
abstract
Shifts in microbial populations of the intestinal tract have been associated with a multitude of nutritional, autoimmune, and infectious diseases. The limited diversity following antibiotic treatments creates a window for opportunistic pathogens, diarrhea, and inflammation as the microbiome repopulates. Depending on the antibiotics used, microbial diversity can take weeks to months to recover. To alleviate this loss of diversity in the intestinal microbiota, supplementation with probiotics has become increasingly popular. However, our understanding of the purported health benefits of these probiotic bacteria and their ability to shape the microbiome is significantly lacking. This study examined the impact of probiotics concurrent with antibiotic treatment or during the recovery phase following antibiotic treatment of mice. We found that probiotics did not appear to colonize the intestine themselves or shift the overall diversity of the intestinal microbiota. However, the probiotic supplementation did significantly change the types of bacteria which were present. In particular, during the recovery phase the probiotic caused a suppression of Enterobacteriaceae outgrowth (Shigella and Escherichia) while promoting a blooming of Firmicutes, particularly from the Anaerotruncus genus. These results indicate that probiotics have a significant capacity to remodel the microbiome of an individual recovering from antibiotic therapy.
Introduction
The gastrointestinal tract of mammals contains a highly diverse and complex community of microorganisms. The species level taxonomy of this community can vary significantly between individuals but the core microbiome is conserved and has high functional redundancy.Citation1-3 The host relies on these microbial denizens for nutrition, regulation of intestinal structures, immune system development, and protection from opportunistic pathogens.Citation4-6 The microbiome plays an essential role in maintaining the host health.
Disturbances to the microbiome can lead to altered states of microbial complexity, also known as dysbiosis, which has been associated with various disease states. Correlations between altered microbiome and disease state has been suggested in several conditions such as obesity, inflammatory bowel disease (IBD), allergy, antibiotic associated diarrhea, or opportunistic pathogen infection.Citation7-10 In particular the antibiotic induced alterations of the gut microbiome can pose a major health problem due to the significant and long lasting changes to the microbial community following treatment. Taking antibiotics can promote the reservoir of antibiotic resistance genes and the emergence of highly resistant strains of bacteria.Citation11,12 Additionally, antibiotic treatment can eliminate non-pathogenic commensal bacteria allowing opportunistic pathogens, such as Clostridium difficile, to flourish.
To treat gastrointestinal distress the consumption of probiotics has received a substantial amount of public enthusiasm, vastly outpacing research based evidence of efficacy. Probiotics are living organisms which have been ascribed health promoting benefits following ingestion, commonly of the genera Lactobacillus, Lactococcus, or Bifidobacterium. These fermented milk product probiotics do not impact the diversity of an intact “healthy” microbial population.Citation13 Meta analysis of trials utilizing probiotics in the context of disease states has shown some efficacy.Citation14-16 However, the impact on a microbiome devastated by antibiotic treatment has not been well characterized. This study sought to determine the impacts of a mixed probiotic on the diversity of cecal and large intestinal contents of mice when taken during antibiotic treatment or throughout the recovery phase following antibiotic treatment.
This study utilized a multi-prong approach of Sanger sequencing 16S rRNA genes from culture dependent, clonal library, or total 16S next generation sequencing analysis. Since a meaningful analysis of the low abundance taxa requires deep sequencing of many thousands of transcripts, our data focus predominantly on the latter technique. We found that following an extensive course of broad spectrum antibiotics the cecal/intestinal contents can be brought effectively to a monoculture of highly antibiotic resistant Xanthomonads. Removal of the antibiotics leads to a flourishing of opportunistic pathogen Enterobacteriaceae (E.coli and Shigella) which were suppressed by the use of a probiotic throughout the recovery period. Interestingly, probiotic treated animals exhibited a diverse microbiome which was largely dominated by a single, relatively uncharacterized, genus of Anaerotruncus bacteria. These changes highlight the power of probiotic administration to circumvent disease following antibiotic treatment. They also provide a reason for caution of use in clinical settings until the impacts of probiotics on “reseeded” gut flora are better characterized.
Methods
Animals and antibiotic/probiotic administration
C57BL/6 mice were obtained from Charles River (Willmington, MA) at 6–8 weeks of age and housed in the Ithaca College animal facility. Groups of animals were co-housed with mixing of bedding between groups for 3 to 4 weeks prior to the start of antibiotic treatment. Ampicillin (Abraxis Pharma), Streptomycin (Abraxis Pharma), and Clindamycin (Pharmacia) were provided into sterile drinking water at a final concentration of 1mg/ml, as previously described.Citation5 These antibiotics were chosen due to the broad spectrum capacity and well documented impacts on intestinal microbiota.Citation17 Animals were allowed to drink ad libitum for the duration of the experiment with water replacement at 3 day intervals. A probiotic blend of L. rhamnosus A 191, Lactobacillus acidophilus, Bifidobacterium breve, Bifidobacterium longum (CVS Maximum Strength Probiotic) were resuspended in 5mls PBS for a total of 4 billion organisms per milliliter. Probiotic was provided to the mice via oral gavage (0.1ml/mouse) every other day for 2 weeks. All experiments and protocols were approved by Ithaca College Institutional Animal Care and Use Committee.
Sample collection and DNA extraction
DNA was extracted from large intestine and cecal contents using the Power Fecal DNA kit (Mo-Bio) according to manufacturer's instructions. DNA was amplified with PCR primers 27F/1492R and was next generation PGM sequenced or cloned into Topo-TA vectors (Life Technologies) for clonal isolation.Citation18 Individual clones were further isolated and Sanger sequenced (Genewiz, NJ, USA). The antibiotic with probiotic group produced 2 usable DNA samples while all other groups were successfully extracted. For culture based growth, the remaining cecal and intestinal contents were diluted in 2mls PBS and serial diluted on trypticase soy agar plates. Both aerobic and anaerobic colonies were streaked to isolation, lysed, and 16S rRNA genes amplified for sequencing (Genewiz, NJ, USA). Clonal and culture based results were concordant with next generation sequencing analysis.
