
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Rare-earth elements (REEs) are critical to modern industry but difficult to separate due to their subtle and monotonic changes in physicochemical properties. MoS2-based two-dimensional (2D) materials offer novel opportunities for enhancing REE separation, exhibiting a distinct volcano-shaped transport performance distribution that peaks at Sm3+. However, the specific contributions of thermodynamic and kinetic factors to ion transport within 2D confinement remain unclear. In this study, we conducted a series of non-equilibrium all-atom molecular dynamics (MD) simulations to explore the effects of interlayer spacing and external pressure on the transport of lanthanide ions in Å-scale acetate functionalized 2D MoS2 (MoS2-COOH) channels. We examined ion entry and permeation rates, water flux, dehydration, and binding modes. The simulation results reveal that the transport trends of lanthanide ions are jointly driven by the dehydration degree and the relative-binding strengths of ions to water and to the acetate within the 2D channels. Notably, the dehydration pattern of lanthanide ions during permeation is closely linked to kinetic factors. Overall, this study provides a detailed atomistic understanding of the mechanisms underlying lanthanide ion transport under confinement. These findings point to the significant potential for tuning confinement and chemical functionalization within Å-scale channels for more efficient REE separation.
GRAPHICAL ABSTRACT
Introduction
Lanthanide elements or rare earth elements (REEs), are critical components in numerous areas, including electronics, renewable energy systems, manufacturing, medical science, and technology, owing to the versatile chemical, magnetic, phosphorescent, and catalytic properties of the lanthanide-ion 4f electronic configurations [Citation1,Citation2]. However, the separation of REE cations poses a significant challenge due to their remarkably similar physicochemical properties, and the so-called “lanthanide contraction” that leads to monotonic shrinking of their ionic radii and monotonic increase in Lewis acidity [Citation2,Citation3].
Traditional separation methods such as solvent extraction and ion exchange, are often inefficient, costly, and environmentally detrimental [Citation4,Citation5]. These techniques struggle with the fine differentiation required to separate lanthanides effectively, leading to a pressing need for more efficient and sustainable separation processes. Inspired by the high efficiency and precision of cell membranes in separating water and ions [Citation6], two-dimensional (2D) nanomembranes have emerged as a promising solution for selective ion transport and separation [Citation7–20]. Materials like MoS2 offer several advantages due to their atomic-scale thickness, tunable surface properties, stability, and good mechanical strength [Citation21–23]. These membranes can be functionalized with specific chemical groups, such as carboxylate groups, to selectively interact with different REE ions. This functionalization provides precise control of the interlayer spacing and influences the migration energy barrier, potentially improving the separation efficiency by exploiting subtle differences in ion dehydration energy and binding affinities [Citation22–28].
We recently reported on the transport of lanthanide ions in Å-scale 2D MoS2-COOH channels, and we observed a well-defined volcano shape in transport peaked at Sm3+ [Citation24]. We examined the thermodynamic and kinetic behaviors of lanthanide ions within the MoS2-COOH channel, noting that the high permeability of Sm3+ can be attributed to its moderate hydration-free energy (HFE) and moderate binding affinity of Sm3+ to acetate. Nonetheless, the specific contributions of thermodynamic and kinetic factors to ion transport performance under 2D confinement were not elucidated. These can be explored by tuning the interlayer-spacing of the membranes and by varying the applied pressure [Citation29–31]. These parameters can be explored through experiments; however, the limited experimental data currently available for MoS2-based membranes with various functional groups restricts our ability to predict what effects these changes might produce.
Here, we perform non-equilibrium all-atom MD simulations to systematically study the transport behavior of lanthanide ions in Å-scale 2D MoS2-COOH channels. We examine the ion entering and permeation rate, as well as water flux, dehydration, and binding mode, and we also investigate the effect of interlayer spacing and external pressure on their performance. The simulation results reveal that altering the interlayer spacing impacts both kinetic and thermodynamic factors. These changes affect the dehydration degree and binding modes of lanthanide ions within the 2D channels, thereby influencing their permeability. Variations in pressure primarily affect kinetic factors by accelerating ion movement, with minimal impact on dehydration degree and binding strength. This aligns with previous findings, suggesting that ion entry and permeability are primarily dependent on pore size rather than pressure [Citation32–34]. Importantly, our work unveils the critical role of dehydration and binding modes in determining the transport trends of lanthanide ions in confinement and points to the great potential for designing confinement dimension and chemical functionalization for REE separation via transport.
