
ABSTRACT
The discovery of enzymes responsible for previously unappreciated microbial metabolic pathways furthers our understanding of host-microbe and microbe-microbe interactions. We recently identified and characterized a new gut microbial glycyl radical enzyme (GRE) responsible for anaerobic metabolism of trans-4-hydroxy-l-proline (Hyp). Hyp dehydratase (HypD) catalyzes the removal of water from Hyp to generate Δ1-pyrroline-5-carboxylate (P5C). This enzyme is encoded in the genomes of a diverse set of gut anaerobes and is prevalent and abundant in healthy human stool metagenomes. Here, we discuss the roles HypD may play in different microbial metabolic pathways as well as the potential implications of this activity for colonization resistance and pathogenesis within the human gut. Finally, we present evidence of anaerobic Hyp metabolism in sediments through enrichment culturing of Hyp-degrading bacteria, highlighting the wide distribution of this pathway in anoxic environments beyond the human gut.
Introduction
The trillions of microorganisms that live in and on the human body play a central role in human physiology.Citation1 Recent research focused on the inhabitants of the human gastrointestinal tract (the human gut microbiota) has highlighted the potential for this community to influence host health and disease.Citation2 While much progress has been made in characterizing community compositions, we know surprisingly little about the biochemical functions of the gut microbiota and the mechanisms by which microbes influence host biology.Citation3 In order to understand the metabolic potential of the gut microbiota and inform mechanistic research, it is imperative that we link the genes present in this habitat to functions. However, the biological roles of most microbial genes remain unidentified, indicating a great need to investigate this uncharacterized genetic information.Citation3,Citation4
Discovery of a glycyl radical enzyme responsible for anaerobic Hyp metabolism
We recently described a functional profiling pipeline that can guide the discovery of novel enzymes in microbiomes.Citation5 We used this workflow to assess the distribution of GREs in Human Microbiome Project (HMP) metagenomes. Members of the GRE superfamily participate in anaerobic primary metabolism, including DNA synthesis (class III ribonucleotide reductase) and keto acid fermentation (pyruvate formate-lyase and ketobutyrate formate-lyase).Citation6,Citation7 Furthermore, GREs represent one of the most abundant protein families in human stool metagenomes and metaproteomes.Citation8,Citation9 A GRE must be posttranslationally modified by a partner radical S-adenosylmethionine (SAM) activating enzyme (AE), which installs a radical on the α-carbon of a conserved glycine residue ().Citation7,Citation10 The use of protein-based radicals enables these enzymes to perform a diverse set of chemically challenging transformations ().Citation6,Citation7,Citation10 Due to their susceptibility to inactivation by molecular oxygen, both GREs and AEs are only present in facultative and obligate anaerobes.Citation10,Citation11
Figure 1. (A) General mechanism of glycyl radical enzymes (GREs). A partner radical S-adenosylmethionine (SAM) activating enzyme (AE) first abstracts a hydrogen atom from the α-carbon of a conserved active site glycine residue on the GRE to generate a carbon-centered radical. The glycine-centered radical is proposed to abstract a hydrogen atom from an essential active site cysteine residue to generate a thiyl radical intermediate that initiates reaction with the substrate. Further reaction of the substrate-based radical generates a product-based radical, which then abstracts a hydrogen atom from the essential cysteine residue to regenerate the thiyl radical. (B) Previously characterized activities of 4-hydroxyproline dehydratase (HypD) and Δ1-pyrroline-5-carboxylate reductase (P5CR) from Clostridioides difficile 70-100-2010.