Sequencing and bioinformatics data analysis
The 16S rDNA V4 variable region PCR primers 515/806 were used in a single-step 30 cycle PCR using the HotStartTaq Plus Master Mix Kit (Qiagen) under the following conditions: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds and 72°C for 1 minute, after which a final elongation step at 72°C for 5 minutes was performed.Citation19 Sequencing was performed at MR DNA (www.mrdnalab.com) on an Ion Torrent PGM following the manufacturer's guidelines. Sequence data were processed using a proprietary analysis pipeline (MR DNA). Briefly, sequences were depleted of barcodes and primers, then sequences <150bp removed, sequences with ambiguous base calls and with homopolymer runs exceeding 6bp were also removed. Sequences were denoised, operational taxonomic units (OTUs) generated and chimeras removed. OTUs were defined by clustering at 3% divergence (97% similarity). Final OTUs were taxonomically classified using BLASTn against a database derived from GreenGenes (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi), RDPII http://rdp.cme.msu.edu) and NCBI (www.ncbi.nlm.nih.gov).
We applied the “Quantitative Insights Into Microbial Ecology” (QIIME) software as part of our analysis.Citation20 To eliminate the differences in sequencing effort between samples, α diversity was calculated based on 44000 reads per individual. We calculated 1) Chao1, 2) phylogenetic diversity, and 3) the Shannon index. Total OTU rarefaction was calculated using R package “vegan” and plotted in excel. To examine β diversity PcoA biplots were produced using the QIIME analysis and Emperor visualization tool. The coordinates of a given taxon are plotted as a weighted average where the size of the sphere is proportional to the mean relative abundance of the taxon across all samples.
Quantitative PCR
The abundance of total and specific probiotic bacterial groups (Bacteroidetes, Enterobactericeae, Firmicutes) and specific genus (Lactobacillus, Bifidobacterium, Anaerotruncus) was measured by qPCR using universal and group-specific 16S rRNA gene primers (Table S1) with the qScript One-Step SYBR Green qRT-PCR kit (Quanta Biosciences).Citation21-23 Bacterial DNA purified from resuspension of Probiotics (CVS Max Strength, Culturelle) via the Power fecal DNA kit (Mo-Bio) following manufacturer directions. qPCR was performed on a Lightcycler 480(Roche). Bacterial DNA was quantified relative to general 16S rRNA primers (Table S1) via the ΔCTmethod.
Statistical analysis
The abundance distributions across groups were compared as Dirichlet-multinomial (D-M) distributions using the HMP package (R software).Citation24 We examined our data for statistical power using the included analysis tools (Table S2). All treatment groups could be differentiated when taxonomic data are included as part of the QIIME diversity analysis. Chao1, phylogenetic diversity, Shannon index and cecal morphology were each compared among treatment groups with Kruskal-Wallis tests. In all cases for which the p < 0.01, we conducted a nonparametric post-hoc test (using R with the package nparcomp) to identify pairwise differences. We used a Student T-test to compare quantitative PCR analysis between CVS and Culturelle probiotics. Cecal lengths and areas were calculated by first sizing images to equivalent scale and then using the measurement tools in Nikon Elements D software for image quantitation. (* = p < 0.05, ** = p < 0.01)
Results
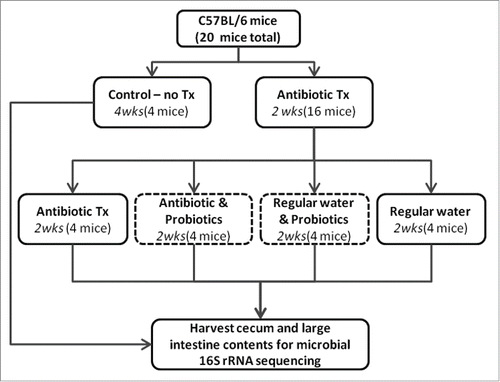
A multitude of diseases can be linked to dysbiosis of the microbiome in the gastrointestinal tract. In particular, antibiotic associated diarrhea and infections from opportunistic pathogens, such as Clostridium difficile, can be common following treatment with antibiotics.Citation25-27 The use of probiotics has become popular among the public despite a general lack of understanding in mechanisms of action by which they provide “health benefits.” We hypothesized that probiotics helped to promote microbial diversity in the gastrointestinal tract following antibiotic treatment. To analyze the impacts of antibiotics and probiotics on composition of gut microbiome, antibiotics were administered to C57BL/6 mice via their water for 2–4weeks (). The recovery period from antibiotic treatment or continued antibiotic treatment was supplemented with probiotic administration. The microbiota from all mice (cecal and large intestine) were simultaneously harvested and assessed based on 16S rRNA sequencing. The 16S sequences were isolated via microbial culture, clonal isolation, and massively parallel PGM sequencing approach. All approaches were congruent in their findings but the PGM based on the V4 hypervariable region of the small-subunit rRNA provided the greatest depth of coverage and is reported here.
Figure 1. Schematic for antibiotic and probiotic administration. Sixteen C57BL/6 mice received antibiotic treated water for 2 weeks in duration. Mice were then split evenly into 4 treatments: 1) continued antibiotics, 2) continued antibiotics with gavaged probiotics 3) regular water with gavaged probiotics, 4) regular water. Control animals were maintained on regular water throughout the experiment. Mice were euthanized and contents of cecum and large intestine collected.
We analyzed the 16S rRNA genes from 5 different treatments of mice yielding a total of 2,006,079 generated operational taxonomic unit (OTU) sequence tags. Based on rank abundance data, not including taxonomic identifiers, a multi-group comparison using Dirichlet-multinomial (D-M) method-of-moments analysis found significant differences among the groups, and a subsequent family of pairwise comparisons determined all groups are unique (p<0.01 after family-wise correction) except for comparisons between control and recovery groups. However, these populations can be further defined by including taxonomic information along with abundance data and will be discussed below. At a significance level of 0.05, power of the multi-group comparison was 100%.Citation24 Pairwise analysis of groups demonstrated the least powerful comparison existed between recovery groups (power > 86%) (Table S2).