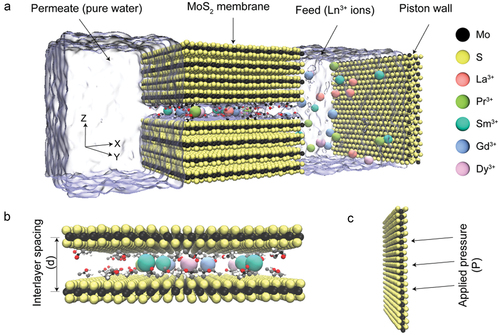
Figure 1. (a) Simulation modeling of the transport of a mixture of five lanthanide ions through 2D MoS2-COOH channels, incorporating six additional MoS2 sheets as a support membrane, with water depicted in transparent ice blue. The lanthanide ions are shown, omitting Cl− anions for clarity, alongside two MoS2 sheets serving as piston walls. (b) Depiction of the MoS2-COOH channel structure explored in this study, featuring lanthanide ions confined within the membrane. The interlayer spacing is defined as the distance between Mo atoms from adjacent layers. (c) Illustration of pressure applied to the piston walls.
Methods
We conducted all-atom molecular dynamics (MD) simulations to investigate the transport of mixed REE (La3+, Pr3+, Sm3+, Gd3+, Dy3+) cations in MoS2-COOH membranes. The system comprised an ion-filled feed region, a multi-layered MoS2 membrane functionalized with acetate groups serving as the ion channel, and a pure water permeate region (). Our simulation setup also included a bilayer MoS2 sheet acting as a rigid piston wall to apply external pressure (which mimics the effect of osmotic pressure but could also represent a system where pressure is added externally). The multi-layered MoS2 membrane was modeled as two MoS2 layers extracted from bulk crystals (2 H-MoS2) and modified with acetate groups, maintaining a 12.5 Å interlayer spacing (Mo–Mo distance), consistent with experimental data [Citation24]. We constructed a rectangular box with dimensions 170 Å × 57.2 Å × 54.0 Å. The OPC3 water model was used, and the setup was visualized using Visual Molecular Dynamics (VMD) [Citation35]. Parameters for the MoS2 membrane were adopted from Heinz [Citation36], while -COOH functional groups were modeled using the general AMBER force field (GAFF) [Citation37]. Nonbonded interactions for monovalent/trivalent ions were described using the 12-6-4 Lennard-Jones-type model [Citation38,Citation39].
Given the experimentally observed pH (~4.7) close to the pKa of acetic acid (~4.76), we deprotonated half of the MoS2-COOH groups, resulting in a functionalization degree of ~25% per MoS2. Twelve lanthanide ions were introduced into the membrane to neutralize its charge, with ion positions based on configurations from our previous work [Citation24]. All the MD simulations were performed using the OpenMM package [Citation40], examining the effects of interlayer spacing (12.5 Å, 13.125 Å, and 13.5 Å) and external pressure (21.6 MPa, 32.4 MPa, and 43.2 MPa) on the REE separation performance of the membrane. It is important to note that, to accelerate the MD simulations and collect sufficient statistics at the nanosecond scale, the applied external pressures are significantly higher than the experimentally measured osmotic pressure (~0.5 MPa).
Each system underwent a 10,000-step energy minimization, followed by a 10 ns NVT ensemble equilibration at 300 K using Langevin dynamics. The piston wall and membrane were positionally restrained, while ions were restrained so that they could not pass through the channel using a flat-well potential [Citation33]. Subsequently, ion constraints were removed, and non-equilibrium production simulations were performed in the NVT ensemble, applying external pressures to the rigid piston wall to simulate ion permeation through the MoS2-COOH membrane. Temperature was maintained at 300 K using a Langevin thermostat with a friction coefficient of 1 ps−1. Electrostatic interactions were calculated using the particle mesh Ewald method with a short-range cutoff of 1.0 nm [Citation41]. Periodic boundary conditions were applied, and simulations were run with a 2 fs timestep, recording atomic coordinates every 20 ps.
Results and Discussion
Volcano-shape ion transport
Our recent study demonstrated that lanthanide ions exhibit a volcano-shaped transport performance within 2D MoS2-COOH channels [Citation24]. All-atom molecular dynamics simulations revealed this result when the interlayer spacing (Mo–Mo distance) was maintained at 12.5 Å, in accordance with experimental conditions, and a specific pressure of 32.4 MPa was applied. The normalized and real fluxes of lanthanide ions entering and traversing the membrane exhibited a volcano-shaped profile, peaking at Sm3+, as shown in and Figure S1, respectively. Interestingly, for most lanthanide ions (except Sm3+), the flux entering the membrane is larger than that traversing it. Therefore, we believe that the rate-determining step here are the ions traversing the membrane, which is also supported by Zhou et al [Citation42].
