalose, the sustainable penetration was improved to 70 mm, which was 35 times the thickness of the tested strip (, and Movie S7, ESI†). The absorbed energy reached 70 mJ (), ~4.7-fold improvement in comparison to the neat PAM hydrogel. To demonstrate the resistance to dynamic puncture, we used a mechanical ejector to shoot at a piece of PAM@Tre hydrogel sheet with a 2.1 g projectile (a steel ball, diameter: 8 mm). A projectile with velocity of 22 m s–1 and kinetic energy of 0.52 J did not penetrate the hydrogel sheet (see the details in Table S2 and Movie S8, ESI†). In contrast, the same projectile could easily penetrate four pieces of common A4 paper.
Experimental insights into hydrogen-bonding effects
The prominent mechanical performance of PAM@Tre hydrogel promoted us to consider if trehalose has been polymerized into the network of PAM. We thus took an ‘infrared spectroscopy method’ to verify it. For the PAM@Tre composite hydrogel, if trehalose had participated the polymerization into network, after washing free trehalose off, there must be a prominent absorption band of ether bond at 987 cm‒1 in the infrared spectrum. With this concept in mind, we carefully treated PAM@Tre hydrogel by washing free trehalose off with water. We then measured neat PAM, unwashed PAM@Tre hydrogel and water-washed PAM@Tre hydrogel.
As shown in Fig. S8, ESI†, the absorption band of ether bonds appeared at 987 cm‒1 for the unwashed PAM@Tre hydrogel, while there was no absorption band of ether group for the neat PAM hydrogel. However, after being washed with water to remove free trehalose out of the network, the absorption band at 987 cm‒1 disappeared, which provided direct evidence to prove that trehalose molecules were free inside the network, rather than being reacted into the network. Therefore, the prominent mechanical performance of PAM@Tre hydrogel should not be attributed to the covalent reaction of AM monomers and trehalose, but probably owing to the non-covalent interaction such as hydrogen bonding.
To get insights into the hydrogen-bonding interactions, we collected the Raman spectra of PAM@Tre and neat PAM hydrogels. The C = O stretching vibration appears at 1662 cm−1 for the neat PAM hydrogel, which shifts to 1670 cm−1 after hybridizing with trehalose. This indicates that the original strong hydrogen-bonding interactions between C = O and H2O are largely weakened, replaced by weaker hydrogen-bonding interactions between C = O and the O-H groups of trehalose () [Citation32,Citation33]. Moreover, the O-H bending vibration of water shifts from 1611 to 1620 cm−1, implying the enhancement of hydrogen-bonding among self-associated water molecules[Citation34]. Both the two wavenumber shifts direct to the same event that trehalose molecules participate in hydrogen-bonding interactions with the C = O groups of PAM network, which causes more water molecules to be self-associated. Such a process can be further evidenced by the spectral changes as recorded by Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. As the number of water molecules are much higher than that of trehalose and PAM units, the vibration between 3650 and 3000 cm−1 is mainly contributed by the O-H stretching vibration of water molecules. In comparison with neat PAM hydrogel, the peak intensity of O-H stretching vibration from water is strengthened at around 3200 cm−1 at the expense of that at 3136 cm−1 and 3395 cm−1, which reveals again the gradually enhanced self-association of water molecules, caused by the direct associations between PAM chains and trehalose molecules (). These data indicated that the hydrogen bonding interaction indeed formed between trehalose and PAM network.
Figure 6. Molecular-level analysis on the hydrogen-bonding interaction between trehalose and PAM network. Ramanspectra of PAM@Tre and neat PAM hydrogels indicating the trehalose effects on hydrogen bonding interaction of C = O groups of PAM. (b) ATR-FTIR spectra of PAM@Tre and neat PAM hydrogels demonstrating the trehalose effects on hydrogen bonding interaction of water in hydrogel system. (c) Raman spectra of NaCl and LiBr treated/non-treated PAM@Tre hydrogels. (d) Stress– profiles of PAM@Tre hydrogels, in which two types of salts (NaCl and LiBr) were respectively tested to break the hydrogen bonds. The tensile results were compared with the neat PAM@Tre hydrogel.