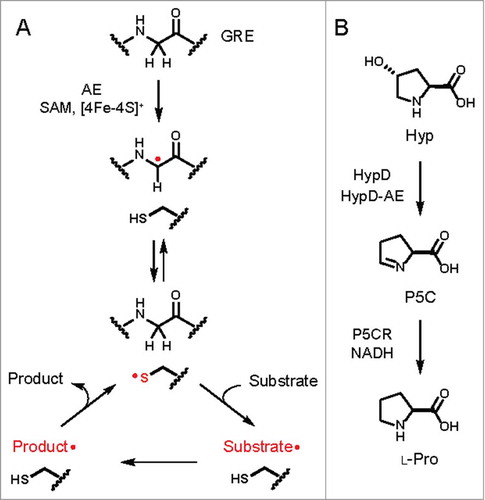
Our chemically guided functional profiling effort identified HypD as a GRE of unknown function that was both prevalent and highly abundant in the HMP stool metagenomes. To predict the biochemical function of HypD, we obtained insights from sequence analyses, structure prediction, and analysis of its genomic context. Sequence alignments of HypD with characterized GREs and construction of a homology model revealed an active site resembling those of GRE dehydratases. Additionally, a gene annotated as P5C reductase (P5CR) is colocalized with the hypD gene in many Clostridial genomes. P5CR catalyzes the reduction of P5C to l-proline (Pro) in Pro biosynthesis. We therefore hypothesized that this new GRE might catalyze the removal of water from Hyp to generate P5C, which could then be reduced to Pro by P5CR (). Using an in vitro coupled enzyme assay, we validated this hypothesis experimentally. In addition, HypD displays a strong preference for the biologically relevant stereoisomer of 4-hydroxyproline, Hyp. Together, these experiments confirmed the activity of this prominent gut microbial GRE and revealed the first anaerobic pathway for Hyp metabolism.
The discovery of HypD has allowed us to identify microbial species capable of anaerobic Hyp metabolism and to study the distribution of this activity across various environments. In this addendum, we discuss possible roles HypD may have in various microbial metabolic pathways, assess the potential biological relevance of anaerobic Hyp utilization within the human gut microbiota, and present evidence that this activity is widespread in additional habitats. We also illustrate the importance of enzyme discovery in microbiota research by highlighting how expanding our knowledge of microbial metabolism has helped to shed light on the roles of an underappreciated, but abundant host-derived amino acid in the gut microbiota.
Strategies for microbial Hyp metabolism
Our previous work examined Hyp metabolizing enzymes, HypD, AE, and P5CR, from Clostridioides difficile 70-100-2010 (formerly classified as Clostridium difficile). The presence of this pathway in the order Clostridiales was notable given that anaerobic metabolism of Hyp was first observed in Clostridiales.Citation13 These organisms carry out Stickland fermentation wherein pairs of amino acids are used as electron donors and acceptors in energy metabolism.Citation12 It was known as early as 1934 that Pro and Hyp can be used as electron acceptors in such fermentations.Citation12 Furthermore, it was shown that Hyp can induce the expression of an enzyme responsible for Pro reduction in C. difficile.Citation13 In contrast to the characterized pathway for Pro reduction (), the enzymes and downstream metabolites involved in Hyp fermentation had not been identified until our work.Citation12
Figure 2. Characterized pathways for Hyp metabolism. (A) Pathway for Stickland fermentation using l-proline (Pro) as an electron acceptor. The product of Pro reduction, 5-aminovalerate, can be further oxidized and reduced by certain Clostridiales to generate short-chain fatty acids and ammonia. (B) HypD provides an anaerobic route for Hyp metabolism. (C) In mammals, Hyp is oxidized to glyoxylate and pyruvate in a multistep pathway. (D) An oxidative pathway for Hyp metabolism generates α-ketoglutarate (α-KG) and ammonia as sources of carbon and nitrogen.