Figure 2. MD simulations with an interlayer spacing of 12.5 Å and an applied pressure of 32.4 MPa. (a) Normalized fluxes of lanthanide ions into the MoS2-COOH channel and into the permeate, respectively. Flux calculations are based on linear fits to the time evolution of ion entry, focusing on periods of maximum rate increase. (b) Distributions of various lanthanide ions within the channel relative to the distance from the center of the channel in the z-axis direction. (c) Dehydration numbers for lanthanide ions during entry into the channel (black curve) and while all ions are inside the membrane with no pressure applied to the piston wall (static condition, red curve). (d) Average changes in the coordination number (CN) in the first solvation shell of lanthanide ions over time, with corresponding integrated CN distribution profiles. (e) Averaged distributions of dwell times for lanthanide ions near the 3.5 Å region of the –COOH/–COO– groups. (f) Average binding mode distributions of lanthanide ions with the –COOH/–COO– groups (3.5 Å cutoff). The insets show the schematic definition of binding to one or two carboxylates. Data represent averaged values derived from three independent simulations, with error bars showing standard errors of the means.
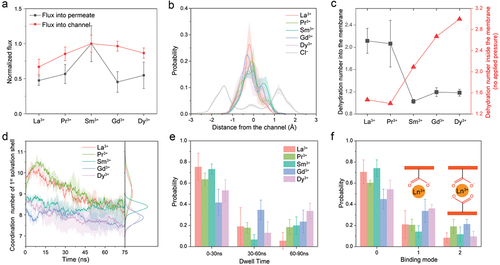
Ion pairing, dehydration, and ion–membrane interactions are recognized as critical factors influencing the mechanism of ion transport [Citation26–28,Citation43,Citation44]. To assess the effect of ion pairing, we performed calculations to determine the distribution of lanthanide ions and Cl− anions within the 2D channels (). Our observations revealed that the pathways of lanthanide ions and Cl− anions through the channel do not overlap; specifically, lanthanide ions tend to traverse through the center of the channel, whereas Cl− anions prefer pathways closer to the channel surface. This suggests that ion pairing does not significantly contribute to the distinct volcano-shaped transport observed, which aligns with our previous findings, indicating that Cl− does not significantly participate in the transport of lanthanide ions [Citation24]. We also calculated radial distribution functions (RDFs) to examine ion interactions with acetate group atoms (Figure S2). The RDF results show that Ln3+ cations are closer to electronegative oxygen atoms, and Cl− anions are closer to electropositive hydrogen atoms. This aligns with binding mode calculations (), showing that the Ln3+ ions are captured by one or two – COOH/–COO− groups, positioning them in the middle of the channel.
We then explored the impacts of dehydration on lanthanide ions as they enter the channel, as shown in . Notably, La3+ and Pr3+ cations exhibit the highest degree of dehydration upon entering the channel, losing approximately 2 water molecules, whereas Sm3+, Gd3+, and Dy3+ cations dehydrate by 1–1.2 water molecules. Coupled with the fact that hydration-free energy (HFE) monotonically increases with atomic number (Table S1) [Citation24], this explains the contribution to the volcano-shaped profile from lanthanide ions entering the channel.
To further elucidate the impact on the dehydration effect during permeation, we examined the variations in coordination number (CN) within the first solvation shell of the lanthanide ions and their corresponding Gaussian distributions (). The initial state refers to all ions being in the feed, while the final state is when the piston wall reaches the membrane. The Gaussian fit parameters, detailed in (referring to the results for d = 12.5 Å, p = 32.4 MPa), indicate that Sm3+ has the narrowest gaussian distribution (σ = 0.2), while La3+ and Pr3+ exhibit a broader CN distribution (σ = 0.6–0.7). This implies that Sm3+ maintains a more stable coordination number during permeation through the channel, whereas La3+ and Pr3+ more readily gain or lose water molecules (~2 water molecules) during this process. Moreover, according to Merz et al. [Citation38], the CN of the first solvation shell for the five lanthanide ions under investigation in bulk water is 9, except for Pr3+, which has a CN of 9.4. Therefore, compared to the bulk solution, Sm3+ loses approximately 0.6 water molecules during permeation, while Gd3+ and Dy3+ each lose around 1.1 water molecules. These variations in water molecule loss correlate with the observed differences in permeability, where Sm3+ demonstrates the highest permeability among the lanthanide ions studied. Note that the methods used to calculate the dehydration number and the changes in the CN are different. The former is determined by calculating the average dehydration number of each lanthanide ion upon entering the membrane, while the latter involves computing the CN changes of all lanthanide ions over time. Consequently, the dehydration numbers obtained from these two calculations differ, but the overall trends remain consistent.
We analyzed the radial distribution function (RDF) between Ln3+ ions and water’s oxygen atoms, comparing the solvation shells in the bulk (feed or permeate) and membrane (Figure S3). The first solvation shell’s peak positions are consistent, with slight differences (~0.1 Å) in the second shell. The main difference is in peak heights, indicating coordination numbers. These findings suggest that hydrated diameters and shapes remain largely unchanged when passing through the channel, with the main variation in dehydration numbers. This is consistent with the subtle physicochemical differences among the lanthanide ions, where dehydration numbers differ by only 1–2 water molecules.
Table 1. Gaussian fit parameters (mean value μ and standard deviation σ) for the integrated CN distribution profiles for the five lanthanides with different interlayer spacing (d) and applied pressure (P).
