
Abstract
Idiopathic inflammatory bowel disease (IBD) is a common cause of chronic gastrointestinal (GI) disease in dogs. The combination of an underlying host genetic susceptibility, an intestinal dysbiosis, and dietary/environmental factors are suspected as main contributing factors in the pathogenesis of canine IBD. However, actual mechanisms of the host-microbe interactions remain elusive. The aim of this study was to compare the fecal microbiota and serum metabolite profiles between healthy dogs (n = 10) and dogs with IBD before and after 3 weeks of medical therapy (n = 12). Fecal microbiota and metabolite profiles were characterized by 454-pyrosequencing of 16 S rRNA genes and by an untargeted metabolomics approach, respectively. Significantly lower bacterial diversity and distinct microbial communities were observed in dogs with IBD compared to the healthy control dogs. While Gammaproteobacteria were overrepresented, Erysipelotrichia, Clostridia, and Bacteroidia were underrepresented in dogs with IBD. The functional gene content was predicted from the 16 S rRNA gene data using PICRUSt, and revealed overrepresented bacterial secretion system and transcription factors, and underrepresented amino acid metabolism in dogs with IBD. The serum metabolites 3-hydroxybutyrate, hexuronic acid, ribose, and gluconic acid lactone were significantly more abundant in dogs with IBD. Although a clinical improvement was observed after medical therapy in all dogs with IBD, this was not accompanied by significant changes in the fecal microbiota or in serum metabolite profiles. These results suggest the presence of oxidative stress and a functional alteration of the GI microbiota in dogs with IBD, which persisted even in the face of a clinical response to medical therapy.
Abbreviations
| 16 S rRNA | = | 16 S ribosomal RNA |
| ANOSIM | = | analysis of similarities |
| CIBDAI | = | canine IBD activity index |
| FDR | = | false discovery rate |
| GI | = | gastrointestinal |
| GC-TOF/MS | = | gas chromatography coupled with time-of-flight mass spectrometry |
| IBD | = | idiopathic inflammatory bowel disease |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes |
| LEfSe | = | linear discriminant analysis (LDA) effect size |
| PCA | = | principal component analysis |
| PCoA | = | principal coordinates analysis |
| PICRUSt | = | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States |
| ROC | = | receiver operating characteristic |
Introduction
The close relationship between the gastrointestinal (GI) microbiota and the host has a crucial impact on the health status of an animal.Citation1 Intestinal dysbiosis, which describes an alteration of the GI microbiota, has been observed in human patients with chronic GI inflammationCitation2 but also in dogs.Citation3–6 Idiopathic inflammatory bowel disease (IBD) is a common cause of chronic GI disease in dogs. Similarly to human IBD, the combination of an underlying host genetic susceptibility, an intestinal dysbiosis, and dietary and/or environmental factors are suspected as main contributing factors in the pathogenesis of canine IBD.Citation7-9 Furthermore, similarly to humans, intestinal inflammation develops spontaneously in dogs living in natural home environments, and the most frequently used treatment modalities for these disorders include antibiotic therapy and immunosuppression.Citation10
Despite well documented evidence that the intestinal microbiota plays a role in the pathogenesis of canine IBD, the actual mechanisms of the host-microbe interactions remain elusive, but are believed to be mediated in part by microbial products (metabolites) derived from the GI microbiota and locally and/or systemically absorbed by the host.Citation11 Therefore, studies that assess functional aspects of the GI microbiota are needed. Metabolomics, the comprehensive study of small molecules present in biological samples, is an emerging method for better understanding of disease pathophysiology and host-microbe interactions, with the potential to develop novel diagnostic and/or treatment approaches.Citation12,13 Metabolomics studies have been reported in human patients with IBD as well as animal models of IBD, and provided new insights into disease pathogenesis.Citation14,15 Using mass spectrometry platforms, an untargeted metabolomics approach can identify hundreds of metabolites in biological samples simultaneously, and therefore provides a comprehensive functional overview of biochemical pathways that are being up- or down-regulated during different physiologic or pathophysiologic state.Citation16 In veterinary medicine, only few studies have utilized this approach in clinical patients,Citation17,18 and to our knowledge, no studies using an untargeted metabolomics approach have been reported in dogs with IBD. Therefore, we aimed to characterize the fecal microbiota and serum metabolite profiles in healthy dogs and compare those to dogs with IBD. We also investigated the effects of 3 weeks of medical intervention on these profiles. To investigate the global differences in GI microbial communities and metabolic profiles, we utilized 454-pyrosequencing of 16 S ribosomal RNA (16 S rRNA) genes and an untargeted metabolomics approach using gas chromatography coupled with time-of-flight mass spectrometry (GC-TOF/MS).
Results
The characteristics of the healthy control dogs (HC) and dogs with IBD (pre-treatment, IBD-PRE; post-treatment, IBD-POST) are summarized in . Significant differences in age and body weight were observed between HC and dogs with IBD. The canine IBD activity index (CIBDAI) was significantly decreased after 3 weeks of medical therapy ().
Table 1. Dog characteristics
Sequence analysis
The 454-pyrosequencing pipeline yielded 297,619 quality sequences with an average of 8,753 sequences per sample. To account for unequal sequencing depth across samples, the subsequent analysis was performed on a randomly selected subset of 2,900 sequences per sample. The pre-treatment sample from one dog with IBD was excluded due to insufficient sequencing depth. A total of 11 phyla and138 genera were identified. The sequencing data have been deposited into the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under accession number SRP040310.
Fecal microbial communities in healthy control dogs and dogs with IBD
To characterize the global differences in fecal microbial communities between groups, a principal coordinates analysis (PCoA) was performed on unweighted UniFrac distances. The PCoA plots showed significant separation between the fecal samples from the HC and IBD-PRE groups (analysis of similarity [ANOSIM]; p = 0.01) (). The Shannon index and the Chao 1 metric were significantly decreased in the IBD-PRE group compared to the HC group (p = 0.014, 0.0146, respectively; ). The number of observed species was also decreased in the IBD-PRE group but did not reach statistical significance (p = 0.0656; ). Linear discriminant analysis (LDA) effect size (LEfSe) was utilized to determine differentially abundant bacterial taxa between the animal groups. A total of 22 bacterial groups were differentially expressed (α = 0.01, LDA score > 3.0) between the HC and IBD-PRE groups. Gammaproteobacteria were overrepresented, while Erysipelotrichia, Clostridia, and Bacteroidia were underrepresented in dogs with IBD-PRE (). Multivariate Analysis by Linear Models (MaAsLin) was utilized to identify associations of microbial abundances with clinical metadata (age, weight, gender), which may be confounding factors. None of these variables were significantly associated with microbial abundance.