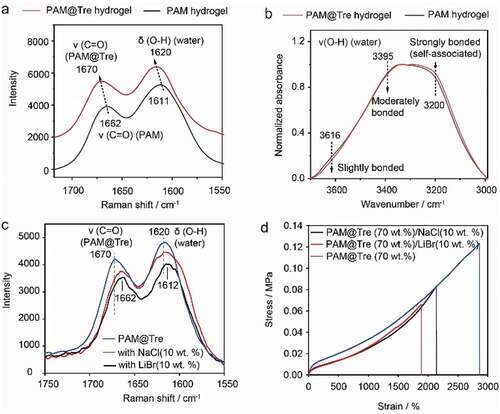
To further demonstrate the contribution of trehalose on the hydrogen bonding interaction, LiBr or NaCl (10 wt.% of PAM) was added as a competitive hydrogen-bonding agent during the preparation of PAM@Tre hydrogel [Citation35,Citation36]. The samples were also analyzed by Raman spectroscopy (), which demonstrate the reverse variation of hydrogen-bonding interactions in comparison to that in . Apparently, the addition of LiBr and NaCl destroyed the hydrogen-bonding interaction in PAM@Tre hydrogel. As a result, the maximum elongation of a PAM@Tre hydrogel strip was reduced by 1.9-fold owing to the addition of 10 wt.% LiBr or NaCl, in comparison to the neat PAM@Tre hydrogel (). The LiBr or NaCl weakened the hydrogen bonding interaction, which in turn proved the existence of hydrogen-bonding interaction between trehalose and crosslinking PAM network.
Motility of trehalose molecules in hydrogel network
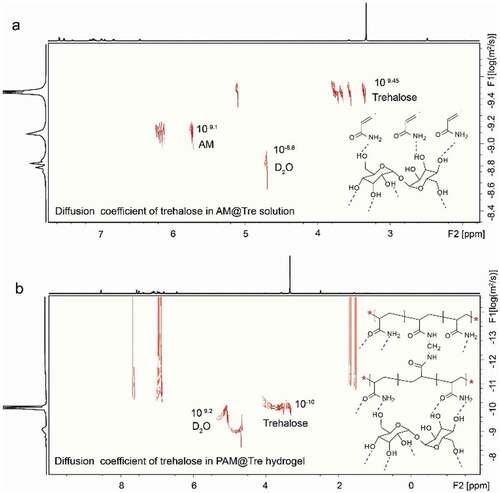
The reversibility of hydrogen-bonding interaction is of great significance to maintain the high shape recoverability of hydrogel. For PAM@Tre hydrogel system, we attribute the reversible hydrogen-bonding interaction to the movements of trehalose molecules inside the network. Upon the hydrogen bonds being destroyed, the trehalose molecules moved and acted as a ‘molecular bridge’ to rebuild the hydrogen-bonding interaction. To characterize the motility of trehalose molecules, the diffusion measurement was carried out on an NMR spectrometer to determine the diffusion coefficient of trehalose molecules in the network of PAM@Tre hydrogel (see the details in Methods). We first measured the diffusion coefficient of trehalose in AM@Tre solution, which was prepared by dissolving trehalose (140 mg, 0.4 mmol) and AM (200 mg, 2.8 mmol) into 1 mL D2O at room temperature. We then measured the diffusion coefficient of trehalose in PAM@Tre hydrogel. For this experiment, we prepared the solution by dissolving trehalose (140 mg, 0.4 mmol), AM (200 mg, 2.8 mmol), ammonium persulfate (2 mg, 0.0087 mmol) and MBA (0.1 mg, 0.0006 mmol) into 1 mL D2O. The solution was injected into a pre-cleaned NMR tube and exposed to UV irradiation (intensity: 300 mW cm‒2, λ = 365 nm) for 40 min prior to the diffusion measurements.
revealed that the diffusion coefficient of trehalose molecules was 10‒9.44 m2 s‒1 in the AM@Tre solution, which decreased to 10‒10 m2 s‒1 after polymerization of the AM@Tre solution into PAM@Tre hydrogel. The diffusion coefficient of deuteroxide molecules was 10‒8.80 m2 s‒1 in the AM@Tre solution, while it was decreased to 10‒9.80 m2 s‒1 in the PAM@Tre hydrogel. Even through the diffusion coefficients of trehalose and deuteroxide molecules decreased, they were still movable inside the network of PAM@Tre hydrogel; otherwise, no data on the diffusion coefficient could be detected. The results thus confirmed that the trehalose molecules were capable of movements inside the network of PAM@Tre hydrogel.
Figure 7. Diffusion ordered spectroscopy. (a) Diffusion coefficient of trehalose in AM@Tre solution, and (b) in PAM@Tre hydrogel. The experiments were carried out at 25°C.