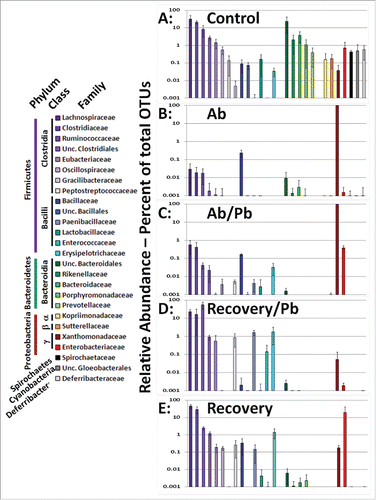
In accordance with multiple microbiome studies, we found that the majority of cecal/intestinal OTUs mapped to the Firmicutes, Bacteroidetes, and Proteobacteria ( and figure S1). All data were rank ordered based on non-treated controls and a bacterial family was included if at least one mouse exhibited greater than one percent of that family as part of its cecal/intestinal microbiome. The treatment of broad spectrum antibiotics drastically reduced the Bacteroidetes populations which never fully recovered following cessation of antibiotic treatment. The cecal and large intestinal microbiome of the 4 control mice were predominantly composed of Firmicutes and Bacteroidetes. In contrast, antibiotic treatment resulted in Xanthomonadaceae becoming the predominant bacterial family ( and Figure S1). The addition of probiotics concurrent with continued antibiotic treatment did cause a rise in microbial diversity leading to small increases in the Firmicutes phylum and an increase in the Enterobacteriaceae family. The cessation of antibiotic treatment resulted in a restoration of Firmicutes as the dominant bacterial phyla (99.8% ± 0 .7 Recovery + Pb and 80% ± 21 Recovery alone). As compared to recovery alone, the addition of probiotics leads to a notable decrease in Proteobacteria which will be addressed in further detail below. Collectively, the broad spectrum antibiotics used in this study efficiently eliminated the majority of gut bacteria complexity allowing for the study of probiotics in the repopulation of microbial diversity during the recovery period.
Figure 2. Comparison of cecal/intestinal microbial community structure by rank ordering relative abundance of bacterial families following antibiotic and probiotic treatment. More than 2 million OTU tags were retrieved from sequenced DNA and relative abundance at the taxonomic family level was calculated. Family level abundance was pooled for each experimental group. Any bacterial family above 1% total in any single animal lead to inclusion in figure for all groups. Control mice did not receive antibiotics. Ab mice were on antibiotics for all 4 weeks. Ab/Pb mice were on antibiotics for 4 weeks and the last 2 weeks were supplemented with gavaged probiotics. Recovery mice were switched to regular water with or without probiotic treatment.
To assess the impact of antibiotics and probiotics on species diversity we used QIIME analysis to calculate multiple measures of α and β diversity (). We calculated Chao1, phylogenetic diversity, and Shannon index of diversity for each group. By all measures, antibiotic treatment lead to a significantly reduced microbial diversity in the cecal and large intestine contents of treated mice (p < 0.01). The antibiotic treatment condition limited the microbiome to predominantly 2 genera of bacteria (Stenotrophomonas and Xanthamonas)(). The addition of probiotics concurrent with antibiotics did not improve species diversity (p > 0.32 across all measures). The recovery period lead to a restoration of microbial diversity (p < 0.01), which was not quite commensurate with untreated control animals (p < 0.01). Thus the diversity under both recovery conditions never quite returned to original levels. Rarefaction curves involves resampling of survey data to generate idealized species abundance curves (). This provides an estimate of depth of sampling in a complex community. Notably, the species depth of recovery animals is higher in the absence of probiotic administration. Comparisons of treatment groups using principal coordinate analysis separated the animals based on 5 keystone taxonomies (). PC1 accounted for 24.4% of the microbiome diversity and separated antibiotic mice on the presence of Xanthamonadacea (Stentrophomonas and Xanthamonas) as compared to the abundance of Lachnospiraceae in all other groups. PC3 accounted for 14% of overall diversity and separates recovery groups based on the presence of Ruminococcaceae (Anerotrouncus) compared to the Enterobacteriaceae (Eschiricia and Shigella). PC2 accounts for 21.8% of overall diversity and separates control animals from all other groups based on the abundance of Bacteroidales. Collectively, the addition of probiotics during recovery did not significantly increase the diversity of bacteria but there was a considerable change in the specific types of bacterial genera present.
Figure 3. Impact of antibiotic treatment and probiotic supplementation on microbial diversity in the mouse intestine. Diversity analysis utilized the QIIME pipeline. Alpha diversity was determined based on rarefied analysis of A) Chao1, B) Phylogenetic diversity, C) Shannon index, and D) Observed species abundance. Standard deviations among group members are provided. Intergroup comparisons were made based on principal coordinate analysis of unweighted UniFrac distances (E). In the biplot, the 5 most prevalent taxa driving the differences in the PCoA are provided (F = family level, O = order level). Taxon specific spheres vary in size based on mean relative abundance in all groups. (** = p < 0.01)
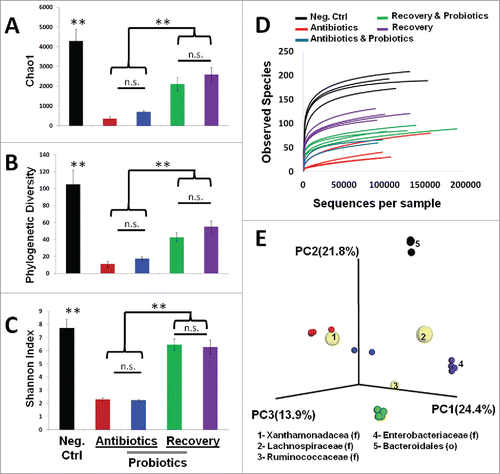
Figure 4. Genus level diversity of dominant 15 taxa among treatment groups. The top 5 genera from each treatment group were consolidated into a pool of 15 dominant genera. These dominant genera make up the majority of OTUs from each mouse, average OTU coverage ± standard deviation displayed above pie charts. A) Control animals B) Antibiotics for 4 weeks C) Antibiotics for 4 weeks supplemented with probiotics D) Recovery from antibiotics supplemented with probiotics E) Recovery from antibiotics on regular water.