Figure 1. Bacterial diversity measures. β diversity: (A) principal coordinates analysis (PCoA) of unweighted UniFrac distances of 16S rRNA genes. PCoA plots along with Principal Coordinates (PC) 1 and PC 3. Analysis of similarity (ANOSIM) revealed clustering between healthy control dogs and dogs with IBD (p = 0.01), but not between IBD pre-treatment and IBD post-treatment groups (p = 0.34). Alpha diversity measures: (B) rarefaction analysis (number of observed species) of 16 S rRNA gene sequences. Lines represent the mean of each group, while the error bars represent the standard deviations. (C) Comparisons of α diversity. Samples from dogs with IBD post-treatment were divided into 2 groups based on antibiotic administration status during 3 weeks of medical intervention. Red lines represent the median for each measure. HC, healthy control dogs; IBD-PRE, dogs with IBD pre-treatment; IBD-POST_NON AB, dogs with IBD post-treatment that did not receive antibiotic; IBD-POST_AB, dogs with IBD post-treatment that received antibiotic *p < 0.05; and **P < 0.01
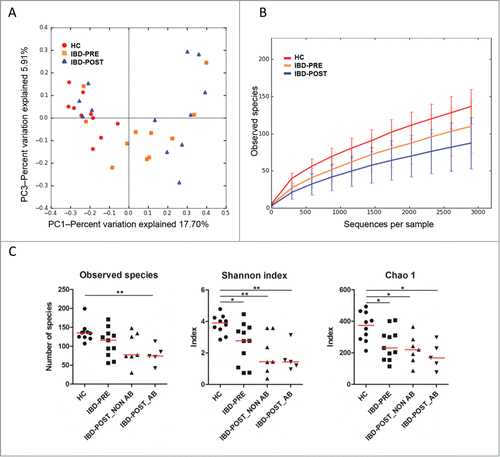
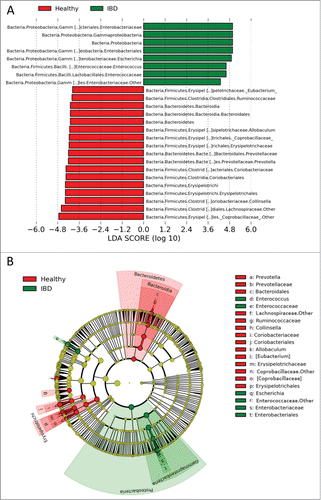
Figure 2. Linear discriminant analysis (LDA) effect size (LEfSe) of 454-pyrosequencing data sets based on 16 S rRNA gene sequences. (A) Histogram of the LDA scores computed for differentially abundant bacterial taxa between healthy control dogs and dogs with IBD (pre-treatment). (B) Taxonomic distribution of bacterial groups significant for IBD. A total of 22 differentially abundant bacterial taxa were detected (α = 0.01, LDA score > 3.0). Of those, 7 bacterial taxa were significantly overrepresented in pretreatment samples from dogs with IBD (green) and 15 bacterial taxa were overrepresented in samples from healthy control dogs (red).
qPCR analysis of fecal microbial communities
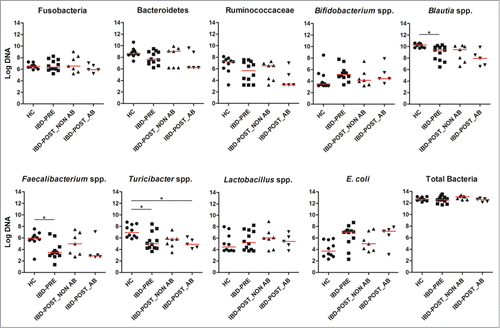
qPCR assays for selected bacterial groups were performed to confirm pyrosequencing results and/or to quantitate bacterial groups that are typically present at very low abundance or underrepresented in 16 S rRNA gene sequencing data based on the authors’ experience from previous studies (i.e., Bifidobacterium spp., Faecalibacterium spp).Citation19–21 The abundances of Blautia spp., Faecalibacterium spp, and Turicibacter spp. were significantly decreased in the IBD-PRE group (q = 0.0277, 0.0480, 0.0380, respectively) compared to those in the HC group. E. coli appeared to be increased in the IBD-PRE group, but this increase did not reach statistical significance when the p-value was adjusted by False Discovery Rate (FDR) of 5% (q = 0.0560; ).
Figure 3. The abundances of selected bacterial groups in healthy dogs and dogs with IBD (pre- and post-treatment) based on qPCR. Samples from dogs with IBD post-treatment were divided into 2 groups based on antibiotic administration status during the 3 weeks of medical intervention. Red lines represent the median of log DNA. HC, healthy control dogs; IBD-PRE, dogs with IBD pre-treatment; IBD-POST_NON AB, dogs with IBD post-treatment that did not receive antibiotic; IBD-POST_AB, dogs with IBD post-treatment that received antibiotic *q < 0.05
Predicted functional composition of fecal microbial communities
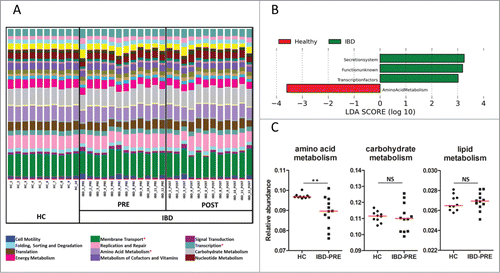
To investigate alterations in GI microbial function, an analysis for Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was applied to the 16 S rRNA gene sequencing data. The PICRUSt results () were then analyzed using LEfSe to identify microbial functions that were significantly different in their abundance between groups. A total of 4 differentially abundant bacterial functions were observed between the HC and IBD-PRE groups (α = 0.01, LDA score > 3.0). Of those, the functions of secretion system and transcription factors were overrepresented in the IBD-PRE group. In contrast, amino acid metabolism was underrepresented in the IBD-PRE group (). To investigate the alteration of some of the bacterial functions of major metabolic systems, a univariate analysis was applied on the PICRUSt data. The relative abundances of lipid, carbohydrate, and energy metabolisms were not significantly different between the HC and IBD-PRE groups ().
Figure 4. Predicted functional composition of metagenomes based on 16 S rRNA gene sequencing data. (A) Relative abundances of predicted functions (second level of the Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog (KO) hierarchy) based on phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) data set. Each stacked bar represents relative abundances of the predicted functions of each dog. (B) LEfSe based on the PICRUSt data set (third level of the KO hierarchy) revealed a total of 4 differentially enriched bacterial functions (enriched in healthy controls: amino acid metabolism, enriched in IBD dogs: secretion system, transcription factors, and a pathway with unknown function) between healthy control dogs and dogs with IBD (pre-treatment; α = 0.01, LDA score > 3.0). None of the bacterial functions were differentially expressed between IBD pre-treatment and IBD post-treatment. (C) Univariate analysis of major metabolisms. Red lines represent the median of relative abundance. HC, healthy control dogs; IBD-PRE, dogs with IBD pre-treatment; IBD-POST, dogs with IBD post-treatment; NS, no significance *metabolic functions that differed significantly between healthy control dogs and dogs with IBD **q < 0.01.
Effects of medical treatment on fecal microbial communities
We further evaluated the effects of 3 weeks of medical intervention on fecal microbial communities in dogs with IBD. All dogs received a standard therapy, which consisted of administration of an immunosuppressive drug (i.e., prednisone [1–2 mg/kg, q24 h, PO], prednisolone [1–2 mg/kg, q24 h, PO], budesonide [1–3 mg/kg, q24 h, PO], cyclosporine [3–5 mg/kg, q12 h or q24 h, PO], or a combination of these) and feeding of an elimination diet (i.e., antigen-restricted or protein hydrolysate diets). Five out of 12 IBD dogs received the antibiotic metronidazole (10 mg/kg, q12 h, PO) in addition to immunosuppressive drug therapy. All dogs responded to their therapies (i.e., GI signs resolved, CIBDAI improved) and none had diarrhea after medical intervention. Although all dogs showed clinical improvement after 3 weeks of therapy, no major differences in microbial communities were observed between the IBD-PRE and IBD-POST groups (ANOSIM, p = 0.34; ). In the IBD-POST group, the diversity indices further decreased compared to the IBD-PRE, but this decrease did not reach statistical significance (). The LEfSe analysis revealed that none of the bacterial taxa were differentially expressed between IBD-PRE and IBD-POST groups. These observations were confirmed by the qPCR assays, as none of the abundances of the evaluated bacterial groups were significantly different between IBD-PRE and IBD-POST (). Consequently, none of the predicted bacterial functions were differently expressed between IBD-PRE and IBD-POST. To address if the administration of metronidazole had a confounding effect on the GI microbiota in this study, the sequencing data within the IBD-POST group was compared between dogs that received antibiotic versus dogs that did not receive antibiotic. The PCoA plots showed no significant separation between the samples from dogs that received antibiotic and those that did not (ANOSIM, p = 0.51; ). Also, there were no significant differences in unweighted UniFrac distances between the pre- and post-treatment samples from dogs that received antibiotic and those that did not (p = 0.5273; ). The results of the qPCR assays were also statistically analyzed in the IBD-POST group based on antibiotic administration. Noteworthy trends were observed, suggesting that dogs that did not receive antibiotic in general had median abundances of Fusobacteria, Bacteroidetes, Ruminococcaceae, Bifidobacterium spp, Blautia spp., Faecalibacterium spp, and E. coli that were more similar to median abundances observed in the HC group (). This was most notable for Turicibacter spp., where dogs that had received antibiotic had significantly decreased abundances of this bacterial group compared to dogs of the HC group. MaAsLin was utilized to identify associations of microbial abundance with antibiotic treatment. No significant association between antibiotic treatment and microbial abundance was identified.