Molecular dynamics simulation of hydrogen-bonding interaction
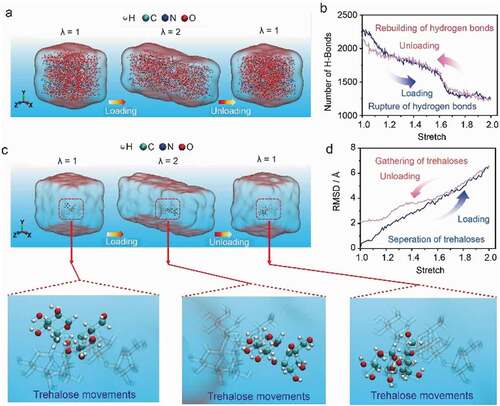
To further reveal the trehalose movements-triggered reversibility of hydrogen bonding interaction, the all-atom molecular dynamics (MD) simulation was carried out to explore the evolution of the hydrogen bonds in PAM@Tre hydrogel in a loading/unloading cycle of uniaxial tension at a strain rate of 109 s−1 (). The simulated system contained 8000 water molecules, 60 trehalose molecules, and 16 PAM chains with 25 monomers each. As expected, the total number of hydrogen bonds in the simulated system monotonically decreased as the uniaxial tensile strain increased. As the tensile load was released after reaching the maximum stretch (λ = 2.0), the number of the hydrogen bonds was restored to 94% of the original value when the deformation was recovered (, ESI†). Such evolution of the number of hydrogen bonds was induced by the external loading as it was not observed in the control system free of loading (Fig. S9, ESI†). The quick recovery of the hydrogen bonds in the simulated systems was attributed to the high motility of small trehalose molecules as shown by the calculated trajectory of a trehalose molecule inside the hydrogel network during the loading/unloading cycle (, ESI†). Moreover, we traced the relative root-mean-square-displacement (RMSD) between two typical adjacent trehalose molecules, which was found to reach ~6.6 Å upon the stretch of 2.0, and then reduce to ~2.0 Å after unloading, indicating the mobility of trehalose molecules (). The above MD simulation was also performed at the lower strain rate of 108 s−1 to 106 s−1 and the more prominent recovering behavior of H-bond number was observed (Figs. S10–S12, ESI†). To verify the repeatability of the MD results, we kept on the loading/unloading process for two more cycles after the first one. The evolution of the H-bonds number in the second and third cycles follows almost the same trend as exhibited in the first one (Fig. S13, ESI†).
Figure 8. Hydrogen bond evolution during the loading and unloading cycle. (a) Snapshots of the PAM@Tre hydrogel with trehalose content of 70 wt% under uniaxial stretching. All atoms and hydrogen bonds are shown. (b) The number of hydrogen bonds evolution during the loading and unloading cycle of the hydrogel, demonstrating the high recoverability of the hydrogen bonding between trehalose and PAM network. (c) Snapshots showing the position of a representative trehalose molecule in the simulated PAM@Tre hydrogel during a loading/unloading cycle. For a clearer visualization of the trajectory, the instant configuration of trehalose molecule is shown together with its ghost configurations at the other two moments, displaying the motility of the trehalose molecule. (d) The evolution of RMSD between the representative trehalose molecules shown in the snapshots and one of its adjacent, which implies the motility of trehalose accounting for the quick recovery of the broken hydrogen bonds in PAM@Tre hydrogel system.
Applications
As a paradigm of application, a PAM@Tre hydrogel film was sandwiched between two glass sheets (100 mm × 100 mm × 1 mm) (Fig. S14a, ESI†). For comparison, a control sandwich panel was prepared by using a neat PAM hydrogel film of the same dimensions as the interlayer. Standard drop-weight tests were carried out on the both sandwich panels. Upon a standard drop-weight impact, both hydrogel-based panels were pierced while the broken glass fragments remained attached to the hydrogel films (Fig. S14b and c, ESI†). The transient variation of the reaction force and energy loss are shown in Fig. S14d, ESI†. For the glass/PAM/glass sandwich panel, peaks P1, P2 on the reaction force profile (blue dotted line) correspond to the impact of the indenter on two glass sheets. The force peak from the PAM interlayer can be barely seen, implying that the PAM interlayer provided little resistance to the piercing. For the glass/PAM@Tre/glass sandwich panel, peaks P3, P4, and P5 on the reaction force profile (blue solid line) correspond to the impact of the indenter on the front glass sheet, the rear glass sheet, and the hydrogel film, respectively. The heights of peaks P4 and P5 are comparable, implying that the PAM@Tre interlayer provided similar resistance to the piercing as the glass sheet did. The whole piecing process lasted for 1.08 ms, which is almost double of the piecing time on the control sample. This was attributed to the tough PAM@Tre hydrogel that could sustain large deformation before being punctured. The large deformation of the PAM@Tre hydrogel enabled the sandwich panel to absorb 0.1 J kinetic energy, which is almost two folds of the value in the control case (see the red solid line in Fig. S14d, ESI†). After being punctured, the fractured glass/PAM@Tre/glass sandwich panel remained stretchable without showing apparent shedding of the glass fragments (Fig. S14e, ESI†). Such excellent resistance to dynamic piercing in combination with its high optical transmittance (Fig. S14 f, ESI†) makes the PAM@Tre hydrogel an excellent candidate for transparent protective shields.