To distinguish whether dehydration is driven by thermodynamic or kinetic factors, we further examined the dehydration behavior of lanthanide ions inside the membrane under static conditions, i.e. with no pressure applied to the piston wall, as depicted in . Under these static conditions, the dehydration trend is determined by the binding affinity, with the largest dehydration being associated with the heaviest lanthanide that has the strongest Lewis acidity and prefers to bind to acetate rather than water. We also see that Sm3+ no longer exhibits the least dehydration. The stark difference between the two curves in shows that the dehydration pattern of lanthanide ions during permeation is closely linked to kinetic factors rather than thermodynamics.
Our previous potential of mean force (PMF) calculations indicate that the binding affinity between Ln3+ ions and the membrane increases with atomic number. However, these results should be viewed only as a reference for evaluating the binding affinity between REE cations and – COOH/–COO− groups, not as an accurate representation of actual permeation phenomena for REE mixtures [Citation24]. Additionally, we investigated the interactions between ions and the membrane during permeation by evaluating the probability distribution of the residence time of lanthanide ions near – COOH/–COO− groups (within 3.5 Å) and the binding modes of lanthanide ions with – COOH/–COO− groups, as illustrated in . Both analyses indicate that Sm3+ exhibits a shorter residence time near the – COOH/–COO− groups and a lower probability of binding with these groups. This observation aligns with the finding that Sm3+ undergoes minimal coordination number change during permeation, supporting the notion that dehydration-enhanced ion–pore interactions dominate ion transport and selectivity in nanochannels, as reported by Qu et al. [Citation45] Similarly, La3+ also shows a shorter residence time and a lower binding probability with – COOH/–COO− groups. However, due to the larger change in CN experienced by La3+ during entry and permeation through the channel, its permeation flux is lower than that of Sm3+. Therefore, the transport and selectivity of lanthanide ions through MoS2-COOH channels is governed by dehydration and ion–membrane interactions, with a correlation between the two effects.
Impact of interlayer spacing on ion and water transport
The interlayer spacing within MoS2–COOH channels, determined by the binding configuration between – COOH/–COO− groups and Ln3+ ions as well as the arrangement of surrounding water molecules, is influenced by both dehydration and ion–membrane interactions. To assess the impact of interlayer spacing on the performance of ion transport, we performed non-equilibrium all-atom MD simulations with adjusted Mo–Mo distances of 13.125 Å and 13.5 Å, representing increases of 5% and 8%, respectively, from the baseline 12.5 Å specified in our original work (as determined by XRD measurements) [Citation24]. All other simulation parameters, including the functional group, density, and the applied pressure, were maintained constant. The normalized and real fluxes of lanthanide ions entering and traversing the membrane are depicted in and Figure S1, respectively.
Figure 3. Transport properties for REE cations at various interlayer spacings under an applied pressure of 32.4 MPa. (a) Normalized fluxes of lanthanide ions into the MoS2-COOH channel and into the permeate at different interlayer spacings, where the flux of ions into the membrane was treated as a negative number. (b) Flux of water molecules into the permeate at varying interlayer spacings. (c) Water density relative to the distance from the channel in the x-axis direction for MoS2-COOH channels with varying interlayer spacing under pressure. (d) Dehydration numbers for lanthanide ions during their entry into the channel for different interlayer spacings. (e) Average changes in the coordination number (CN) within the first solvation shell of lanthanide ions over time for MoS2-COOH channels with an interlayer spacing of 13.5 Å, including corresponding integrated CN distribution profiles. (f) Average binding mode distributions of lanthanide ions with the –COOH/–COO– groups in MoS2-COOH channels at an interlayer spacing of 13.5 Å. Data represent averaged values derived from three independent simulations, with error bars showing standard errors of the means. Note that the Pr3+ bar is essentially zero.
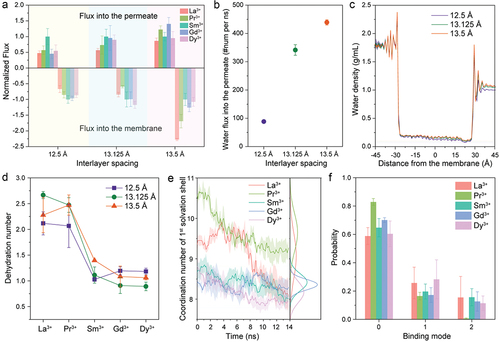
Our results indicate that increasing the interlayer spacing in the MoS2–COOH channels leads to deviations from the volcano-shaped flux profile shown in . When the interlayer spacing is increased by 5% to 13.125 Å, the most notable change in the shape of the ion distribution refers to the flux of lanthanide ions entering the membrane, and then for 13.5 Å there are larger changes in the shape of the distribution and in the magnitude of the flux. These deviations underscore the sensitivity of ion transport to structural modifications, highlighting the potential to alter peak transport trends of Ln3+ ions by adjusting interlayer spacing and functional group modifications.