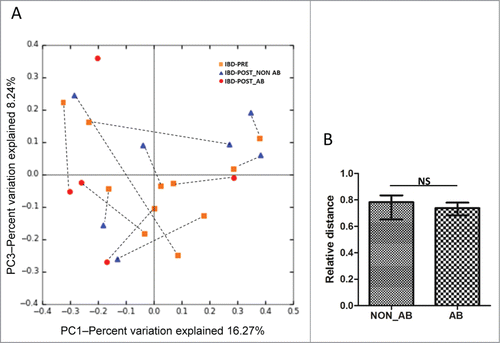
Figure 5. Effect of antibiotic administration on the fecal microbiota. (A) PCoA of unweighted UniFrac distances of 16 S rRNA genes from dogs with IBD pre-treatment and post-treatment. Dogs in the IBD post-treatment group were divided into 2 groups based on the antibiotic administration status during 3 weeks of medical intervention. ANOSIM revealed no clustering either between IBD-PRE and IBD-POST_AB, or IBD-PRE and IBD POST-NON_AB (ANOSIM p = 0.26, p = 0.51, respectively). Dashed lines connect the pre-treatment and the post-treatment samples from each dog. The pre-treatment sample from one dog with IBD was excluded due to insufficient sequencing depth. (B) Median unweighted UniFrac distance between pre- and post-treatment samples from dogs with and without antibiotic treatment. Whiskers represent interquartile ranges. IBD-PRE, dogs with IBD pre-treatment; IBD-POST_NON AB, dogs with IBD post-treatment that did not receive antibiotic; IBD-POST_AB, dogs with IBD post-treatment that received antibiotic; NS, no significance
Untargeted metabolomics analysis
A total of 359 metabolites were detected. Of those, 157 were identified metabolites, while 202 lacked full structural identification (unknown metabolites). These unknown metabolites are listed by their Binbase identification numbers (http://fiehnlab.ucdavis.edu/projects/binbase_setupx)Citation22 and the complete dataset is shown in Table S1.
Serum metabolite profiles in healthy control dogs and dogs with IBD
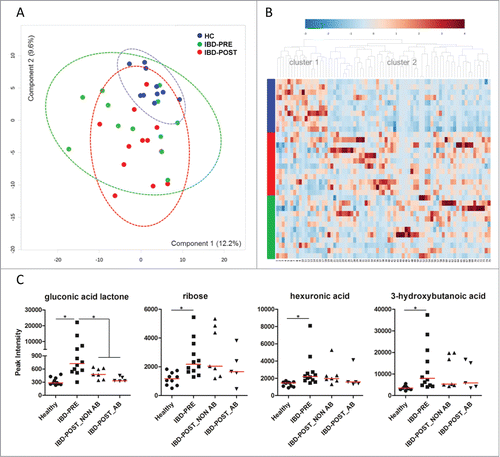
To characterize the global metabolic differences between groups, a principal component analysis (PCA) was applied. Serum metabolite profiles associated with the HC group were tightly clustered, whereas those in the IBD-PRE group were much more scattered (). There was considerable overlap in metabolite profiles observed between the 2 IBD groups. Similarly to the results of the PCA analysis, no distinct pattern was observed when the data were plotted by heatmap analysis (). However, 2 major groups of metabolites clustered in the dendrogram, with cluster 1 being more abundant in healthy animals and cluster 2 appearing to be more abundant in the IBD group. Although many unknown metabolites are included on this heatmap, cluster 1 contains a number of amino acids, whereas cluster 2 is composed of a wide variety of metabolites. Nonparametric univariate analysis using FDR of 5% revealed a total of 9 metabolites (4 identified and 5 unknown) that differed significantly between the HC and IBD-PRE groups. Identified metabolites are shown at the top of and in . Complete statistical results for all metabolites with FDR less than 25% are shown in Table S2. The 4 identified metabolites include gluconic acid lactone (also known as gluconolactone), hexuronic acid (also known as ascorbic acid or vitamin C), 3-hydroxybutanoic acid (ketone body) and ribose. All 4 metabolites were significantly increased in the IBD-PRE group ().
Table 2. Identified metabolites with altered peak intensity (q < 0.25) between healthy control dogs and dogs with IBD
Figure 6. Serum metabolite profiles. (A) PCA score plots of metabolites in serum from healthy control dogs, dogs with IBD pre-treatment, and dogs with IBD post-treatment. Ellipses represent the 95% confidence interval of metabolite profiles for each group. No clear separations between groups were identified. (B) Hierarchical clustering and heatmap of the top 75 metabolites that were different in their peak intensity between groups. The 75 most abundant metabolites are provided on the x-axis (see detail in Table S4). Each row represents the serum metabolite profile of each dog and sorted by group (colored bars on the x-axis represent groups (blue, healthy control dogs; red, dogs with IBD post-treatment; green, dogs with IBD pre-treatment) (C) Comparisons of the peak intensity for differentially expressed serum metabolites. Samples from dogs with IBD post-treatment were divided into 2 groups based on antibiotic administration status during 3 weeks of medical intervention. Gluconic acid lactone was the only identified metabolite that differed significantly between dogs with IBD pre-treatment and post-treatment. Red lines represent the median of peak intensity. HC, healthy control dogs; IBD-PRE, dogs with IBD pre-treatment; IBD-POST_NON AB, dogs with IBD post-treatment that did not receive antibiotic; IBD-POST_AB, dogs with IBD post-treatment that received antibiotic *q < 0.05
Effects of medical treatment on serum metabolite profiles
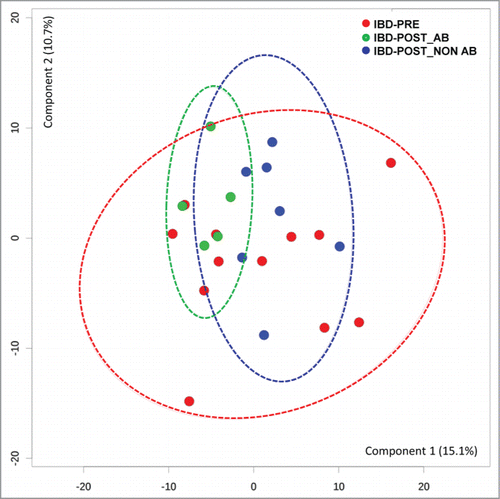
Three metabolites differed significantly between the IBD-PRE and IBD-POST groups. Of these, only one, gluconic acid lactone, was an identified metabolite ( and Table S2). As performed in the analysis of fecal microbial communities, serum metabolite profiles were also evaluated based on antibiotic administration. The PCA plot showed no clear separation between samples from dogs that received antibiotic and those that did not (). The univariate analysis of the selected metabolites based on antibiotic administration status also revealed no significant differences in any of the significantly different metabolites between samples from dogs that received antibiotic and those that did not ().