Experimental Methods
Materials
Acrylamide (AM, >99%), trehalose (98%), N,N’-methylenebisacrylamide (MBA, >99%), ammonium persulfate (>99%), NaCl (99%), LiBr (99%), trifluoromethanesulfonic anhydride (>99%), deuterium oxide (D2O), deuterium chloroform (CDCl3), (99.9%), and deuterium dimethyl sulfoxide (DMSO-d6) were purchased from commercial suppliers (Aldrich). A transparent spherical mold was purchased from an online shop (https://www.taobao.com/). The ball-like mold was made of transparent plastics, and its diameter was 2 cm.
Design and fabrication of transparent template for hydrogel synthesis
A silicone sheet with dimensions of 75 mm × 25 mm × 1.5 mm was prepared and cleaned by acetone. Its middle area of 70 mm × 20 mm was removed by a graver to give a rectangular framework. This silicone framework was then sandwiched between two pre-cleaned glass slides to form a hollow transparent template. The hollow area was vacuumized prior to the use.
Preparation of neat PAM hydrogel
The mixture of AM (2 g, 28 mmol), ammonium persulfate (0.02 g, 0.087 mmol) and MBA (1 mg, 0.0065 mmol) was dissolved in 10 mL deionized water to form a clear and transparent solution. The solution was injected into the hollow transparent template and exposed to ultraviolet (UV) light (intensity: 300 mW cm ‒2) of 365 nm for 40 min. The crosslinked hydrogel was taken out and cut to customized shapes for mechanical tests and other applications.
Preparation of PAM@Tre hydrogel
The hydrogel was prepared by photo-introduced crosslinking reaction, and initiator was selected according to reported method[Citation37]. We dissolved AM (2 g, 28 mmol), trehalose (0.6 g, 1.6 mmol), ammonium persulfate (0.02 g, 0.087 mmol) and MBA (1 mg, 0.0065 mmol) in 10 mL deionized water. The formed solution was injected into the hollow transparent template. After being irradiated by UV light (intensity: 300 mW cm‒2, λ = 365 nm) for 40 min, the sealed template was opened to give PAM@Tre hydrogel. By keeping the contents of AM, ammonium persulfate, and MBA constant, changing the trehalose to 0.6 g, 1.4 g, 2 g, 4 g, and 6 g produced a series of PAM@Tre composite hydrogels with following the similar reaction processes.
Preparation of PAM@Tre hydrogel ball
We dissolved AM (2 g, 28 mmol), trehalose (1.4 g, 3.7 mmol), ammonium persulfate (0.02 g, 0.087 mmol), and MBA (1 mg, 0.0065 mmol) in 10 mL deionized water. The formed solution was injected into the transparent spherical plastic mold with diameter of 2 cm. After irradiation with UV light (intensity: 300 mW cm‒2, λ = 365 nm) for 40 min, a solidified PAM@Tre hydrogel ball was obtained.
Quasi-static puncturing testing
A quasi-static puncturing test was carried out with a universal testing machine equipped with a needlelike indenter with diameter of 1 mm (Model: HY-0580, Shanghai Hengyi Test Instrument Co., Ltd.). To prepare a sample for puncturing, we applied a piece of hydrogel sheet (50 mm × 50 mm × 1.5 mm) to wrap the mouth of a glass bottle (mouth diameter: 3 cm), and fixed it tightly with a rubber band. The bottle was placed beneath the indenter. The overhung portion of the hydrogel sheet was punctuated by the indenter moving downwards slowly at a speed of 50 mm min‒1. The whole puncturing process was recorded by a camera (JVC, GC-PX100). The force–displacement curve was plotted to show the puncture-resistance of the hydrogel sheet. The area beneath the obtained force–displacement curve was calculated as the puncturing energy.