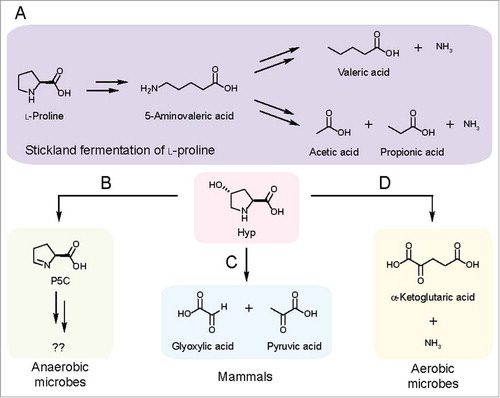
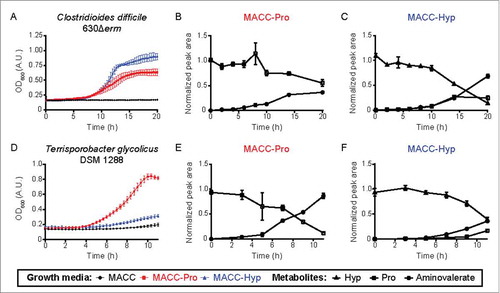
The discovery of HypD suggested that Stickland fermentation of Hyp involves the generation and reduction of Pro. Indeed, HypD is present in species previously demonstrated to use Hyp and Pro as electron acceptors (C. difficile, Clostridium sporogenes).Citation12,Citation13 Notably, ∼65% of the Clostridiales species that possess the prd operon, which is essential for Pro reduction, also encode HypD.Citation13,Citation14 In these species, the conserved proC gene (encoding P5CR) adjacent to hypD provides a strong indication that Hyp is metabolized to Pro and used as an electron acceptor. In Stickland fermentation, Pro is metabolized via a 2-step pathway to 5-aminovalerate, which often accumulates as an end product ().Citation14,Citation15 To test whether HypD-encoding Clostridiales consume Hyp via this pathway, we examined two strains encoding both hypD and the prd operon (C. difficile 630Δerm and Terrisporobacter glycolicus DSM 1288). We confirmed that they generate 5-aminovalerate when grown in the presence of Hyp (). In some species, 5-aminovalerate can be further metabolized for energy production ().Citation16,Citation17 Though enzymes involved in these transformations have been purified and characterized, their sequences have not been reported and thus the distribution of this pathway in microbes remains to be determined.
Figure 3. Growth curves and metabolite analysis of two HypD-encoding Clostridiales cultures reveal an increase in downstream Stickland fermentation metabolites accompanying Hyp consumption. (A, D) Enhanced growth was observed for both C. difficile 630Δerm and T. glycolicus DSM 1288 upon supplementation of a previously reported medium with either Pro or Hyp (20 mM final concentration).Citation5 Downstream metabolites were quantified at various timepoints in cultures supplemented with Pro (B, E) or Hyp (C, F). Experiments were set up as previously describedCitation5 with the following modifications: 200 µL cultures were grown in 96-well plates and OD600 measurements were taken using a PowerWave HT Microplate Spectrophotometer (BioTek). Samples were collected at various time points for metabolite quantification by liquid chromatography tandem-mass spectrometry.Citation5 5-Aminovalerate was measured using the Hyp detection method by monitoring precursor and product ions of m/z 118.1 → m/z 101.1. Sample peak areas are normalized to the corresponding standard. Data points are shown as mean ± standard deviation with n = 5.
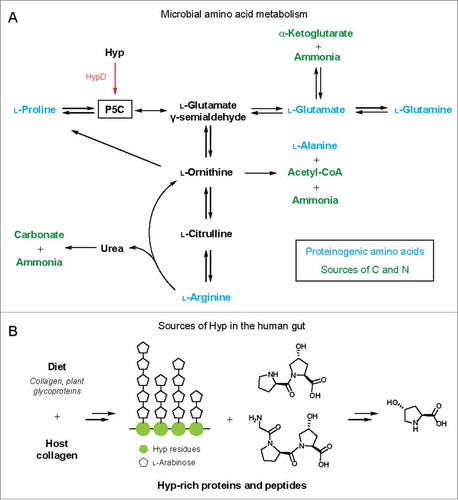
We next wondered whether HypD was important for metabolic pathways beyond Stickland fermentation. A multigene BLASTCitation18 search revealed that only a small subset of HypD-encoding genomes has a neighboring proC gene. This suggests that the majority of HypD-containing species do not reduce Hyp to Pro. In some organisms, including Oscillibacter valericigenes, a gene encoding a putative dehydrogenase is located in close proximity to HypD, indicating that P5C may be oxidized to l-glutamate.Citation19 Additionally, P5C can be converted into several other amino acids ().Citation20–Citation23 Interfacing HypD with these pathways could allow microbes to access building blocks for protein synthesis, substrates for fermentation, and sources of carbon and nitrogen. Overall, the precise role of Hyp and its downstream metabolites remain unclear in most of the anaerobes that encode HypD ().
Figure 4. (A) Hyp metabolism by HypD interfaces with microbial amino acid metabolic pathways. Arrows can represent multiple steps and only key metabolites are shown. P5C is a central intermediate in amino acid metabolism. The downstream metabolites α-KG, carbonate, acetyl-CoA, and ammonia can serve as sources of carbon and nitrogen. (B) Sources of Hyp in the human gut from diet or endogenous collagen turnover. Major collagen-derived peptides and Hyp repeats from extensin, a plant cell wall glycoprotein, are shown as examples.