Figure 7. Principal component analysis (PCA) score plots of serum metabolomics data from dogs with IBD pre-treatment, dogs with IBD post-treatment that received antibiotic, and dogs with IBD post-treatment that did not received antibiotic. Ellipses represent the 95% confidence interval of the metabolite profile of each group. No clear separations between groups were identified. IBD-PRE, dogs with IBD pre-treatment; IBD-POST_NON AB, dogs with IBD post-treatment that did not receive antibiotic; IBD-POST_AB, dogs with IBD post-treatment that received antibiotic
Evaluation of the performance of potential biomarkers
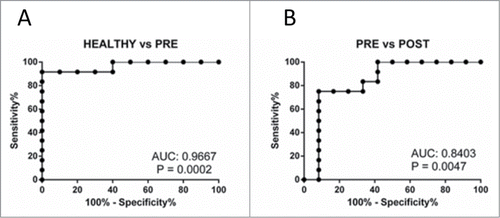
To evaluate the utility of the differentially abundant metabolites as potential biomarkers, we conducted a receiver operating characteristic (ROC) analysis. All metabolites, both identified and unknown, as well as metabolite ratios, were screened for their ability to discriminate between the HC and IBD-PRE groups using ROCCET.Citation23 Complete results for all metabolites with area under the curve (AUC) greater than 0.75 are shown in Table S3. Gluconic acid lactone had the highest discrimination ability among 157 identified metabolites. With an optimal cut-off peak intensity value of 497, gluconic acid lactone could be used to differentiate between the HC and IBD-PRE groups with 91.67% sensitivity and 100% specificity (). It could also be used discriminate between the IBD-PRE and IBD-POST groups with 75.0% sensitivity and 91.67% specificity at a cutoff value of 495 ().
Figure 8. Receiver operating characteristic (ROC) analysis for gluconic acid lactone. (A) Discrimination ability between healthy control dogs and dogs with IBD. (B) Discrimination ability between dogs with IBD pre- and post-treatment. AUC, area under the curve
Metabolite set enrichment analysis
The pentose phosphate pathway was significantly enriched (q = 0.0012) in the IBD-PRE group compared to the HC group. Catecholamine biosynthesis (enriched in HC) and nicotinate and nicotinamide metabolism (enriched in IBD-PRE) were also differentially expressed, but did not differ statistically when p-values were adjusted by FDR of 5% (q = 0.1288, 0.1602, respectively). No significantly enriched pathways were observed between the IBD-PRE and IBD-POST groups.
Network analysis
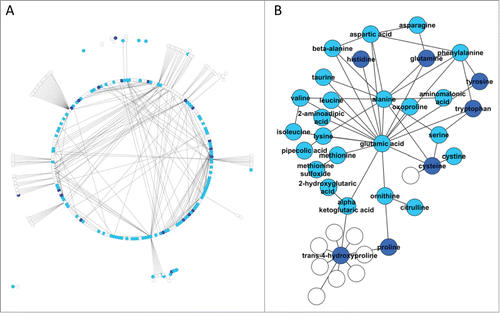
Due to the low number of identified metabolites that were significantly different between groups using an FDR of 5%, we used an FDR of 25% (roughly corresponding to p < 0.05) in order to evaluate the biochemical relationships among the metabolites. This resulted in a set of 33 identified metabolites that were differentially abundant between groups. A total of 120 metabolites, 20 of which differed significantly between groups, were mapped to a modified MetaMapp network,Citation17,24 which included 1,070 relationships among 293 metabolites (). In this network, nodes represent metabolites and edges denote relationships within a biochemical pathway. Differentially abundant metabolites were distributed throughout the network and none were directly connected to each other, except for trans-4-hydroxyproline and proline. Both of these metabolites were reduced in IBD animals, which could be reflective of the increased turnover of collagen often seen in IBD and a depletion of the substrates required for its synthesis. Creation of a sub-network using only metabolites identified in the present study as well as first neighbors showed a group of amino acids that clustered around glutamic acid (), suggesting a central role for glutamic acid or its associated pathways in IBD.
Figure 9. Network analysis of serum metabolites. (A) Overview of the network of relationships among metabolites. (B) Clustered network detected by network analysis. White nodes indicate metabolites that were not detected in this study, light blue nodes indicate metabolites that were detected but were not different (q > 0.25) between groups, dark blue nodes indicate metabolites that were detected and were different among groups. The edges indicate biochemical relationship among the metabolites.
Correlation analysis
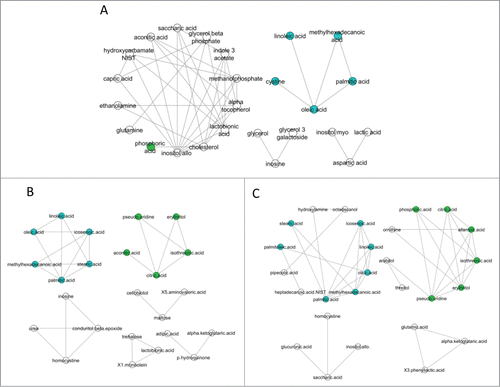
We next explored the metabolic networks associated with each group using a correlation analysis. Metabolites in equilibrium are positively correlated; the loss or gain of correlations thus suggests an alteration in the flow of substrates through a given pathway. The IBD groups had 2 clusters of correlated metabolites. One of the clusters, highlighted in green in , was centered around citric acid and its metabolites (). The second cluster, highlighted in blue in , was composed of saturated, monounsaturated, and polyunsaturated fatty acids (). No relationships between biochemical pathways were identified for either cluster using network analysis. In the healthy group, there was only one minor cluster which was composed of fatty acids ().
Figure 10. Correlation analysis of serum metabolites. Metabolic networks associated with (A) healthy control dogs, (B) dogs with IBD pre-treatment, and (C) dogs with IBD post-treatment. The IBD groups had 2 clusters of correlated metabolites. One of the clusters, highlighted in green, was centered around citric acid and its metabolites. The second cluster, highlighted in blue, was composed of saturated, monounsaturated, and polyunsaturated fatty acids. The healthy group had only one minor cluster which was composed of fatty acids.