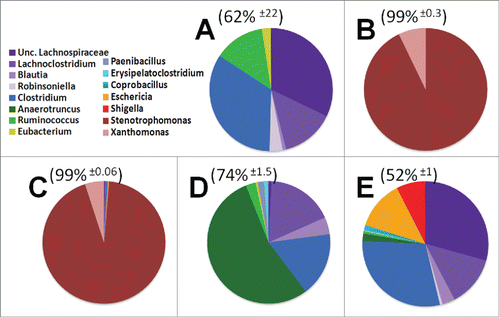
Examination of the cecal and large intestine microbiome identified 15 dominant genera of bacteria when different experimental treatments were taken into account. These dominant genera are graphically depicted in . This collection of dominant genera constitute the majority (62% ± 22 ) of the bacteria in control untreated mice. Dominant bacterial families were Lachnospiraceae(purple), composed of genus Lachnoclostridum, Blautia, Robinsoniella, and a population which could not be subclassified down to the genus level (Unc. Lachnospiraceae). The family of Clostridiaceae were predominantly composed of a single genus, Clostridium (dark blue). Treatment with broad spectrum antibiotics for 4 weeks in duration lead to the outgrowth of 2 dominant Proteobacteria genus, Stenotrophomonas (93% ± 0 .5) and Xanthamonas (7% ± 0 .3). Concurrent treatment of antibiotics and probiotics for 2 weeks lead to a decrease of the Xanthamonas population (5% ± 0 .3) while the Stenotrophomonas maintained its overall percentage of microbial abundance. Probiotic treatment did cause an increase in Firmicutes, such as the Lachnospiraceae family (0.5%), and other Proteobacteria such as the genus of Eschericia (0.2%) and Shigella (0.1%). These data indicate the antibiotics used in this study effectively eliminated nearly all bacterial diversity leading to the dominance of the Xanthomonadaceae and eliminating variation in the microbiome. Concurrent antibiotic and probiotic treatment exhibited a marginal shift in the bacterial community compared to antibiotic alone treatment ()
The recovery from antibiotics can occur rapidly or require a long duration, depending on the antibiotics used in the study.Citation28 In our previous experiments we had identified that 2 weeks treatment of our antibiotic mixture resulted in essentially a monoculture of Stenotrophomonas while diversity at 2 and 4 weeks recovery indicated a fairly rapid return to highly diverse microbiome(, previous data not shown). Commensurate with the changes in the microbiome are changes in the gross anatomy of the mouse. Antibiotic treatment causes the ceca to significantly enlarge and develop a deeper shade of brown (supplemental ). Removal of antibiotics causes the ceca to return to more normal size and color although still significantly larger compared to control animals (p < 0.01).
To test the impact of probiotics on the recovery of the microbiome we examined the composition of cecal and large intestine contents from probiotic fed mice. In line with several other reports, we rarely found any OTUs which associated with the bacterial genera in the probiotic mix. However, the mice fed probiotics throughout recovery exhibited a bloom of Anaerotruncus (53% ± 23 ) with the microbiome dominated by this single bacterial genus in 3 out of 4 mice (). This is in stark contrast to animals which were allowed to recover from antibiotic therapy without further manipulation. These animals exhibited an outgrowth of opportunistic pathogens, specifically Eschericia (12.3% ± 13 ) and Shigella (7% ± 7 .6)(). Where one mouse appeared to be highly susceptible to these opportunistic pathogens (49%), 2 mice were intermediate for their abundance (13%), and one mouse was largely resistant to colonization (1%). These data suggest that probiotics suppress opportunistic pathogens but promote the significant outgrowth of a relatively unknown bacterial genus, Anaerotruncus.
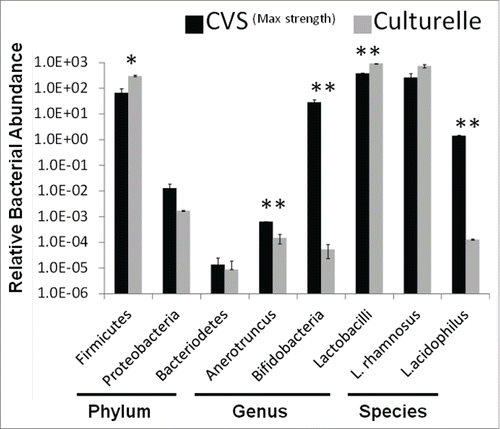
The bacteria found in probiotics are not always consistent with their package descriptions.Citation29,30 Thus, we used quantitative PCR to determine if the Anaerotruncus bloom was introduced by the probiotic vs. an outgrowth of the recovering microbiome. To control for background vs. purposefully added bacteria in the probiotic mix, we examined the probiotic Culturelle™ and compared it to the CVS Maximum Strength probiotic used in this study. The CVS brand probiotic contains Lactobacillus rhomnosus, Lactobacillus acidophilus, and Bifidobacteria whereas the Culturelle probiotic should only contain Lactobacillus rhamnosus. Based on 16S primers specific for major bacterial phylum, both probiotics contain predominantly Firmicutes with a minor contamination of Proteobacteria (<0 .01%) and Bacteroidetes (<10−5%) (). At the genus level, both probiotics had robust amounts of lactobacilli but only the CVS probiotic exhibited a significant quantity of Bifidobacteria. The genus specific primers for Anaerotruncus were also of very low abundance in the bacterial mix. Although there is statistically more Anaerotruncus in the CVS brand probiotic (p < 0.01) compared to Culturelle™, the levels of abundance were commensurate with the negligible background levels of Bifidobacteria amplification found in the Culturelle™ probiotic. As a second control for background levels we also examined the levels of abundance for L. acidophilus which should be absent from the Culturelle™ probiotic. The relative abundance of L. rhamnosus was high for both probiotics and only the CVS brand gave a robust signal for the presence of L. acidophilus. Levels of Anaerotruncus were commensurate with the background levels of L. acidophilus found in Culturelle™. Collectively, the Anaerotruncus observed in the recovering mice treated with probiotics is highly unlikely to have been acquired from the probiotic itself, although this cannot be definitively ruled out.