Ballistic impact testing
A square hydrogel sheet (100 mm × 100 mm × 1 mm) was fixed vertically by two clamps (Movie S8, ESI†). A home-made mechanical ejector was applied to eject projectile (steel balls, weight: 2.1 g, diameter: 8 mm) with controllable speed to shoot at the hydrogel sheet (Table S1, ESI†). Projectiles with different speeds were tried to determine the critical speed at which the hydrogel sheet was just penetrated by the projectile.
Fracture-resistant testing
The fracture energy of hydrogels was measured by following the method reported in the literature[Citation27]. First, two identical sheets (30 mm × 30 mm × 1.5 mm) of the same hydrogel were prepared. Then, a notched sample was prepared by using a razor blade to cut into one sample a 6 mm-long notch. Each sample was gripped by two clamps with initial distance of 2 mm and pulled with a universal tensile tester (HY0508, Shanghai Hengyi Test Instrument Co., Ltd.). The pulling force was recorded as a function of the inter-clamp distance (L). The fracture energy of the tested hydrogel was calculated from , where
is the area beneath the force–distance curve of the unnotched sample at
,
stands for the critical distance at which the notched sample starts to crack, and
is the cross-sectional area (perpendicular to the loading direction) of the unnotched hydrogel sheet.
Tensile testing
The tensile property of PAM@Tre hydrogel was measured on a tensile tester (HY-0580, Shanghai Hengyi Test Instrument Co., Ltd.) A series of hydrogel strips with same dimensions (25.0 mm × 10.0 mm × 1.5 mm) were prepared. They were, respectively, tested at a loading rate of 50.0 mm min‒1 at room temperature. With the same instrument, compression test was also carried out on a PAM@Tre hydrogel ball (diameter: 2 cm). The loading rate was 50.0 mm min‒1 and the maximum compression was 90% of the ball’s diameter. A 50.0 N load cell was used for all tests.
Scanning Electron Microscopy (SEM)/element mapping analysis
The hydrogels were dried in a freeze-dryer (Model: FD-1A-50, Shanghai Bilon Instrument Manufacturing Co., Ltd). The surface was observed with scanning electron microscope (S-4800, SYST TA PRO 1156) using primary electron energy of 5 kV. The samples were attached to a silicon wafer with adhesive carbon tape without any other treatments prior to the SEM imaging, and the energy-dispersive spectroscopy (EDS) mapping analysis. The micrographs were recorded at room temperature and at a pressure of 8.8 × 10−7 Pa. The element mapping analysis was carried out with focus on ‘F’ and ‘S’ elements for all samples.
Infrared spectroscopy
An attenuated total reflectance Fourier transformed infrared spectrometer (Thermo Scientific Nicolet IS10) was used to measure the water-swollen PAM@Tre and neat PAM hydrogels at the range of 4000 to 800 cm–1. All ATR-FTIR data were collected at a spectral resolution of 4 cm–1. Thirty-two scans were accumulated at room temperature. The measurement was repeated three times for each sample and the air background was subtracted.
Raman spectroscopy
Raman spectroscopy was conducted on Renishaw inVia Reflex with 532 nm laser as a light source (power: 12 mW; exposure time: 4 s). All Raman data were collected at a spectral resolution of 1 cm−1, and five scans were accumulated. After all spectra were collected, the baselines of original spectra were uniformly corrected.
Temperature-variable Infrared spectroscopy
The PAM@Tre hydrogel containing 70 wt.% trehalose was kept in a chamber at room temperature and relative humidity (RH) 60% for 24 h to reach equilibrium. The sealed hydrogel sample was heated from 30 to 60°C with an interval of 2°C in the transmission mode on a Nicolet iS50 FTIR spectrometer for temperature-controlled measurements.
Measurement of diffusion coefficient
Diffusion ordered spectroscopy (Dosy) using stimulated echo (STE) pulse sequence was performed on a Bruker Avance NEO 600.41 MHz spectrometer (Bruker Biospin, Switzerland), equipped with a Z-gradient probe. The temperature was controlled at 25°C. The diffusion coefficients were determined using the relationship between the echo signal intensities and the field-gradient parameters [Citation38]: , where I(g) and I(0) are the echo signal intensities with and without the field gradient pulse, respectively; is the gyromagnetic ratio of [Citation1]H;
is the duration of the gradient pulses; g is the field gradient strength;
is the diffusion time; and D represents the diffusion coefficient. Here,
and
were fixed to 1.5 ms and 0.4 s, respectively. Standard decay curves were recorded with 16 increments and the gradient strength linearly increased from 2% to 95% of the probe’s maximum value. The maximum gradient strength was 5.35 G/mm.