Discussion
In this study, we applied a dual-omics approach to improve our current understanding of the pathogenesis of canine IBD, the host-microbial interactions, and the functional aspects of the canine GI microbiota. Microbiota analysis revealed significant differences in fecal microbial communities between dogs with IBD and healthy dogs, and also significantly decreased α-diversity indices in dogs with IBD. On higher phylogenetic levels, the major differences were decreases in Bacteroidetes and various taxa within Firmicutes and concurrent increases in Proteobacteria in dogs with IBD. Similar compositional shifts have also been observed in intestinal biopsies and/or fecal samples from human IBD patients. Citation25–28 In this study, the increased abundance of Gammaproteobacteria was mainly due to increases in Enterobacteriaceae. The qPCR results suggested that this increase was due to E. coli, and this group is of particular interest, as previous studies have reported an increased virulent potential of E. coli, such as adhesive capacity, invasive capacity, toxin production, and inflammatory cytokine stimulation, in human patients with IBD.Citation29,30 Adherent and invasive E. coli strains have been reported specifically in Boxer dogs with granulomatous colitis.Citation31 Whether the virulent potential of E. coli plays a similar role in other forms of canine inflammatory bowel disease remains to be determined. Various members of the Firmicutes (Blautia spp, Faecalibacterium spp., and Turicibacter spp) were decreased in IBD. Most of these bacterial groups belong to Clostridium clusters IV and XIVa and are believed to be major producers of short chain fatty acids (SCFA) and various other metabolites. Therefore, a decrease in these groups may have a significant effect on the host health. These differences are similar to those of previous studies evaluating the GI microbiota in duodenal mucosal/luminal content and feces in dogs with IBD.Citation3–6,32,33
Based on PICRUSt analysis, the relative abundances of genes associated with a given pathway may indicate an increased metabolic capacity of the GI microbiota with regard to that pathway. Inferences of the functional gene content were grouped by function according to the 3-level Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog (KO) hierarchy and compared using LEfSe with the significance threshold set at α of 0.01 and a LDA score of 3.0. In dogs with IBD, the pathways for secretion system and transcription factors were overrepresented. This may reflect the activation of bacterial defense mechanisms responding to the change in intestinal environmental conditions such as availability of nutrients, which may lead to a competitive advantage/disadvantage for microbial communities. Pathways representing amino acid metabolism were, on the other hand, significantly underrepresented in IBD. It has been suggested that commensal microbiota play an important role in the extraction, regulation of absorption, or synthesis of some amino acids.Citation34 Recently, an association between intestinal dysbiosis and protein energy malnutrition (e.g., kwashiorkor disease) has been reported.Citation35 In this study, the metabolomics data also suggested alterations in amino acid metabolism and this might be, in part, affected by metabolic dysfunction of bacteria. No other pathways were significantly differently expressed between healthy control dogs and dogs with IBD. This was surprising for carbohydrate metabolism, as many of the decreased bacterial taxa in dogs with IBD are associated with the production of SCFA from carbohydrates. Faecalibacterium spp., one of the main butyrate producing bacteria,Citation36 are prominent members of the canine GI microbiome.Citation37 Therefore, it was surprising that the carbohydrate metabolism did not differ between groups. A limitation of this study was that we used a prediction model based on 16 S rRNA genes rather than a true metagenomic approach with shot gun sequencing of fecal DNA. Future studies are warranted to evaluate functional properties of the canine microbiota in more detail in canine IBD.
The untargeted metabolomic approach brought new insights in regards to metabolic differences in canine IBD. Gluconic acid lactone is an oxidized derivative of glucose and is capable of scavenging free radicals. Hexuronic acid is a biologically active form of vitamin C and considered an antioxidant due to its ability to donate electrons. 3-hydroxybutanoic acid is a ketone body, and increased synthesis of this metabolite reflects an energy insufficiency. Ribose is vital for biological systems and an important source for further metabolism. These findings are somewhat different than those in human IBD studies which have shown altered, for the most part, lipid and amino acid metabolism.Citation14 One of the potential explanations is that most of the studies in human IBD applied nuclear magnetic resonance spectroscopy. We, on the other hand, applied GC-TOF/MS which may not be sensitive enough for lipid molecules. Therefore, the difference in the analytical method can contribute to the observed differences. Metabolomics is a relatively new field and still at a descriptive stage even in the study of human IBD. Therefore, large scale multi-center studies in both human and canine IBD are needed to address this species differences. Although gluconic acid lactone showed high discriminate ability for disease status, this may simply reflect inflammation status and may not be specific for IBD. Therefore, further studies that evaluate specific metabolites in different disease cohorts will be useful to see if those can be useful non-invasive maker for IBD. However, gluconic acid lactone has a possible utility as a biomarker for treatment efficacy. The enrichment analysis indicated that the pentose phosphate pathway was more activate in IBD, mainly due to increased abundance of gluconic acid lactone and ribose. The pentose phosphate pathway is an alternative to glycolysis and a critical pathway in cell redox balance and proliferation. This pathway produces NADPH, ribose, and energy sources for further metabolism and is required for glutathione reduction. Therefore, this pathway has an important role in the protection from oxidative stress and DNA/RNA synthesis.Citation38 These metabolic alterations may suggest the presence of oxidative stress. This is further supported by the decreased abundance of aminomalonic acid, and the increased abundances of 2-hydroxybutanoic acid and cysteine in dogs with IBD, although the differences of these metabolites did not reach statistical significance set at FDR of 5%. Taken all together, these results suggest that dogs with IBD are affected by oxidative stress due to their state of inflammation, and additional therapeutic interventions aimed toward mitigating oxidative stress could be beneficial. The association between the alteration of GI microbial communities and the presence of oxidative stress in GI tract has also been reported in human IBD patients, and E. coli seems to gain competitive advantage in an oxidative stress environment.Citation39,40 To validate this hypothesis, large scale studies with different treatment regimens (e.g., traditional standard therapy vs. additional antioxidant drug intervention) evaluating the difference in the abundance of antioxidants and byproducts of oxidative stress related metabolism (e.g., gluconic acid lactone) are needed.
We also evaluated the effects of 3 weeks of medical treatment on both the GI microbial and serum metabolite profiles. Although all dogs with IBD in this study showed clinical improvement, this did not correlate with recovery from dysbiosis. This suggests an ongoing pathological process. Therefore, these dogs may need more time to recover and/or additional therapy such as antioxidant supplementation, and these dogs may have a potential risk of relapse of clinical signs. However, administration of metronidazole may also contribute to this inconsistency in some extent. Antibiotics have been shown to be a strong driver to shift GI microbial composition.Citation40 In this study, the qPCR assays suggested that antibiotic administration might delay a recovery from the dysbiosis, as those dogs that did not receive antibiotic had median abundances of bacterial groups more similar to those of healthy dogs. A recent large multicenter study also observed similar effects of antibiotics on human Crohn's disease patients, and the authors hypothesized that the use of antibiotics has the potential to impact the overall GI microbial community and increase the potential for exposure to dysbiosis.Citation41 Studies also reported different effects on the composition of the microbiome with different antibiotics. A study reported increased abundances of Actinobacteria and Bacteroidetes after metronidazole treatment in an model of experimental colitis.Citation42 While this was not the case in our study, we observed similar effects as observed in a study reporting the relative resistance of Bifidobacterium spp to metronidazole.Citation43 Limited information is available with regards to the effect of antibiotics on dogs with IBD, although one study evaluating the effects of single and combination drug regimens showed that oral prednisone monotherapy is as effective as combination therapy with prednisone and metronidazole for treatment of dogs with IBD.Citation10 An increased susceptibility to pathogens and bacterial community shifts (abundance change and functional change) were observed by administration of antibiotics in a rodent model of intestinal inflammation.Citation44 These results may suggest potential negative effects of indiscriminate administration of antibiotics on the intestinal microbiota. The effect of antibiotics on the clinical outcome itself requires further studies, as in one study, there was no clear benefit of adding metronidazole to treatment regimens, but there was also no disadvantage.Citation10 In our study, all 5 dogs that received metronidazole showed clinical improvement. Metronidazole treatment was initiated at the time of diagnosis in 4 of 5 dogs due to clinician's preference. Therefore, it remains unknown whether this improvement was achieved by the effect of the drug combination, or achieved solely by the effect of the immunosuppressive drug. In one dog, metronidazole was added later to the treatment regimen due to an incomplete response to immunosuppressive mono-therapy. Although, this dog showed improvement, we cannot verify whether the improvement was achieved by antimicrobial activity of metronidazole independent of their immunomodulatory effects. Consequently, further studies are needed to correlate changes in microbial communities and clinical outcome of patients, especially when antibiotics are administered.