Figure 5. Relative abundance of bacterial taxa in probiotics. Quantitative PCR was used to determine the relative abundance of bacteria in probiotic supplements to determine ‘purity’. Probiotic mixture CVSMax Strength was compared to mono-culture probiotic Culturelle™. Probiotics were screened at the phylum, genus, and species level using taxa specific PCR primers. (* = p < 0.05, ** = p < 0.01) Representative of 3 independent experiments.
Discussion
The intestinal microbiota of vertebrates has coevolved with each host species and is shaped by the types of foods eaten, physiology of the gastrointestinal tract, immune status, and competition between commensal bacteria.Citation31,32 Once established the microbiota of each individual is relatively unique, with many shared major taxa, and fairly stable throughout life.Citation11 These microbial denizens have helped shape their host as evidenced by several immune defects and physiology changes identified in germ-free mice.Citation32 Studies to examine the colonization of the intestinal tract have looked at introduction of microbiota to these germ free mice.Citation32,33 An alternative to these measures would be to eliminate the microbiota via antibiotic administration and then manipulate the bacterial constituents during re-population to achieve a healthy “normal” microbiota.
The disruption of intestinal microbiota, dysbiosis, has been associated with a multitude of diseases. The abundance of Bacteroidetes vs Firmicutes/Actinobacteria has been associated with host obesity.Citation34,35 The microbial diversity has been hypothesized to impact the host immune response enhancing susceptibility to allergy.Citation36 The microbiota can enhance, or suppress, immune status of the intestines during inflammatory bowel disease.Citation26 Most importantly, the commensal flora is able to keep opportunistic pathogens at bay.Citation7,37 This defensive capacity is most prevalent following a course of antibiotics taken by the host. Although protection from infection can be attributed to a single commensal bacterial species in some cases, it is now well accepted that the overall diversity of the microbial flora is the best indicator of protective capacity.Citation38 Two specific areas of intense investigation have emerged regarding microbial diversity as a mechanism of protection, antibiotic induced diarrheal disease (ADD) and infections with Clostridium difficile (C. diff).Citation26 In both situations manipulation of the microbiome has proven to be a promising method for treating the disease.Citation14-16 In particular, the use of fecal transplant has shown significant efficacy for C.diff infections which have been resistant to several rounds of antibiotic treatment.Citation39 In a similar mechanism, mice treated with antibiotics recover significantly faster and with greater microbial diversity when co-housed with a normal control mouse, due to natural coprophagic activity.Citation28 Multiple meta-analysis of ADD have found that concurrent intake of probiotics along with antibiotics result in the reduction in ADD onset.Citation15,40
Probiotics, live microbes with health promoting benefits, are becoming increasingly used as a manipulation of the host microbiome. Following ingestion, these probiotic strains are rarely identified as established members of the fecal microbiota and thus their impact may require constant administration.Citation41-43 In “healthy flora” the addition of a probiotic does not readily impact the resident microbial populations.Citation43,44 However, in a situation exhibiting dysbiosis the addition of probiotics could have a significant impact on reshaping the microbiota. Since there is no regulatory body which has oversight of probiotics it requires the research community investigate this over the counter treatment.Citation45 Mislabeled products can significantly impact the interpretation of probiotic safety and efficacy.Citation29,30 Commonly used probiotic strains have also been documented to contain antibiotic resistance genes which could create a potential source for transfer of resistance to other members of the microbial flora.Citation46 In some rare incidents, such as patients with underlying medical conditions, the probiotic strain can become the pathogen causing an adverse event.Citation47-49 Collectively, a better understanding of the impacts of probiotics on the microbial flora is required to ascertain how best to use them to treat disease.
This study examined the impact of probiotics under 2 separate conditions, probiotic concurrent administration with antibiotics and probiotic administration during the recovery phase. The sample size in this study was small, but intergroup variation in the biomes was marginal relative to across group comparison making the biomes very distinguishable among treatments. The broad spectrum antibiotic treatments effectively reduced the microbial flora to 2 genera, Stenotrophomonas and Xanthomonas. Stenotrophomonas being the predominant genus is of note since this highly antibiotic resistant bacteria is also a potential emerging opportunistic pathogen.Citation50,51 Stenotrophomonas infections have been associated with poor prognosis for cystic fibrosis treatment.Citation52 We found that concurrent treatment with antibiotics and probiotics lead to an increase in microbial diversity, albeit a small percentage and not statistically significant. The probiotics themselves did not persist in the ceca or large intestine of antibiotic treated mice, but it did promote the presence of more Firmicutes and Enterobacteriaceae. The increase in Escherichia and Shigella was small but could be considered relevant in the context that this enhances the starting populations for these genera as the host enters the recovery phase. Future experiments should examine recovery following concurrent Antibiotic/Probiotic treatment to determine the impact of these increases in Enterobacteriaceae microbial diversity as this could result in an adverse outcome and should be cautioned against in a clinical setting.