We further computed the water flux by fitting the slope of the filtered water molecules versus the simulation time curve. Under an external pressure of 32.4 MPa, the water flux through MoS2-COOH channels with interlayer spacings of 12.5 Å, 13.125 Å, and 13.5 Å is 88.45, 341.44, and 439.11 molecules/ns, respectively. These results suggest a strong correlation between water flux and interlayer spacing, with the fractional increase in water flux being significantly larger than the fractional increase in interlayer spacing. Additionally, the water density distribution during permeation follows a distinct pattern (). At the entrance, the water density is higher due to higher applied pressure on the feed side of the membrane [Citation34]. Within the channel, the water density decreases to 0.1–0.2 g/ml, indicating a significant difference in ion solvation compared to bulk water. Finally, the water density is approximately 1 g/ml after exiting the channel, as expected for bulk water with no applied pressure. Here, the feed inlet has a higher water density than the permeation side, mainly due to the applied pressure. Under static conditions (no pressure applied), the water density on both sides of the membrane (away from the inlet and outlet) is close to 1 g/ml (Figure S5). shows that membranes with different interlayer spacings exhibit similar density distributions, with a slight increase in water density with increased spacing.
To explain the changes in fluxes into the membrane, we examined the dehydration numbers for lanthanide ions entering the channel, as shown in . When the interlayer spacing of MoS2-COOH channels is increased from 12.5 Å to 13.125 Å, La3+ and Pr3+ cations exhibit a higher degree of dehydration upon entering the channel, with an increase of 0.5 dehydrated water molecules. This appears to contradict the common understanding that increasing interlayer spacing should reduce ion dehydration numbers. We attribute this to the increased water density at the membrane entrance (), which facilitates the loss or gain of water molecules by the lighter lanthanide ions (due to their lower hydration-free energy). This results in a slight increase in the dehydration numbers of La3+ and Pr3+ ions. Nevertheless, the overall CN distribution trend () shows that larger interlayer spacing generally leads to reduced water loss for lanthanide ions. The dehydration of Sm3+ ions remains relatively unchanged, while the heavier lanthanide ions, Gd3+ and Dy3+, experience a decrease of 0.3 dehydrated water molecules. This results in the flux of various lanthanide ions entering the membrane becoming more uniform, except for Pr3+, which likely faces a higher dehydration energy barrier to remove additional 0.5 water molecules. However, the transport of lanthanide ions into the permeate remains a volcano-shaped profile, which suggests that the 5% increase in interlayer spacing affects the dynamics of ions entering the membrane more than those traversing the membrane.
Interestingly, when the interlayer spacing is further increased to 13.5 Å, the dehydration number for Sm3+ upon entering the channel increases from 1 (at 12.5 Å) to 1.4, whereas the dehydration numbers for Gd3+ and Dy3+ decrease from 1.2 (at 12.5 Å) to 1.07. Consequently, the entry rate of Sm3+ decreases, while the entry rates of Gd3+ and Dy3+ increase. Due to the significantly accelerated water flux, La3+ and Pr3+ exhibit higher flux into the membrane despite greater dehydration, consistent with the HFE trend. Additionally, the volcano-shaped distribution of lanthanide ions permeating through the channel disappears, with Pr3+ and Gd3+ becoming the peak positions. This can be explained by the CN distributions. According to and , Gd3+ has the narrowest distribution, losing 0.6 water molecules during permeation. In contrast, Pr3+ exhibits a broader distribution, with dehydration occurring mainly at the beginning of the simulation. The final coordination number stabilizes at 9.4, matching its coordination number in the bulk solution. This indicates that Pr3+ undergoes minimal dehydration during membrane permeation.
To further validate this, we investigated the binding configuration between lanthanide ions and – COOH/–COO− groups in the MoS2-COOH channel with an interlayer spacing of 13.5 Å, as shown in . We found that the probability of Pr3+ cations binding with – COOH/–COO− groups is only about 20%, with a single binding mode predominating. Overall, our results suggest that while La3+ has a much larger flux into the channel, Pr3+ results in a much higher permeation rate compared to La3+ due to less dehydration and weaker binding affinity to the – COOH/–COO− groups. Furthermore, our study on interlayer spacing demonstrates that altering the spacing simultaneously affects the thermodynamic and kinetic properties of ion transport, thereby influencing their performance in entering and traversing the membrane.
Figure 4. REE transport results for different pressures with an interlayer spacing of 12.5 Å. (a) Averaged fluxes of lanthanide ions into the MoS2-COOH channel and into the permeate under different pressures, where the flux of ions into the membrane was treated as a negative number. (b) Flux of water molecules into the permeate as a function of the applied pressure. (c) Water density relative to the distance from the channel in the x-axis direction for MoS2-COOH channels as a function of applied pressure. (d) Average changes in the coordination number (CN) within the first solvation shell of lanthanide ions over time for MoS2-COOH channels under an applied pressure of 43.2 MPa, including corresponding integrated CN distribution profiles. Data represent averaged values derived from three independent simulations, with error bars showing standard errors of the means.