Drop-weight tests
Drop-weight tests were carried out with an instrumented drop tower system (Dynatup 9250HV, Instron, Norwood, USA). The drop weight was 17.34 kg and the drop height was 0.5 m. The impacting indenter was a 304 stainless steel hemisphere-ended cylinder with a diameter of 5 mm. The tested specimens, which were square plates with in-plane dimensions of 100 mm × 100 mm, were firmly fixed at the sample stage by a pneumatically driven frame, leaving a circular overhang region with a diameter of 80 mm in the center for impacting. During the testing, the reaction force exerted by the specimen on the indenter and the energy loss of the drop weight, which is assumed equal to the energy absorbed by the specimen, was recorded in real-time.
Molecular dynamics (MD) simulation
The PAM@Tre hydrogel structures were constructed by Monte Carlo random method, which was imported into LAMMPS code to relax. MD simulation was performed 500 ps under NPT ensemble after the energy minimization to get the equilibrated structure of PAM@Tre hydrogel. The equilibrium structure was used as the initial testing sample for the loading and unloading cycling deformation simulations. The deformation simulation was conducted by controlling the displacement in the x dimension of the simulation box at a constant engineering strain rate, and the y and z dimensions could fluctuate under NPT ensemble. The strain rate in the deformation simulations was set to 109 s−1. The Nose–Hoover thermostat and barostat were used to regulate and control the temperature and pressure, respectively.
The OPLS-AA and SPCE force fields were used for PAM@Tre network and water, respectively. The total potential energy of the system consisted of non-bond energy, bond stretching energy, angle bending energy, torsional energy and hydrogen bond energy. The non-bond energy included the Coulomb and Lennard-Jones potentials. The bond stretching, angle bending and torsional energies were described by the force field parameters and equations obtained from the OPLS-AA force field. A pair of hydroxyl-amino groups or a pair of hydroxyl-amino group and water molecule was defined hydrogen-bonded if the distance and angle satisfied the following conditions: RDA ≤ 4.5 Å, RDH ≤ 2.75 Å, φ ≤ 135°, where RDA is the distance between the donor and acceptor, RDH is the bond length between the donor and hydrogen atom, φ is the D-H-A angle formed by the donor, hydrogen atom and acceptor. The hydrogen bonding interaction could be rebuilt after debonding if the above conditions are satisfied again. All equilibrium and deformation simulations were implemented under the periodic boundary condition. The time step was set as 1 fs. The global cutoff distance was 9 Å for the Lennard-Jones potential and 12 Å for the Coulomb potential.
Conclusions
Hydrogen-bonding interaction can be easily constructed by electron donors and acceptors in various chemical structures, which is an important non-covalent force to improve the mechanical properties of materials[Citation34]. The formation of hydrogen bonds doesn’t have to overcome high activation energy (<10 kcal/mol); therefore, they can go with reversible association and disassociation kinetics with the change of external conditions. However, the distance between electron donors and acceptors is a critical parameter to determine their association and disassociation rates. The broken hydrogen bonds cannot be rapidly re-built, mostly because of an inappropriate distance of electron donors and acceptors, which thus decreases the hydrogen-bonding interaction kinetics.
To improve the hydrogen-bonding interaction kinetics, this work developed a molecular movement mechanism of trehalose inside a single network of PAM. The movements of trehalose molecules were capable of increasing the association rates of the electron donors and acceptors, enabling the rapid rebuilding of broken hydrogen-bonding interaction. The molecular dynamics analysis demonstrated that the number of broken hydrogen bonds could be restored to 94% of the original value in a few seconds. Such a rapid hydrogen-bonding association rate resulted in the resultant PAM@Tre hydrogel capable of full recovery after 10,000 loading/unloading cycles at a strain of 500%. Even after being stretched at a strain of 2500%, it can recover to its original shape within 10 seconds. The high recoverability of the PAM@Tre hydrogel rendered it immune to damage even under severe compression, distortion, or hammering. Moreover, the PAM@Tre hydrogel exhibited superior resistance to static and dynamic piercing and crack propagation due to its higher fracture energy (~9000 J m–2) and toughness (~1600 kJ m–3). When being used as the interlayer in sandwiched structures, the PAM@Tre hydrogel exhibited the higher enhancement to the impact resistance of the structure as compared to a neat PAM counterpart. This facile yet effective protocol might provide an important information to synthesize highly recoverable tough hydrogels for applications in water-free systems, such as explosion-protection devices, bionic skin, soft actuators, and stretchable electronics.