In addition to anaerobic Hyp metabolism mediated by HypD, a different pathway allows aerobic bacteria to use Hyp as a sole source of carbon and nitrogen ().Citation24 The enzymes from this pathway have been characterized in Pseudomonas and Sinorhizobium.Citation20,Citation25 Aerobic Hyp metabolism involves sequential oxidations of Hyp to generate α-ketoglutarate (α-KG) and ammonia, providing carbon and nitrogen for anabolic pathways (). cis-4-Hydroxy-d-proline dehydrogenase carries out a key step in this pathway and shares high sequence similarity with hydrogen cyanide synthase, which is predicted to use O2 as the terminal electron acceptor.Citation20,Citation26 Given its distribution and the potential requirement for O2, we hypothesize that this metabolism occurs only in aerobes. To assess the presence of this pathway in the human microbiota, we performed TBLASTNCitation27 searches of the HMP reference genomes (NCBI BioProject: PRJNA28331) using two characterized cis-4-hydroxy-d-proline dehydrogenasesCitation20 as query. This search identified hits mostly in organisms isolated from the skin and respiratory tract. Therefore, we can conclude that this alternative pathway likely does not contribute significantly to Hyp consumption in the gut.
To summarize, there are two characterized pathways for Hyp metabolism in microbes (, D). Although we cannot rule out the possibility of additional, yet-to-be-discovered pathways, findings from culture-based studies have clearly implicated these two routes. The logic underlying these pathways is distinct, with the oxidative metabolism of Hyp providing essential nutrients for aerobic bacteria, while the anaerobic pathway uses Hyp as an electron sink to regenerate NAD+ as part of Stickland fermentation in Clostridiales, but likely has distinct roles in other species.
HypD is widely distributed across microbial species
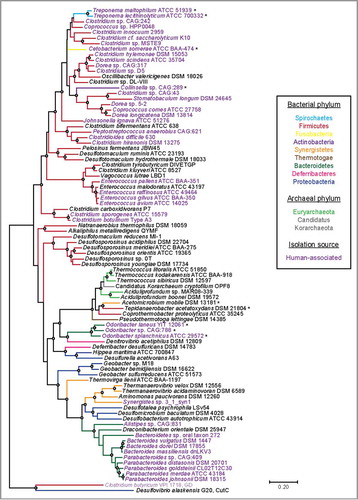
A particularly striking feature of HypD is its wide distribution in microbial genomes. Previous BLAST searches revealed HypD in >850 genomes from the NCBI database, with the majority of hits found in Firmicutes and Bacteroidetes.Citation5 HypD is not just widely distributed across bacterial phyla but is also present in several archaeal genomes. Notably, we found HypD in many common oral and gut species from genera Treponema, Clostridium, Bacteroides, Parabacteroides, and Enterococcus (), leading us to propose that HypD is likely responsible for the anaerobic Hyp degradation previously observed in human fecal and gingival samples.Citation28,Citation29 In addition to human commensals, HypD is also encoded in several notable human pathogens including C. difficile and Clostridium botulinum (). To understand evolutionary relationships between HypDs from different bacteria and archaea, we constructed a phylogenetic tree using representative protein sequences from the UniProt database (). HypD sequences mostly grouped according to phylum with a few exceptions. Cetobacterium somerae, Collinsella sp., and Treponema species represent three distinct phyla, but their HypD sequences all claded with sequences from Firmicutes. Interestingly, the hypD genes in these three organisms are colocalized with an adjacent proC gene, sharing the genomic organization commonly found in Clostridiales. This shared genomic context may indicate a recent horizontal gene transfer event. Additionally, HypDs from Odoribacter species form a distinct clade from the HypDs found in other Bacteroidetes. Archaeal HypDs formed their own clade and are closely related to HypDs from thermophilic bacteria. Overall, the presence of HypD in eleven different phyla suggests that Hyp utilization is an important metabolic function in anaerobes.