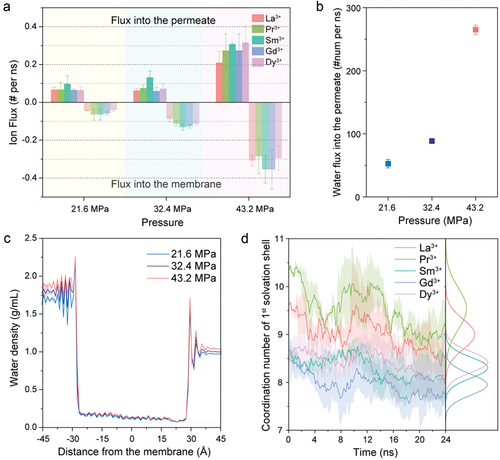
Impact of applied pressure on Ion and water transport
To further differentiate the effects of thermodynamic and kinetic factors on ion transport, we investigated the effect of varying external pressure on the results. Besides the previously applied external pressure of 31.2 MPa, we also introduced pressures of 21.6 MPa and 43.2 MPa. The average fluxes of lanthanide ions entering and traversing the membrane under these different external pressures are depicted in .
Surprisingly, the volcano-shaped trend remains largely unchanged under all three pressures, with Sm3+ still at the peak, while the fluxes for Gd3+ and Dy3+ exhibit slight fluctuations. This consistency with previous findings indicates that the entry and permeability of ions is primarily dependent on the pore size rather than pressure [Citation32–34]. The water flux through MoS2-COOH channels shows a positive correlation with external pressure (), with values of 52.61, 88.45, and 265.08 molecules/ns for external pressures of 21.6 MPa, 31.2 MPa, and 43.2 MPa, respectively. Indeed, from 21.6 MPa to 43.2 MPa, the water flux increased over fivefold, while the ion flux only increased by a factor of three to four. This suggests that ion mobility has important diffusive components rather than being solely governed by the applied pressure (hydrodynamics), consistent with the competing contributions of diffusion and advection in the extended Nernst−Planck equation [Citation46,Citation47]. The water density distribution for the three different pressures also exhibits a similar trend, with the water density slightly increasing as the applied external pressure increases.
To elucidate the impact on the dehydration effect during permeation, we examined the variations in CN within the first solvation shell of the lanthanide ions and their corresponding Gaussian distributions (). The Gaussian fit parameters, detailed in , indicate that Sm3+ and Dy3+ have the narrowest gaussian distribution (σ = 0.2) with mean CN value is 8.3 and 8.4, respectively. In contrast, though La3+ and Pr3+ have mean CN values around the CN value in bulk solution, they exhibit a broader CN distribution (σ = 0.3–0.4). This implies that Sm3+ and Dy3+ maintain a more stable coordination number while traversing through the channel, whereas La3+ and Pr3+ more readily gain or lose water molecules during this process, which is similar to the cases observed at 32.4 MPa () with the minor exception of Dy3+.
Conclusion
In summary, we have used molecular dynamics calculations to systematically study the transport behavior of lanthanide ions in Å-scale 2D MoS2-COOH channels in an aqueous environment. We explained the experimentally observed volcano-shaped trend in lanthanide ion transport through dehydration and ion–membrane interactions. We have also analyzed the effects of interlayer spacing and external pressure on ion transport performance under 2D confinement. The simulation results indicate that altering the interlayer spacing affects both kinetic and thermodynamic factors, altering the dehydration degree and binding strength of lanthanide ions within the 2D channels and thus influencing their transport trends. This implies the potential for achieving rare earth element separations in confinement through precise functionalization. Pressure variations primarily impact kinetic factors by changing the speed of ions moving in the channels. Although pressure has little effect on the dehydration degree and binding strength, it reduces the thermodynamic contribution, thereby decreasing the separation efficiency of lanthanide elements. An important conclusion from this work is that even though the pressures used in our simulations are well above the osmotic pressures available in the experiments, the volcano behavior of the transport distribution is not dependent on pressure, which means that our approach to simulating the experiments is justified. In addition, our simulations suggest that the transport trends of lanthanide ions can be predicted by calculating dehydration and binding properties using molecular dynamics. These findings will facilitate the reverse engineering of materials to achieve selective separation of lanthanide elements.
Associated content
Supporting Information. Figures S1-S5 are included in the supporting information.