The administration of probiotics throughout the recovery phase had highly significant impacts. Comparisons based on rank abundance in the D-M analysis were unable to differentiate the control and recovery populations as they were equivalently complex. However, using taxonomic identifiers during Qiime analysis it is clear that the recovery groups were composed of drastically different types of bacteria. The mice lacking probiotic supplementation were susceptible in varying degrees to infections with the Enterobacteriaceae, genus Escherichia and Shigella. In contrast, these opportunistic pathogens were highly suppressed in the mice supplied probiotics. In line with current therapies for AAD, it is evident that the probiotic supplementation leads to an intestinal microbiota more resistant to opportunistic pathogens.Citation15,40 In our study, the intestinal microbiome of probiotic treated mice was largely dominated by the genus Anaerotruncus. The Anaerotruncus have twice been described, in the context of fecal samples from 2 boys with late onset autism and isolated from a fecal sample of an elderly women with bacteraemia.Citation53,54 The Anaerotruncus were included as one of 17 strains shown to induce a suppressive environment capable of inducing regulatory T-cells.Citation55 This group of 17 strains have also been associated with improved outcomes from fecal transplant to treat inflammatory bowel disease.Citation56 The mislabeling or “pure” clonality of probiotics has been problematic in previous reports and thus we determined if the Anaerotruncus were being introduced directly via the probiotic.Citation29,30 Based on the real-time PCR analysis, it is highly unlikely that the probiotic was the cause of the microbial bloom (). The recovery alone mouse also had a significant bump in the genus Anaerotruncus (1.9%±0 .7), a 4fold increase from control mice never treated with antibiotics. Thus, it is most likely that the probiotic enhanced the growth of the Anaerotruncus from the resident flora during the recovery process. Additional studies on the intestinal health impacts of Anaerotruncus are warranted due to their very significant domination of the recovering microbiota. It was also noted that the supplementation of probiotics effectively eliminated Bacteroidetes from the cecal and intestinal microbiome (). The loss of this phylum of bacteria due to probiotic supplementation could have significant impact on their capacity to repopulate as part of the host microbiota. Since the Firmicutes/Bacteroidetes ratio can be tied to obesity, probiotic supplementation could have far reaching consequences in health and nutrition following antibiotic treatment. Citation9,34
The mechanism by which these probiotics are exerting their ‘healthy benefits’ are still largely unknown. Two potential mechanisms have been purported. First, probiotics can act directly on the intestinal mucous layer and underlying epithelium to impact intestinal barrier function and mucosal immunity. Secondly, the probiotic could have an indirect impact on the host by causing a modulation in the resident microbial population structure or enzymatic activity.Citation16 To suppress local inflammation at the intestine mucosal epithelium, bacteria can express super oxide dismutases (SOD). This reduction in local inflammation, driven by probiotic expression of SOD genes, has been correlated with improvements in mouse models of colitis.Citation57,58 Interestingly, subsets of mice were more responsive to this probiotic treatment based on the composition of the existing microbiota. The addition of the probiotic caused a metabolic shift in the microbiota which resulted in a non-permissive environment for opportunistic pathogens.Citation59 A more recent report by Eloe-Fadrosh et al. Citation43 examined the dynamics of the gut transcriptome following probiotic consumption. They identified an increase in flagellar motility, chemotaxis, and adhesion genes from resident commensal bacteria which were positively regulated by the addition of the probiotic to the host diet. These particular genes are hypothesized to improve the commensal bacteria's ability to colonize the mucosal epithelium. Thus, the probiotic can have both direct and indirect impacts on host biology and further studies should examine the transcriptome of both the host and microbiota to tease out these direct and indirect impacts.
It has been well established that disruption of the intestinal microbial community, such as antibiotic use, can lead to deleterious health consequences for the host organism. The use of probiotics is one mechanism which can be used to help reshape the microbial flora as it repopulates. Although the probiotic bacteria themselves do not appear to colonize and become part of the fecal waste, these probiotics have very significant impacts on the types of bacteria which repopulate the intestine. Probiotics as treatment to purposefully manipulate the microbiome will require further study to determine the mechanisms by which they promote a healthier intestinal microbiota.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KGMI_A_1138197.zip
Download Zip (1.3 MB)Acknowledgments
The authors would like to thank Dr. Brooks Miner and Dr. Ruth Ley for input on statistical tests and ecological data presentation. In particular the authors would like to thank the students of Ithaca College Microbiology Spring 13′ (D. Cimino, K.Dau, N.Dragotakes, A. Finley, A. Fonseca, K. Harney, S. Haugen, J. Hetterich, J. Messinger, D. Mills, S.Rabice, J. Schmidt, E. Seekamp, M. Sherry, L. Soderstrom, E. Stoyanova, K.Tumas, M. Vogel) and Ithaca College Microbiology Spring 15′(C. Brownell, B. Capirci, S. Casola, B.Champey, M. Clouden, G. Cordaro, N. Czajak, E. Edgerton, M. Getachew-Zewde, E. Gildersleeve, T.Kenney, J. Lisack, L. Metzger, B. Pattavina, A. Schultze, A. Wilks, A. Wishart, B. Zionkowski) for their work in collecting these data.
Funding
This manuscript was funded by Ithaca College and grants from the Ford Fund.