Similarly to the effects on the GI microbial communities, antibiotic intervention did not significantly affect the serum metabolite profiles. However, in contrast to their effects on the GI microbiota, dogs that received antibiotic had median abundances of differentially expressed metabolites that were more similar to median abundances observed in healthy dogs. This may potentially be due to an immunomodulatory effect of metronidazole. These data suggest that more extensive studies are needed to study the impact of antibiotics in canine IBD.
There are limitations to this study that need to be addressed. Firstly, the small sample size of animals used in this study limited the statistical power. Therefore, we may have missed some differences in bacterial and metabolite profiles between animal groups, especially when evaluating the effects of differences in treatment (i.e., use of antibiotic). Secondly, this study was conducted at only one institution and no standardization of diet and environment was conducted. However this study cohort represents the patients commonly seen in clinical veterinary practice. Moreover, the PCA and PCoA plots showed that healthy control dogs were tightly clustered, even though their diets varied more than the diets fed to dogs with IBD. This suggests that the dietary effects were not a confounding factor in this study. Similar findings were also observed in a previous study.Citation3 Finally, we did not have matching control dogs. In this study cohort, there were significant differences in body weight and age between the healthy control group and dogs with IBD. The evaluation of GI microbial communities using the UniFrac distance matric did not indicate any clustering based on body weight and age in this and previous studies.Citation3,4 Furthermore, the multivariate statistical analysis also did not identify any associations of microbial abundances and these variables. Therefore, these significant differences in age and body weight may not be a major bias in this study.
In conclusion, this study demonstrates intestinal dysbiosis and altered serum metabolite profiles in dogs with IBD. These alterations may suggest the presence of oxidative stress and functional alteration of GI microbiota in dogs with IBD. Ongoing dysbiosis and bacterial dysfunction were suspected to exist in dogs with IBD even after clinical improvement. Due to the small sample size, further large, multicenter studies are needed to validate these findings.
Materials and Methods
Animals and sample collection
The collection and analysis of serum and fecal samples from healthy control dogs and dogs with IBD were reviewed and approved by the Iowa State University Institutional Animal Care and Use Committee. Written informed consent was obtained from all owners of enrolled dogs. A blood sample was obtained during initial physical examination and serum was frozen within a few hours of collection and stored at -80°C until analysis. Left-over naturally-passed feces collected for routine fecal examination were frozen within a few hours of collection at -80°C, and were stored frozen until processing of samples for DNA extraction.
Healthy control dogs
Ten healthy dogs were enrolled. All dogs were privately owned, lived in diverse home environments, and were fed a variety of commercial diets (Table S5). All dogs were judged to be healthy on the basis of normal clinical examination findings and routine laboratory testing. None of the dogs had exhibited clinical signs of GI disease for at least 6 weeks prior to clinical evaluation. None of the dogs had received antibiotics or other drug therapy for at least 2 months prior to clinical examination. All dogs were eating a commercial canine maintenance diet (Table S5).
Dogs with IBD
Dogs with clinical signs of chronic GI disease (i.e., vomiting, diarrhea, anorexia, weight loss, etc.) were diagnosed with idiopathic inflammatory bowel disease (IBD) by a board certified veterinary internist (AEJ) based on the World Small Animal Veterinary Association (WSAVA) criteria: (i) chronic (i.e., > 3 weeks) GI signs; (ii) histopathologic evidence of mucosal infiltration with inflammatory cell; (iii) inability to document other causes of GI inflammation; (iv) inadequate response to dietary, antibiotic, and anthelmintic therapies, and (v) clinical response to anti-inflammatory or immunosuppressive agents. Histological samples were obtained endoscopically. The clinical status of each dog was evaluated using a published clinical canine IBD activity index (CIBDAI).Citation9 Twelve dogs that showed clinical improvement after therapy were enrolled this study. All medications had been discontinued at least 2 weeks prior to the first sample collection time point (IBD-PRE). After the first sample collection, dogs were then treated with immunosuppressive drug therapy (i.e., prednisone [1–2 mg/kg, q24 h, PO], prednisolone [1–2 mg/kg, q24 h, PO], budesonide [1–3 mg/kg, q24 h, PO], cyclosporine [3–5 mg/kg, q12 h or q24 h, PO], or a combination of these), and 5 of 12 dogs also received the antibiotic metronidazole (10 mg/kg, q12 h, PO). A second set of serum and fecal samples was obtained from these dogs after 21 d of treatment (IBD-POST). In addition to medical therapy, all IBD dogs were fed various commercial diets (Supplementary Table 5).
DNA extraction
An aliquot of 100 mg (wet weight) of each fecal sample was extracted by a bead-beating method using the ZR Fecal DNA KitTM (Zymo Research Corporation, Irvine, CA) following the manufacturer's instructions. The bead-beating step was performed on a homogenizer (FastPrep-24, MP Biomedicals, Santa Ana, CA) for 60 s at a speed of 4 m/s. Fecal DNA was stored at -80°C until analysis.
454-pyrosequencing
Bacterial tag-encoded FLX-titanium amplicon pyrosequencing (bTEFAP) based the V4-V6 region (E. coli position 530 – 1100) of the 16 S rRNA gene was performed to evaluate the relative abundances of bacterial taxa with primers forward 530F: GTGCCAGCMGCNGCGG and reverse 1100R: GGGTTNCGNTCGTTR. Raw sequence data were screened, trimmed, denoised, chimera depleted, and filtered using the QIIME pipeline version 1.8.0 (http://qiime.sourceforge.net)Citation45 with the following settings: minimum read length of 300 bp; no ambiguous base calls; no homopolymeric runs longer than 8 bp; average quality value.q25 within a sliding window of 50 bp. Operational taxonomic units (OTUs) were defined as sequences with at least 97% similarity using QIIME. For classification of sequences on a genus level the naıve Bayesian classifier within the Ribosomal Database Project (RDP, v10.28) was used. The confidence threshold in RDP was set to 80%.
Quantitative PCR (qPCR)
The qPCR assays for selected bacterial groups were performed to validate pyrosequencing result and/or to evaluate bacterial groups that are typically present at very low abundance or underrepresented in 16 S rRNA gene based sequencing (total bacteria; Phylum level – Fusobacteria, Bacteroidetes; Family level – Ruminococcaceae; Genus level –Bifidobacterium spp., Blautia spp, Faecalibacterium spp., Turicibacter spp, and Lactobacillus spp.; and species level – Escherichia coli (E. coli). The qPCR cycling, the oligonucleotide sequences of primers and probe, and respective annealing temperatures for selected bacterial groups were described previously.Citation4,46 A commercial real-time PCR thermal cycler (CFX 96 Touch™ Real-Time PCR Detection System; Biorad Laboratories, Hercules, CA) was used for all qPCR assays and all samples were run in duplicate fashion.