SI anonymous.docx
Download MS Word (868.7 KB)Acknowledgments
This work is supported by the Department of Energy, Office of Basic Energy Science, under grant DE-SC0022231.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19475411.2024.2387926
Additional information
Funding
References
- Balaram V. Rare earth elements: a review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci Front. 2019;10(4):1285–1303. doi: 10.1016/j.gsf.2018.12.005
- Cheisson T, Schelter EJ. Rare earth elements: Mendeleev’s bane, modern marvels. Science. 2019;363(6426):489–493. doi: 10.1126/science.aau7628
- Sholl DS, Lively RP. Seven chemical separations to change the world. Nature. 2016;532(7600):435–437. doi: 10.1038/532435a
- Opare EO, Struhs E, Mirkouei A. A comparative state-of-technology review and future directions for rare earth element separation. Renewable And Sustain Energy Rev. 2021;143:110917. doi: 10.1016/j.rser.2021.110917
- Liu T, Chen J. Extraction and separation of heavy rare earth elements: a review. Separation And Purif Technol. 2021;276:119263. doi: 10.1016/j.seppur.2021.119263
- Doyle DA, Cabral JM, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69
- Kang Y, Xia Y, Wang H, et al. 2D laminar membranes for selective water and ion transport. Adv Funct Mater. 2019;29(29):1902014. doi: 10.1002/adfm.201902014
- Cheng L, Liu G, Zhao J, et al. Two-dimensional-material membranes: manipulating the transport pathway for molecular separation. Acc Mater Res. 2021;2(2):114–128. doi: 10.1021/accountsmr.0c00092
- Kim S, Wang H, Lee YM. 2D nanosheets and their composite membranes for water, gas, and ion separation. Angew Chem. 2019;131(49):17674–17689. doi: 10.1002/ange.201814349
- Wang S, Yang L, He G, et al. Two-dimensional nanochannel membranes for molecular and ionic separations. Chem Soc Rev. 2020;49(4):1071–1089. doi: 10.1039/C9CS00751B
- Karahan HE, Goh K, Zhang C, et al. Mxene materials for designing advanced separation membranes. Adv Mater. 2020;32(29):1906697. doi: 10.1002/adma.201906697
- Cao T-X, Xie R, Ju X-J, et al. Biomimetic two-dimensional composited membranes for ion separation and desalination. Ind Eng Chem Res. 2023;62(37):14772–14790. doi: 10.1021/acs.iecr.3c02048
- Chen Y, Zhu Z, Tian Y, et al. Rational ion transport management mediated through membrane structures. Exploration. 2021;1(2):20210101. doi: 10.1002/EXP.20210101
- Ding Z, Gu T, Zhang R, et al. Plasma‐oxidized 2D MXenes subnanochannel membrane for high‐performance osmotic energy conversion. Carbon Energy. 2024;2024:e509. doi:10.1002/cey2.509
- Liu Z, Liu C, Ni A, et al. Tortuosity regulation of two-dimensional nanofluidic films for water evaporation-induced electricity generation. Nano Res. 1722;17(7):6192–6202. doi: 10.1007/s12274-024-6642-1
- Huang L, Ding L, Caro J, et al. Mxene‐based membranes for drinking water production. Angew Chem Int Ed. 2023;62(52):e202311138. doi: 10.1002/anie.202311138
- Huang L, Chen S, Ding L, et al. Lateral-size-dependent ion transport behavior of the MXene membrane facilitates efficient ion Sieving. ACS Appl Nano Mater. 2024;7(8):9482–9489. doi: 10.1021/acsanm.4c00919
- Huang L, Wu H, Ding L, et al. Shearing liquid‐crystalline MXene into lamellar membranes with super‐aligned nanochannels for Ion Sieving. Angew Chem Int Ed. 2024;63(6):e202314638. doi: 10.1002/anie.202314638
- Cheng C, Jiang G, Garvey CJ, et al. Ion transport in complex layered graphene-based membranes with tuneable interlayer spacing. Sci Adv. 2016;2(2):e1501272. doi: 10.1126/sciadv.1501272
- Bi K, Jiang X, Sun H, et al. Tuning the water interlayer spacer of microwave-synthesized holey graphene films towards high performance supercapacitor application. 2D Mater. 2024;11(3):035021. doi: 10.1088/2053-1583/ad42ae
- Hirunpinyopas W, Prestat E, Worrall SD, et al. Desalination and nanofiltration through functionalized laminar MoS2 membranes. ACS Nano. 2017;11(11):11082–11090. doi: 10.1021/acsnano.7b05124
- Hoenig E, Strong SE, Wang M, et al. Controlling the structure of MoS2 membranes via covalent functionalization with molecular spacers. Nano Lett. 2020;20(11):7844–7851. doi: 10.1021/acs.nanolett.0c02114
- Ries L, Petit E, Michel T, et al. Enhanced sieving from exfoliated MoS2 membranes via covalent functionalization. Nat Mater. 2019;18(10):1112–1117. doi: 10.1038/s41563-019-0464-7