References
- Krych L, Hansen CHF, Hansen AK, van den Berg FWJ, Nielsen DS. Quantitatively different, yet qualitatively alike: a meta-analysis of the mouse core gut microbiome with a view towards the human gut microbiome. PLoS One 2013; 8(5):e62578; PMID:23658749; http://dx.doi.org/10.1371/journal.pone.0062578
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature 2012; 489(7415):220-30; PMID:22972295; http://dx.doi.org/10.1038/nature11550
- Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, Ugarte E, Muñoz-Tamayo R, Paslier DL, Nalin R, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 2009; 11(10):2574-84; PMID:19601958; http://dx.doi.org/10.1111/j.1462-2920.2009.01982.x
- Hooper L V. Bacterial contributions to mammalian gut development. Trends Microbiol 2004; 12(3):129-34; PMID:15001189; http://dx.doi.org/10.1016/j.tim.2004.01.001
- Lamousé-Smith ES, Tzeng A, Starnbach MN. The intestinal flora is required to support antibody responses to systemic immunization in infant and germ free mice. PLoS One 2011; 6(11):e27662; PMID:Can't; http://dx.doi.org/10.1371/journal.pone.0027662
- Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A 2008; 105(6):2117-22; PMID:18252821; http://dx.doi.org/10.1073/pnas.0712038105
- Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, Lupp C, Finlay BB. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect Immun 2008; 76(10):4726-36; PMID:18678663; http://dx.doi.org/10.1128/IAI.00319-08
- Knoop KA, McDonald KG, Kulkarni DH, Newberry RD. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut 2015; Jun 4:pii PMID:26045138; http://dx.doi.org/10.1136/gutjnl-2014-309059
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444(7122):1022-3; PMID:17183309; http://dx.doi.org/10.1038/4441022a
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491(7422):119-24; PMID:23128233; http://dx.doi.org/10.1038/nature11582
- Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J 2007; 1(1):56-66; PMID:18043614; http://dx.doi.org/10.1038/ismej.2007.3
- Salyers AA, Gupta A, Wang Y. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol 2004; 12(9):412-6; PMID:15337162; http://dx.doi.org/10.1016/j.tim.2004.07.004
- McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. The Impact of a Consortium of Fermented Milk Strains on the Gut Microbiome of Gnotobiotic Mice and Monozygotic Twins. Sci Transl Med 2011; 3(106):106ra106-106ra106; PMID:22030749; http://dx.doi.org/10.1126/scitranslmed.3002701
- Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog 2012; 8(10):e1002995; PMID:23133377; http://dx.doi.org/10.1371/journal.ppat.1002995
- Hempel S, Newberry SJ, Maher AR, Wang Z, Miles JN, Shanman R, Johnsen B, Shekelle PG. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA 2012; 307(18):1959-69; PMID:22570464; http://dx.doi.org/10.1001/jama.2012.3507
- Gerritsen J, Smidt H, Rijkers GT, de Vos WM. Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutr 2011; 6(3):209-40; PMID:21617937; http://dx.doi.org/10.1007/s12263-011-0229-7
- Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010; 156(Pt 11):3216-23; PMID:20705661; http://dx.doi.org/10.1099/mic.0.040618-0
- Janssen PH, Yates PS, Grinton BE, Taylor PM, Sait M. Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl Environ Microbiol 2002; 68(5):2391-6; PMID:11976113; http://dx.doi.org/10.1128/AEM.68.5.2391-2396.2002
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 2011; 108 Suppl:4516-22; PMID:20534432; http://dx.doi.org/10.1073/pnas.1000080107
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7(5):335-6; PMID:20383131; http://dx.doi.org/10.1038/nmeth.f.303
- Matsuki T, Watanabe K, Fujimoto J, Kado Y, Takada T, Matsumoto K, Tanaka R. Quantitative PCR with 16S rRNA-gene-targeted species-specific primers for analysis of human intestinal bifidobacteria. Appl Environ Microbiol 2004; 70(1):167-73; PMID:14711639; http://dx.doi.org/10.1128/AEM.70.1.167-173.2004
- Byun R, Nadkarni MA, Chhour K-L, Martin FE, Jacques NA, Hunter N. Quantitative analysis of diverse Lactobacillus species present in advanced dental caries. J Clin Microbiol 2004; 42(7):3128-36; PMID:15243071; http://dx.doi.org/10.1128/JCM.42.7.3128-3136.2004
- Bacchetti De Gregoris T, Aldred N, Clare AS, Burgess JG. Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J Microbiol Methods 2011; 86(3):351-6; PMID:21704084; http://dx.doi.org/10.1016/j.mimet.2011.06.010
- La Rosa PS, Brooks JP, Deych E, Boone EL, Edwards DJ, Wang Q, Sodergren E, Weinstock G, Shannon WD. Hypothesis testing and power calculations for taxonomic-based human microbiome data. PLoS One 2012; 7(12):e52078; PMID:23284876; http://dx.doi.org/10.1371/journal.pone.0052078
- Schubert AM, Sinani H, Schloss PD. Antibiotic-induced alterations of the murine gut microbiota and subsequent effects on colonization resistance against clostridium difficile. MBio 2015; 6(4):e00974; PMID:26173701; http://dx.doi.org/10.1128/mBio.00974-15
- Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 2013; 13(11):790-801; PMID:24096337; http://dx.doi.org/10.1038/nri3535
- Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 2012; 80(1):62-73; PMID:22006564; http://dx.doi.org/10.1128/IAI.05496-11
- Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, Young VB. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun 2009; 77(6):2367-75; PMID:19307217; http://dx.doi.org/10.1128/IAI.01520-08
- Huys G, Vancanneyt M, D'Haene K, Vankerckhoven V, Goossens H, Swings J. Accuracy of species identity of commercial bacterial cultures intended for probiotic or nutritional use. Res Microbiol 2006; 157(9):803-10; PMID:16919915; http://dx.doi.org/10.1016/j.resmic.2006.06.006
- Masco L, Huys G, De Brandt E, Temmerman R, Swings J. Culture-dependent and culture-independent qualitative analysis of probiotic products claimed to contain bifidobacteria. Int J Food Microbiol 2005; 102(2):221-30; PMID:15992621; http://dx.doi.org/10.1016/j.ijfoodmicro.2004.11.018
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 2010; 107(33):14691-6; PMID:20679230; http://dx.doi.org/10.1073/pnas.1005963107
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012; 149(7):1578-93; PMID:22726443; http://dx.doi.org/10.1016/j.cell.2012.04.037