Figure 5. A maximum likelihood tree of 86 full-length HypD amino acid (aa) sequences from the UniProt database. One sequence per species was included. Sequences were aligned with MAFFT using the G-INS-I refinement methodCitation66 and the alignment was trimmed to yield a final length of 783 aa. The tree was constructed in MEGA 767 using the Jones–Taylor–Thorton matrix-based model.Citation68 HypD sequences from species that clade differently from 16S rDNA phylogeny are indicated with an asterisk. Bootstrap values of 70 to 100% are indicated by open circles. Choline trimethylamine-lyase (CutC) and glycerol dehydratase (GD) are included as outgroups.
Hyp is an abundant amino acid in the eukaryotic world
The broad distribution of HypD suggests that Hyp is a widely available substrate for microbial metabolism, which is in agreement with the prevalence of Hyp biosynthesis. Hyp is an abundant non-proteinogenic amino acid generated by prolyl 4-hydroxylase and proline 4-hydroxylase enzymes. These mononuclear non-heme iron and α-KG dependent dioxygenases are found in all domains of life.Citation30 Eukaryotes encode prolyl 4-hydroxylases, which posttranslationally modify Pro residues in proteins and peptides.Citation30 In mammals, Hyp is almost exclusively found in collagen, which is the most abundant protein in the human body.Citation30 Hyp formation is also critical for the function of additional structural and regulatory proteins, including elastin and hypoxia inducible factor.Citation30 Overall, Hyp is estimated to make up ∼4% of the amino acid pool in the human proteome, making it more abundant than several proteinogenic amino acids.Citation30 In addition to animals, plants and algae also produce proteins enriched in Hyp that are important for cell wall rigidity, growth, and signaling.Citation30,Citation31 Indeed, Hyp-rich glycoproteins (HRGPs) are a major component of the plant cell wall, with the 4-hydroxyl groups of Hyp residues serving as sites for O-glycosylation.Citation32
In contrast to its prevalence in eukaryotes, Hyp biosynthesis is uncommon in bacteria and archaea. Most Hyp-generating species use proline 4-hydroxylases to hydroxylate free Pro.Citation30 Free Hyp is synthesized by halophiles and acts as an osmolyte under high salt concentrations.Citation33 Hyp is also generated for use as a building block in the biosynthesis of non-ribosomal peptides, including actinomycin and etamycin.Citation34,Citation35 There are also two reported examples of bacterial prolyl 4-hydroxylases that form Hyp through posttranslational modification of peptidyl Pro.Citation36,Citation37 Overall, it appears that eukaryotes are the primary source of Hyp with bacteria contributing to a much lesser extent.
Sources of Hyp and its metabolism by the human host
The prevalence of HypD in the human gut microbiota raises the fascinating question of how these organisms obtain Hyp. Because Hyp is highly abundant in both mammalian and plant proteins, a wide variety of dietary substrates contain this amino acid (). In meat products, Hyp content correlates with the amount of connective tissue and can be detected at up to 1% in weight.Citation38,Citation39 Hyp is also enriched in dietary fibers that contain plant cell wall polysaccharides as a major component. For example, Hyp makes up over 4% of the protein content in dietary fibers extracted from grape pomace.Citation40 Arabinogalactan proteins, a class of HRGPs, are also a major component in gum arabic, which is used as a thickening and stabilizing agent in the food industry and is found in products like candies, gum, and beverages.Citation41 In addition to dietary sources, it is estimated that endogenous collagen turnover in humans releases ∼300 mg of Hyp daily.Citation42 Overall, both diet and endogenous collagen are candidate sources of Hyp for microbial metabolism in the human body ().