Analysis of serum metabolites
Untargeted metabolomics analysis was performed by the West Coast Metabolomics Center at the University of California (Davis, CA). Serum aliquots were extracted by degassed acetonitrile. Internal standards C08-C30 FAMEs were added and the samples were derivatized by methoxyamine hydrochloride in pyridine and subsequently by N-methyl-N-trimethylsilyltrifluoroacetamide for trimethylsilylation of acidic protons. Analytes were separated using an Agilent 6890 gas chromatograph (Santa Clara, CA) and mass spectrometry was performed on a Leco Pegasus IV time of flight mass spectrometer (St. Joseph, MI) following the published protocol.Citation47
Statistical analysis
Sequence data and qPCR data
Differences in bacterial abundances between the HC and IBD-PRE groups were evaluated using a Mann-Whitney test. Samples collected before and after treatment from each IBD dog were compared using a Wilcoxon signed rank test. The Benjamini & Hochberg's False Discovery Rate was used to correct for multiple comparisons and an adjusted p < 0.05 (i.e., q < 0.05) was considered to be statistically significant. To evaluate differences in overall microbiota composition (i.e., β-diversity) between the groups, the analysis of similarities (ANOSIM) was performed on the unweighted UniFrac distance matrixes. The PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States: http://picrust.github.io/picrust/)Citation40 was used to predict the functional capabilities of bacteria based on the 16 S rRNA gene data set. Linear discriminant analysis (LDA) effect size (LEfSe: http://huttenhower.sph.harvard.edu/galaxy/)Citation48 was utilized to evaluate differentially abundant bacterial taxa and predicted function between the animal groups. To identify associations of microbial abundances with the use of antibiotic and other clinical metadata (age, weight, gender), which may be confounding factors, MaAsLin (Multivariate Analysis by Linear Models: http://huttenhower.sph.harvard.edu/maaslin)Citation40 was utilized
Untargeted metabolomics data
Univariate analysis
Differences in the abundance of serum metabolites between the HC and IBD-PRE groups were evaluated using a Mann-Whitney test. Serum samples collected before and after treatment from each affected dog (IBD-PRE vs. IBD-POST) were compared using a Wilcoxon signed rank test. Univariate analysis was performed using Prism version 5 (Graph Pad Software, La Jolla, CA).
Multivariate analysis
Data were normalized to the sum of the total spectral integral, log transformed, mean centered, and divided by the standard deviation of each variable prior to multivariate analysis. PCA was performed and a heatmap was generated using MetaboAnalyst.Citation49
Correlation and biochemical relationship networks
Correlation analyses were performed in R using the “cor” function. Relationships with a Pearson's correlation coefficient greater than 0.9, or smaller than -0.9, were used to build correlation networks. For biochemical relationship analysis, named metabolites were mapped onto a modified MetaMapp network.Citation24 The network originally published by Barupal et al.Citation24 was expanded by the inclusion of additional nodes and edges as described by Steelman et al.Citation17 Correlation and biochemical relationship networks were visualized in CytoscapeCitation50 by mapping metabolite data onto the expanded MetaMapp network. Data included alphanumeric name, level of significance (q > or < 0.25), and PubChem ID as the unique identifier. A custom visual style was created using the following color scheme: white nodes represent metabolites that were not detected in the present study, light blue nodes represent metabolites that were detected but did not differ significantly among groups, and dark blue nodes represent metabolites that did differ significantly among groups.
Evaluation of the performance of potential biomarkers
ROC analysis was performed using the Classical Analysis module in ROC Curve Explorer & Tester (ROCCET).Citation23
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
KGMI_A_997612_Supplementary_Table_5.docx
Download MS Word (20.8 KB)KGMI_A_997612_Supplementary_Table_4.xlsx
Download MS Excel (14.4 KB)KGMI_A_997612_Supplementary_Table_3.xlsx
Download MS Excel (14.4 KB)KGMI_A_997612_Supplementary_Table_2.xlsx
Download MS Excel (11.5 KB)KGMI_A_997612_Supplementary_Table_1.xlsx
Download MS Excel (361.6 KB)Acknowledgments
This study was supported by the Comparative Gastroenterology Society/Waltham Research Grant.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- DuPont AW, DuPont HL. The intestinal microbiota and chronic disorders of the gut. Nat Rev Gastroenterol Hepatol 2011; 8:523-31; PMID:21844910; http://dx.doi.org/10.1038/nrgastro.2011.133
- Packey C, Sartor R. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis 2009; 22:292-301; PMID:19352175; http://dx.doi.org/10.1097/QCO.0b013e32832a8a5d
- Suchodolski JS, Dowd SE, Wilke V, Steiner JM, Jergens AE. 16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease. PLoS One 2012; 7:e39333; PMID:22720094; http://dx.doi.org/10.1371/journal.pone.0039333
- Suchodolski JS, Markel ME, Garcia-Mazcorro JF, Unterer S, Heilmann RM, Dowd SE, Kachroo P, Ivanov I, Minamoto Y, Dillman EM, et al. The fecal microbiome in dogs with acute diarrhea and idiopathic inflammatory bowel disease. PLoS One 2012; 7:e51907; PMID:23300577; http://dx.doi.org/10.1371/journal.pone.0051907
- Suchodolski JS, Xenoulis PG, Paddock CG, Steiner JM, Jergens AE, Steiner JM. Molecular analysis of the bacterial microbiota in duodenal biopsies from dogs with idiopathic inflammatory bowel disease. Vet Microbiol 2010; 142:394-400; PMID:19959301; http://dx.doi.org/10.1016/j.vetmic.2009.11.002
- Xenoulis PG, Palculict B, Allenspach K, Steiner JM, Van House AM, Suchodolski JS. Molecular-phylogenetic characterization of microbial communities imbalances in the small intestine of dogs with inflammatory bowel disease. FEMS Microbiol Ecol 2008; 66:579-89; PMID:18647355; http://dx.doi.org/10.1111/j.1574-6941.2008.00556.x
- Simpson KW, Jergens AE. Pitfalls and progress in the diagnosis and management of canine inflammatory bowel disease. Vet Clin North Am Small Anim Pract 2011; 41:381-98; PMID:21486642; http://dx.doi.org/10.1016/j.cvsm.2011.02.003
- Overstreet A, Ramer-Tait A. The Role of the Microbiota in Gastrointestinal Health and Disease. In: Szabo I, editor. Inflammatory Bowel Disease. InTech Published; 2012
- Washabau R, Day M. Endoscopic, biopsy, and histopathologic guidelines for the evaluation of gastrointestinal inflammation in companion animals. J Vet Intern Med 2010; 24:10-26; PMID:20391635; http://dx.doi.org/10.1111/j.1939-1676.2010.0520.x
- Jergens a E, Crandell J, Morrison J a, Deitz K, Pressel M, Ackermann M, Suchodolski JS, Steiner JM, Evans R. Comparison of oral prednisone and prednisone combined with metronidazole for induction therapy of canine inflammatory bowel disease: a randomized-controlled trial. J Vet Intern Med 2010; 24:269-77; PMID:20051005; http://dx.doi.org/10.1111/j.1939-1676.2009.0447.x
- Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 2013; 14:676-84; PMID:23778795; http://dx.doi.org/10.1038/ni.2640
- Nicholson JK, Lindon JC. Systems biology: Metabonomics. Nature 2008; 455:1054-6; PMID:18948945; http://dx.doi.org/10.1038/4551054a
- Marchesi J, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, Wilson I, Wang Y. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res 2007; 6:546-51; PMID:17269711; http://dx.doi.org/10.1021/pr060470d