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 2011; 108(15):6252-7; PMID:21436049; http://dx.doi.org/10.1073/pnas.1102938108
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature 2009; 457(7228):480-4; PMID:19043404; http://dx.doi.org/10.1038/nature07540
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341(6150):1241214; PMID:24009397; http://dx.doi.org/10.1126/science.1241214
- Shreiner A, Huffnagle GB, Noverr MC. The “Microflora Hypothesis” of allergic disease. Adv Exp Med Biol 2008; 635:113-4; PMID:18841708; http://dx.doi.org/10.1007/978-0-387-09550-9_10
- Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, Young VB. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun 2015; 83(3):934-41; PMID:25534943; http://dx.doi.org/10.1128/IAI.02768-14
- Ursell LK, Van Treuren W, Metcalf JL, Pirrung M, Gewirtz A, Knight R. Replenishing our defensive microbes. Bioessays 2013; 35(9):810-7; PMID:23836415; http://dx.doi.org/10.1002/bies.201300018
- Van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013; 368(5):407-15; PMID:23323867; http://dx.doi.org/10.1056/NEJMoa1205037
- McFarland L V. Meta-analysis of probiotics for the prevention of antibiotic associated diarrhea and the treatment of Clostridium difficile disease. Am J Gastroenterol 2006; 101(4):812-22; PMID:16635227; http://dx.doi.org/10.1111/j.1572-0241.2006.00465.x
- Alander M, Satokari R, Korpela R, Saxelin M, Vilpponen-Salmela T, Mattila-Sandholm T, von Wright A. Persistence of colonization of human colonic mucosa by a probiotic strain, Lactobacillus rhamnosus GG, after oral consumption. Appl Environ Microbiol 1999; 65(1):351-4; PMID:9872808
- Lee YK, Ho PS, Low CS, Arvilommi H, Salminen S. Permanent colonization by Lactobacillus casei is hindered by the low rate of cell division in mouse gut. Appl Environ Microbiol 2004; 70(2):670-4; PMID:14766540; http://dx.doi.org/10.1128/AEM.70.2.670-674.2004
- Eloe-Fadrosh EA, Brady A, Crabtree J, Drabek EF, Ma B, Mahurkar A, Ravel J, Haverkamp M, Fiorino AM, Botelho C, et al. Functional dynamics of the gut microbiome in elderly people during probiotic consumption. MBio 2015; 6(2):pii: e00231-15; PMID:25873374; http://dx.doi.org/10.1128/mBio.00231-15
- Lahti L, Salonen A, Kekkonen RA, Salojärvi J, Jalanka-Tuovinen J, Palva A, Orešič M, de Vos WM. Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ 2013; 1:e32; PMID:23638368; http://dx.doi.org/10.7717/peerj.32
- Venugopalan V, Shriner KA, Wong-Beringer A. Regulatory oversight and safety of probiotic use. Emerg Infect Dis 2010; 16(11):1661-5; PMID:21029521; http://dx.doi.org/10.3201/eid1611.100574
- Sanders ME, Akkermans LMA, Haller D, Hammerman C, Heimbach J, Hörmannsperger G, Huys G, Levy DD, Lutgendorff F, Mack D, et al. Safety assessment of probiotics for human use. Gut Microbes 1(3):164-85; PMID:21327023; http://dx.doi.org/10.4161/gmic.1.3.12127
- Land MH, Rouster-Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005; 115(1):178-81; PMID:15629999
- Rautio M, Jousimies-Somer H, Kauma H, Pietarinen I, Saxelin M, Tynkkynen S, Koskela M. Liver abscess due to a Lactobacillus rhamnosus strain indistinguishable from L. rhamnosus strain GG. Clin Infect Dis 1999; 28(5):1159-60; PMID:10452653; http://dx.doi.org/10.1086/514766
- Doern CD, Nguyen ST, Afolabi F, Burnham C-AD. Probiotic-associated aspiration pneumonia due to Lactobacillus rhamnosus. J Clin Microbiol 2014; 52(8):3124-6; PMID:24899028; http://dx.doi.org/10.1128/JCM.01065-14
- Brooke JS. Stenotrophomonas maltophilia: an Emerging Global Opportunistic Pathogen. Clin Microbiol Rev 2012; 25(1):2-41; PMID:22232370; http://dx.doi.org/10.1128/CMR.00019-11
- Looney WJ, Narita M, Mühlemann K. Stenotrophomonas maltophilia: an emerging opportunist human pathogen. Lancet Infect Dis 2009; 9(5):312-23; PMID:19393961; http://dx.doi.org/10.1016/S1473-3099(09)70083-0
- Waters V, Atenafu EG, Lu A, Yau Y, Tullis E, Ratjen F. Chronic Stenotrophomonas maltophilia infection and mortality or lung transplantation in cystic fibrosis patients. J Cyst Fibros 2013; 12(5):482-6; PMID:23294530; http://dx.doi.org/10.1016/j.jcf.2012.12.006
- Lau SKP, Woo PCY, Woo GKS, Fung AM, Ngan AH, Song Y, Liu C, Summanen P, Finegold SM, Yuen K. Bacteraemia caused by Anaerotruncus colihominis and emended description of the species. J Clin Pathol 2006; 59(7):748-52; PMID:16467163; http://dx.doi.org/10.1136/jcp.2005.031773
- Lawson PA, Song Y, Liu C, Molitoris DR, Vaisanen ML, Collins MD, Finegold SM. Anaerotruncus colihominis gen. nov., sp. nov., from human faeces. Int J Syst Evol Microbiol 2004; 54(Pt 2):413-7; PMID:15023953; http://dx.doi.org/10.1099/ijs.0.02653-0
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013; 500(7461):232-6; PMID:23842501; http://dx.doi.org/10.1038/nature12331
- Satokari R, Fuentes S, Mattila E, Jalanka J, de Vos WM, Arkkila P. Fecal transplantation treatment of antibiotic-induced, noninfectious colitis and long-term microbiota follow-up. Case Rep Med 2014; 2014:913867; PMID:25548572
- Watterlot L, Rochat T, Sokol H, Cherbuy C, Bouloufa I, Lefèvre F, Gratadoux JJ, Honvo-Hueto E, Chilmonczyk S, Blugeon S, et al. Intragastric administration of a superoxide dismutase-producing recombinant Lactobacillus casei BL23 strain attenuates DSS colitis in mice. Int J Food Microbiol 2010; 144(1):35-41; PMID:20452077; http://dx.doi.org/10.1016/j.ijfoodmicro.2010.03.037
- Ballal SA, Veiga P, Fenn K, Michaud M, Kim JH, Gallini CA, Glickman JN, Quéré G, Garault P, Béal C, et al. Host lysozyme-mediated lysis of Lactococcus lactis facilitates delivery of colitis-attenuating superoxide dismutase to inflamed colons. Proc Natl Acad Sci U S A 2015; 112(25):7803-8; PMID:26056274; http://dx.doi.org/10.1073/pnas.1501897112
- Veiga P, Gallini CA, Beal C, Michaud M, Delaney ML, DuBois A, Khlebnikov A, van Hylckama Vlieg JE, Punit S, Glickman JN, et al. Bifidobacterium animalis subsp. lactis fermented milk product reduces inflammation by altering a niche for colitogenic microbes. Proc Natl Acad Sci U S A 2010; 107(42):18132-7; PMID:20921388; http://dx.doi.org/10.1073/pnas.1011737107