The availability of Hyp to the gut microbiota may also be influenced by host pathways for processing this amino acid. A metabolomics study in humans identified Hyp as a top plasma biomarker for distinguishing omnivores from vegans due to its higher abundance in omnivores.Citation43 This finding supports the proposal that diet is a significant source of Hyp and is potentially in agreement with the higher Hyp content of meat as compared to plant material. This result may also suggest that dietary changes affect the degree to which gut microbes and/or the host metabolize Hyp. In humans, 80–90% of free Hyp is efficiently metabolized to glyoxylate and pyruvate in the kidneys and liver ().Citation21,Citation42 Glyoxylate produced from host Hyp metabolism can be oxidized to oxalate, the accumulation of which can lead to kidney stone formation.Citation44 Up to 95% of Hyp excreted in the urine is incorporated into peptides, with Pro-Hyp as the major species.Citation42 Interestingly, this peptide is not efficiently hydrolyzed by host peptidases.Citation45,Citation46
In addition to Hyp-containing foods, collagen itself is often recommended as a dietary supplement and there is evidence that collagen-derived peptides have beneficial effects on host cells, including antioxidant, anticancer, and proliferative activities.Citation46 Collagen-derived peptides have been detected in plasma after ingestion of collagen.Citation46 Contribution of the gut microbiota to hydrolysis of both host and dietary collagen and collagen-derived peptides has not been studied in detail and may have a significant impact on the availability of these bioactive peptides for the host. In addition, we propose that the gut microbiota could complement host protein metabolism by hydrolyzing recalcitrant short peptides to release free amino acids for further use by both microbes and host. This process would parallel the roles of gut microbes in metabolizing indigestible polysaccharides.
Biological implications of HypD activity in the human gut microbiota
The conditions under which Hyp metabolism occurs in the human gut and the effects of this microbial metabolic activity on host biology are not clear. As Hyp is rarely included in amino acid measurements, Hyp concentrations within the gastrointestinal tract are largely unknown, with one study detecting free Hyp as the most abundant amino acid in the large intestine of one human subject.Citation47 Likewise, Hyp metabolism by gut microbes has not been studied in detail. However, metabolomics experiments in animal models provide evidence that gut microbes can use this amino acid.Citation48-Citation50 Specifically, free Hyp and Hyp-containing peptides (Pro-Hyp, Gly-Pro-Hyp) were detected at higher levels in the urine and feces of germ-free rodents.Citation48-Citation50 Collectively, data from these studies strongly suggest that a significant amount of free Hyp and Hyp-containing peptides reaches the gut and that the gut microbiota plays a major role in Hyp metabolism.
Given the prominence of HypD in the healthy human gut microbiome, we propose that Hyp utilization plays a vital role in this microbial community. This primary metabolic pathway may influence many aspects of the gut microbiota and its interactions with the host. Specifically, Hyp metabolism may play a role in shaping microbiota composition by enabling organisms to access nutrients an energy source. Consumption of Hyp by gut commensals may deplete levels of this amino acid and potentially contribute to colonization resistance against HypD-encoding pathogens. Hyp metabolism may also be important in diseases that involve pathogen infiltration of host tissues and collagen degradation. Finally, the end products of Hyp metabolism may serve as signals to host cells. Here, we explore these potential roles of Hyp metabolism within the human body.
Increased Hyp uptake from the diet or increased turnover of endogenous collagen may promote the growth and expansion of gut microbes possessing HypD. Because the product of HypD, P5C, can be metabolized in various ways, we expect the fitness advantages conferred upon HypD-encoding species to depend on the overall nutrient landscape of the gut environment. For example, Stickland fermentation of Hyp is an energy-yielding process and could provide a significant growth advantage to Clostridiales over other HypD-encoding species when alternative electron acceptors are scarce.
In addition to Clostridiales, HypD is present in many common human gut Bacteroides and Parabacteroides species, including Bacteroides vulgatus and Parabacteroides distasonis (). Bacteroidetes are known for their ability to catabolize complex dietary and host-derived polysaccharides, which are often covalently linked to protein residues.Citation51 The prevalence of HypD in gut Bacteroidetes suggests that HRGPs may provide both carbohydrates and amino acids to gut microbes, and HypD-encoding species may possess proteases and hydrolases to access Hyp and other amino acids from these large glycoprotein substrates. The identification of gut bacterial enzymes that release free Hyp from either dietary plant glycoproteins or collagen presents an important challenge for future investigations.