- Wang M, Xiong Q, Wang M, et al. Lanthanide transport in angstrom-scale MoS2-based two-dimensional channels. Sci Adv. 2024;10(11):eadh1330. doi: 10.1126/sciadv.adh1330
- Xiong Q, Lee O-S, Mirkin CA, et al. Ethanol-induced condensation and decondensation in DNA-linked nanoparticles: a nucleosome-like model for the condensed state. J Am Chem Soc. 2022;145(1):706–716. doi: 10.1021/jacs.2c11834
- Wu Z-Q, Li C-Y, Ding X-L, et al. Synergistic effect of electrostatic interaction and ionic dehydration on asymmetric ion transport in nanochannel/ion channel composite membrane. J Phys Chem Lett. 2022;13(23):5267–5274. doi: 10.1021/acs.jpclett.2c01166
- Mo R-J, Chen S, Huang L-Q, et al. Regulating ion affinity and dehydration of metal-organic framework sub-nanochannels for high-precision ion separation. Nat Commun. 2024;15(1):2145. doi: 10.1038/s41467-024-46378-6
- Epsztein R, DuChanois RM, Ritt CL, et al. Towards single-species selectivity of membranes with subnanometre pores. Nat Nanotechnol. 2020;15(6):426–436. doi: 10.1038/s41565-020-0713-6
- Liu Y, Xia Z, Wang Y, et al. Montmorillonite membranes with tunable ion transport by controlling interlayer spacing. ACS Appl Mater Interfaces. 2023;15(49):57144–57152. doi: 10.1021/acsami.3c13678
- Xia Z, Chen W, Shevate R, et al. Tunable ion transport with freestanding vermiculite membranes. ACS Nano. 2022;16(11):18266–18273. doi: 10.1021/acsnano.2c05954
- Chen L, Shi G, Shen J, et al. Ion sieving in graphene oxide membranes via cationic control of interlayer spacing. Nature. 2017;550(7676):380–383. doi: 10.1038/nature24044
- Abal JPK, Dillenburg RF, Kohler MH, et al. Molecular dynamics simulations of water anchored in multilayered nanoporous MoS2 membranes: implications for desalination. ACS Appl Nano Mater. 2021;4(10):10467–10476. doi: 10.1021/acsanm.1c01982
- Heiranian M, Farimani AB, Aluru NR. Water desalination with a single-layer MoS2 nanopore. Nat Commun. 2015;6(1):8616. doi: 10.1038/ncomms9616
- Meidani K, Cao Z, Barati Farimani A. Titanium carbide MXene for water desalination: a molecular dynamics study. ACS Appl Nano Mater. 2021;4(6):6145–6151. doi: 10.1021/acsanm.1c00944
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5
- Liu J, Zeng J, Zhu C, et al. Interpretable molecular models for molybdenum disulfide and insight into selective peptide recognition. Chem Sci. 2020;11(33):8708–8722. doi: 10.1039/D0SC01443E
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035
- Li Z, Song LF, Li P, et al. Parametrization of trivalent and tetravalent metal ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB water models. J Chem Theory Comput. 2021;17(4):2342–2354. doi: 10.1021/acs.jctc.0c01320
- Sengupta A, Li Z, Song LF, et al. Parameterization of monovalent ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB water models. J Chem Inf Model. 2021;61(2):869–880. doi: 10.1021/acs.jcim.0c01390
- Eastman P, Swails J, Chodera JD, et al. OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol. 2017;13(7):e1005659. doi: 10.1371/journal.pcbi.1005659
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. doi: 10.1063/1.464397
- Zhou X, Wang Z, Epsztein R, et al. Intrapore energy barriers govern ion transport and selectivity of desalination membranes. Sci Adv. 2020;6(48), eabd9045. doi: 10.1126/sciadv.abd9045
- Richards LA, Schäfer AI, Richards BS, et al. The importance of dehydration in determining ion transport in narrow pores. Small. 2012;8(11):1701–1709. doi: 10.1002/smll.201102056
- Wang K, Wang X, Januszewski B, et al. Tailored design of nanofiltration membranes for water treatment based on synthesis–property–performance relationships. Chem Soc Rev. 2022;51(2):672–719. doi: 10.1039/D0CS01599G
- Lu C, Hu C, Chen Z, et al. Dehydration-enhanced ion-pore interactions dominate anion transport and selectivity in nanochannels. Sci Adv. 2023;9(27):eadf8412. doi: 10.1126/sciadv.adf8412
- Oren Y, Biesheuvel P. Theory of ion and water transport in reverse-osmosis membranes. Phys Rev Appl. 2018;9(2):024034. doi: 10.1103/PhysRevApplied.9.024034
- Wang L, Cao T, Dykstra JE, et al. Salt and water transport in reverse osmosis membranes: beyond the solution-diffusion model. Environ Sci Technol. 2021;55(24):16665–16675. doi: 10.1021/acs.est.1c05649