Author contributions
X. H. and H. T. with assistance from M. X. conducted the experiments. J. F. carried out the molecular dynamic simulation, and Y. X. helped with drop-weight testing. S. T. helped with spectroscopic analysis. L. Z. and H. Y. supervised the study, analyzed the results, and wrote the manuscript. L. Z. conceived the idea and designed the experiments. All authors contributed to the discussion and interpretation of the results
Supplemental Material
Download Zip (34.3 MB)Acknowledgments
The authors are grateful to the National Natural Science Foundation of China (Grant No. 51873064, 51603068, 11772283), and the Natural Science Foundation of Shanghai (Grant No. 20ZR1418200; 17ZR1440600) for financial support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19475411.2022.2116735
Additional information
Funding
Related Research Data
References
- Huang WJ, Wang YX, McMullen LM, et al. Stretchable, tough, self-recoverable, and cytocompatible chitosan/cellulose nanocrystals/polyacrylamide hybrid hydrogels. Carbohyd Polym. 2019;222:114977.
- Lin ST, Yuk H, Zhang T, et al. Stretchable Hydrogel Electronics and Devices. Adv Mater. 2016;28(22):4497–4505.
- Ecaterina SD. Design and applications of interpenetrating polymer network hydrogels. A review. Chem Eng J. 2014;243:572–590.
- Chen Q, Chen H, Zhu L, et al. Fundamentals of double network hydrogels. J Mater Chem B. 2015;3(18):3654–3676.
- Gong JP. Why are double network hydrogels so tough? Soft Matter. 2010;6(12):2583–2590.
- Liu RQ, Liang SM, Tang XZ, et al. Tough and highly stretchable graphene oxide/polyacrylamide nanocomposite hydrogels. J Mater Chem. 2012;22(28):14160–14167.
- Hua MT, Wu SW, Ma YF, et al. Strong tough hydrogels via the synergy of freeze-casting and salting out. Nature. 2021;590(7847):594–599.
- Morelle XP, Illeperuma WR, Tian K, et al. Highly Stretchable and Tough Hydrogels below Water Freezing Temperature. Adv Mater. 2018;30(35):1801541.
- Xu JJ, Jin RN, Ren XY, et al. Cartilage-inspired hydrogel strain sensors with ultrahigh toughness, good self-recovery and stable anti-swelling properties. J Mater Chem A. 2019;7(44):25441–25448.
- Takahiro M, Runa K, Ryo N, et al. Mechanoresponsive self-growing hydrogels inspired by muscle training. Science. 2019;363(6426):504–508.
- Wu F, Chen L, Wang YD, et al. Tough and stretchy double-network hydrogels based on in situ interpenetration of polyacrylamide and physically cross-linked polyurethane. J Mater Sci. 2019;54(18):12131–12144.
- Bai RB, Yang JW, Morelle XP, et al. Fatigue Fracture of Self-Recovery Hydrogels. ACS Macro Lett. 2018;7(3):312–317.
- Lei H, Dong L, Li Y, et al. Stretchable hydrogels with low hysteresis and anti-fatigue fracture based on polyprotein cross-linkers. Nat Commun. 2020;11(1):4032.
- Sun WX, Xue B, Fan QY, et al. Molecular engineering of metal coordination interactions for strong, tough, and fast-recovery hydrogels. Sci Adv. 2020;6(16):eaaz9531.
- Jeon I, Cui JX, Illeperuma WRK. Extremely Stretchable and Fast Self-Healing Hydrogels. J Aizenberg and J J Vlassak Adv Mater. 2016;28(23):4678–4683.
- Fang X, Li YX, Li X, et al. Dynamic Hydrophobic Domains Enable the Fabrication of Mechanically Robust and Highly Elastic Poly(vinyl alcohol)-Based Hydrogels with Excellent Self-Healing Ability. ACS Mater Lett. 2020;2(7):764–770.
- Sun GX, Li ZJ, Liang R, et al. Super stretchable hydrogel achieved by non-aggregated spherulites with diameters <5 nm. Nat Commun. 2016;7(1):12095.