- Lin H-M, Helsby N, Rowan D, Ferguson L. Using metabolomic analysis to understand inflammatory bowel diseases. Inflamm Bowel Dis 2011; 17:1021-9; PMID:20629098; http://dx.doi.org/10.1002/ibd.21426
- De Preter V, Verbeke K. Metabolomics as a diagnostic tool in gastroenterology. World J Gastrointest Pharmacol Ther 2013; 4:97-107; PMID:24199025
- Patti GJ, Yanes O, Siuzdak G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol 2012; 13:263-9; PMID:22436749; http://dx.doi.org/10.1038/nrm3314
- Steelman SM, Johnson P, Jackson A, Schulze J, Chowdhary BP. Serum metabolomics identifies citrulline as a predictor of adverse outcomes in an equine model of gut-derived sepsis. Physiol Genomics 2014; 46:339-47; PMID:24619519; http://dx.doi.org/10.1152/physiolgenomics.00180.2013
- Zhang J, Wei S, Liu L, Nagana Gowda G a, Bonney P, Stewart J, Knapp DW, Raftery D. NMR-based metabolomics study of canine bladder cancer. Biochim Biophys Acta 2012; 1822:1807-14; PMID:22967815; http://dx.doi.org/10.1016/j.bbadis.2012.08.001
- Ritchie L, Burke K, Garcia-Mazcorro J, Steiner J, Suchodolski J. Characterization of fecal microbiota in cats using universal 16S rRNA gene and group-specific primers for Lactobacillus and Bifidobacterium spp. Vet Microbiol 2010; 144:140-146; PMID:20092970; http://dx.doi.org/10.1016/j.vetmic.2009.12.045
- Handl S, Dowd SE, Garcia-Mazcorro JF, Steiner JM, Suchodolski JS. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol Ecol 201; 76:301-10; http://dx.doi.org/10.1111/j.1574-6941.2011.01058.x
- Ritchie L, Steiner J, Suchodolski J. Assessment of microbial diversity along the feline intestinal tract using 16S rRNA gene analysis. FEMS Microbiol Ecol 2008; 66:590-598; PMID:19049654; http://dx.doi.org/10.1111/j.1574-6941.2008.00609.x
- Fiehn O, Wohlgemuth G, Scholz M. Setup and annotation of metabolomic experiments by integrating biological and mass spectrometric metadata. Data Integr life Sci 2005;224-39; http://dx.doi.org/10.1007/11530084_18
- Xia J, Broadhurst DI, Wilson M, Wishart DS. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics 2013; 9:280-99; PMID:23543913; http://dx.doi.org/10.1007/s11306-012-0482-9
- Barupal DK, Haldiya PK, Wohlgemuth G, Kind T, Kothari SL, Pinkerton KE, Fiehn O. MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinformatics 2012; 13:99; PMID:22591066; http://dx.doi.org/10.1186/1471-2105-13-99
- Kabeerdoss J, Sankaran V, Pugazhendhi S, Ramakrishna BS. Clostridium leptum group bacteria abundance and diversity in the fecal microbiota of patients with inflammatory bowel disease: a case-control study in India. BMC Gastroenterol 2013; 13:20; PMID:23351032; http://dx.doi.org/10.1186/1471-230X-13-20
- Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJO. Differences between tissue-associated intestinal microfloras of patients with Crohn's disease and ulcerative colitis. J Clin Microbiol 2006; 44:4136-41; PMID:16988016; http://dx.doi.org/10.1128/JCM.01004-06
- Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 2006; 55:205-11; PMID:16188921; http://dx.doi.org/10.1136/gut.2005.073817
- Frank DN, St Amand AL, Feldman R a, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104:13780-5; PMID:17699621; http://dx.doi.org/10.1073/pnas.0706625104
- De la Fuente M, Franchi L, Araya D, Díaz-Jiménez D, Olivares M, Alvarez-Lobos M, Golenbock D, González M-J, López-Kostner F, Quera R, et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int J Med Microbiol 2014; :1-9; PMID:24183099; http://dx.doi.org/10.1155/2014/489569
- Kotlowski R, Bernstein CN, Sepehri S, Krause DO. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut 2007; 56:669-75; PMID:17028128; http://dx.doi.org/10.1136/gut.2006.099796
- Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun 2006; 74:4778-92; PMID:16861666; http://dx.doi.org/10.1128/IAI.00067-06
- Suchodolski JS, Camacho J, Steiner JM. Analysis of bacterial diversity in the canine duodenum, jejunum, ileum, and colon by comparative 16S rRNA gene analysis. FEMS Microbiol Ecol 2008; 66:567-78; PMID:18557939; http://dx.doi.org/10.1111/j.1574-6941.2008.00521.x
- Rossi G, Pengo G, Caldin M, Palumbo Piccionello A, Steiner JM, Cohen ND, Jergens AE, Suchodolski JS. Comparison of microbiological, histological, and immunomodulatory parameters in response to treatment with either combination therapy with prednisone and metronidazole or probiotic VSL#3 strains in dogs with idiopathic inflammatory bowel disease. PLoS One 2014; 9:e94699; PMID:24722235; http://dx.doi.org/10.1371/journal.pone.0094699
- Blachier F, Mariotti F, Huneau JF, Tomé D. Effects of amino acid-derived luminal metabolites on the colonic epithelium and physiopathological consequences. Amino Acids 2007; 33:547-62; PMID:17146590; http://dx.doi.org/10.1007/s00726-006-0477-9
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 2013; 339:548-54; PMID:23363771; http://dx.doi.org/10.1126/science.1229000
- Duncan SH, Hold GL, Harmsen HJM, Stewart CS, Flint HJ. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int J Syst Evol 2002; 52:2141-6; http://dx.doi.org/10.1099/ijs.0.02241-0
- Garcia-Mazcorro JF, Dowd SE, Poulsen J, Steiner JM, Suchodolski JS. Abundance and short-term temporal variability of fecal microbiota in healthy dogs. Microbiologyopen 2012;1:340-7; PMID:23170232; http://dx.doi.org/10.1002/mbo3.36
- Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med 2012; 53:421-36; PMID:22580150; http://dx.doi.org/10.1016/j.freeradbiomed.2012.05.006
- Bertin Y, Girardeau JP, Chaucheyras-Durand F, Lyan B, Pujos-Guillot E, Harel J, Martin C. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ Microbiol 2011; 13:365-77; PMID:20849446; http://dx.doi.org/10.1111/j.1462-2920.2010.02334.x
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward D V, Reyes J a, Shah S a, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13:R79; PMID:23013615; http://dx.doi.org/10.1186/gb-2012-13-9-r79
- Gevers D, Kugathasan S, Denson L a, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe 2014; 15:382-92; PMID:24629344; http://dx.doi.org/10.1016/j.chom.2014.02.005
- Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, Gallini CA, Beal C, van Hylckama-Vlieg JE, Ballal S a, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J 2014; 1-15; PMID:24152717
- Moubareck C, Gavini F, Vaugien L, Butel MJ, Doucet-Populaire F. Antimicrobial susceptibility of bifidobacteria. J Antimicrob Chemother 2005; 55:38-44; PMID:15574479; http://dx.doi.org/10.1093/jac/dkh495
- Theriot CM, Koenigsknecht MJ, Carlson PE, Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 2014; 5:3114; PMID:24445449; http://dx.doi.org/10.1038/ncomms4114
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. correspondEnce QIIME allows analysis of high- throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat Publ Gr 2010; 7:335-6
- Garcia-Mazcorro JF, Suchodolski JS, Jones KR, Clark-Price SC, Dowd SE, Minamoto Y, Markel M, Steiner JM, Dossin O. Effect of the proton pump inhibitor omeprazole on the gastrointestinal bacterial microbiota of healthy dogs. Fems Micriobiology Ecol 2012; 80:642-636
- Fiehn O, Garvey WT, Newman JW, Lok KH, Hoppel CL, Adams SH. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS One 2010; 5:e15234; PMID:21170321; http://dx.doi.org/10.1371/journal.pone.0015234
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60; PMID:21702898; http://dx.doi.org/10.1186/gb-2011-12-6-r60
- Xia J, Wishart DS. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc 2011; 6:743-60; PMID:21637195; http://dx.doi.org/10.1038/nprot.2011.319
- Saito R, Smoot ME, Ono K, Ruscheinski J, Wang P, Lotia S, Pico AR, Bader GD, Ideker T. A travel guide to Cytoscape plugins. 2012; 9:1069-76; PMID:23132118