- Miao Y, Xu MD, Zhang LD. Electrochemistry-Induced Improvements of Mechanical Strength, Self-Healing, and Interfacial Adhesion of Hydrogels. Adv Mater. 2021;33(40):2102308.
- Olsson C, Jansson H, Youngs T, et al. Structure of Aqueous Trehalose Solution by Neutron Diffraction and Structural Modeling. J Phys Chem B. 2016;120(49):12669–12678.
- Peng H, Gao XJ, Sun KJ, et al. Physically cross-linked dual-network hydrogel electrolyte with high self-healing behavior and mechanical strength for wide-temperature tolerant flexible supercapacitor. Chem Eng J. 2021;422:130353.
- Su Q, Wang YJ, Guan S, et al. Rapid formation of highly stretchable and notch-insensitive hydrogels. RSC Adv. 2016;6(36):30570–30576.
- Luo F, Sun TL, Nakajima T, et al. Free Reprocessability of Tough and Self-Healing Hydrogels Based on Polyion Complex. ACS Macro Lett. 2015;4(9):961–964.
- Guan QF, Yang HB, Han ZM, et al. Sustainable Cellulose-Nanofiber-Based Hydrogels. ACS Nano. 2021;15(5):7889–7898.
- Cui W, King DR, Huang YW, et al. Fiber-Reinforced Viscoelastomers Show Extraordinary Crack Resistance That Exceeds Metals. Adv Mater. 2020;32(31):1907180.
- Liu YX, Liu J, Chen SC, et al. Soft and elastic hydrogel-based microelectronics for localized low-voltage neuromodulation. Nat Biomed Eng. 2019;3(1):58–68.
- Lin ST, Liu J, Liu XY, et al. Muscle-like fatigue-resistant hydrogels by mechanical training. Proc Natl Acad Sci USA. 2019;116(21):10244–10249.
- Sun JY, Zhao XH, Illeperuma WRK, et al. Highly stretchable and tough hydrogels. Nature. 2012;489(7414):133–136.
- Lin ST, Liu XY, Liu J, et al. Anti-fatigue-fracture hydrogels. Sci Adv. 2019;5(1):eaau8528. M. Mooney, J Appl Phys., 1940, 11, 582–592.
- Rivlin RS, Saunders DW Large elastic deformations of isotropic materials VII. Experiments on the deformation of rubber . Philos Trans R Soc London, Ser A. 1951;243:251–288.
- Lake GJ, Thomas AG The strength of highly elastic materials . P Roy Soc A-Math Phy. 1967;300:108–119.
- Zhang YW, Huang W, Zhou YF, et al. A physical gel made from hyperbranched polymer gelator. Chem Commun. 2007;25:2587–2589. DOI:10.1039/b701043e
- Sun ST, Wu PY. On the Thermally Reversible Dynamic Hydration Behavior of Oligo(ethylene glycol) Methacrylate-Based Polymers in Water. Macromolecules. 2013;46(1):236–246.
- Sun ST, Wu PY. Role of Water/Methanol Clustering Dynamics on Thermosensitivity of Poly(N-isopropylacrylamide) from Spectral and Calorimetric Insights. Macromolecules. 2010;43(22):9501–9510.
- Shen Y, Wu PY. Two-Dimensional ATR−FTIR Spectroscopic Investigation on Water Diffusion in Polypropylene Film: water Bending Vibration. J Phys Chem B. 2003;107(18):4224–4226.
- Moringo NA, Bishop LDC, Shen H, et al. A mechanistic examination of salting out in protein–polymer membrane interactions. Proc Natl Acad Sci USA. 2019;116(46):22938–22945.
- Shubina ES, Belkova NV, Krylov AN, et al. Spectroscopic Evidence for Intermolecular M−H···H−OR Hydrogen Bonding: interaction of WH(CO) 2 (NO)L 2 Hydrides with Acidic Alcohols. J Am Chem Soc. 1996;118(5):1105–1112.
- Chen F, Zhou D, Wang JH, et al. Rational Fabrication of Anti-Freezing, Non-Drying Tough Organohydrogels by One-Pot Solvent Displacement. Angew Chem Int Edit. 2018;57(22):6568–6571.
- Zhao QH, Breneer T, Matsukawa S. Molecular mobility and microscopic structure changes in κ-carrageenan solutions studied by gradient NMR. Carbohyd Polym. 2013;95(1):458–464.