In addition to supporting the growth of commensal microbes, Hyp metabolism may be important for colonization of opportunistic pathogens and toxin production. For example, C. difficile can utilize both Hyp and Pro as electron acceptors.Citation13 Although the regulation of toxin production in C. difficile is not well understood, fermentation of Pro and other amino acids are thought to indirectly influence this process by modulating NADH/NAD+ ratios in the cell.Citation52 Since metabolism of Hyp to Pro generates NAD+, this transformation may also affect virulence in C. difficile. Metabolomics and transcriptomics experiments in a mouse model for C. difficile infection have concluded that Stickland pathways are critical for host colonization.Citation53,Citation54 Of particular interest, Pro reduction was highly ranked among pathways important for C. difficile colonization.Citation53 Hyp and Pro were also efficiently metabolized by C. difficile upon infection in antibiotic-treated mice.Citation53 Many studies have indicated that a diverse gut microbiota can confer colonization resistance against C. difficile.Citation55 One mechanism by which this may occur is through restricting the metabolic niches available for the pathogen.Citation53 We propose that HypD-encoding commensals in the healthy human gut may consume Hyp, reducing its availability to C. difficile. Disruption of the gut microbiota with antibiotics could reduce Hyp metabolism, increasing the concentrations of this key substrate. This hypothesis can be tested by studying colonization resistance in mice using a defined microbiota containing or lacking HypD. Understanding the role of Hyp utilization in C. difficile could have implications for efforts to develop defined microbial consortia as therapeutics in patients with recurrent C. difficile infection.
It is likely that gut microbial acquisition of Hyp from host collagen could also affect host biology. Collagen is the principle source of Hyp in the human body and a major component of the extracellular matrix in connective tissues, including the basement membrane underlying the gut epithelium.Citation56 In the context of various diseases, the host inflammatory response and certain bacterial pathogens can degrade collagen, resulting in a breach of this barrier.Citation57 The resulting release of peptides and amino acids could increase nutrient availability for microbes and potentially alter the composition and functions of the microbiota. Microbial collagenases are not well understood mechanistically, but have been linked to diseases such as periodontitis.Citation58 Identification and characterization of collagenases from gut microbes will enable efforts to understand the contribution of collagen degradation in diseases involving impaired gut barrier function, including inflammatory bowel disease.
The end products of anaerobic Hyp metabolism may also interact with host cells. Notably, 5-aminovalerate, a product of Stickland fermentations involving Hyp and Pro, has been detected in the cecum and feces of mice, suggesting that Pro reduction is an active pathway in the gut microbiota.Citation48,Citation50,Citation59 Intriguingly, 5-aminovalerate is a chemical analog of γ-aminobutyric acid (GABA) and a weak antagonist of GABAB receptor.Citation60 These inhibitory receptors are activated upon GABA binding. In animals, GABAB receptors are expressed in the enteric nervous system and play a role in regulating gastrointestinal motility.Citation61 They are also expressed in immune cells, and recent studies have linked GABAergic signaling to inflammation.Citation61 The prominence of Hyp metabolism in the gut raises the intriguing possibility that HypD-mediated Stickland fermentation may contribute significantly to 5-aminovalerate production, potentially altering gut motor and immune functions.
Overall, the discovery of HypD has not only provided a more comprehensive understanding of a previously observed microbial metabolic activity, but also revealed its wide distribution in sequenced microbial genomes and potential to influence host health and disease. The many exciting research directions highlighted throughout this section illustrate the importance of incorporating enzyme discovery efforts into microbiota research. We anticipate that future investigations of these hypotheses will reveal a significant contribution of anaerobic Hyp metabolism in shaping gut microbiota composition and influencing host physiological and pathological states.
Beyond the human gut: Exploring anaerobic Hyp metabolism in other environments
In addition to human-associated microbes, we located HypD in the genomes of many environmental isolates mostly from the phyla Firmicutes, Proteobacteria, and Synergistetes (). The wide distribution of anaerobic Hyp utilization in these environmental strains suggests that this metabolic activity is important in a diverse range of microbial habitats.
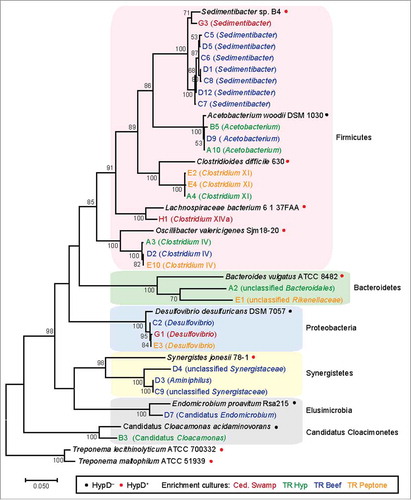
To gain insights into Hyp consumption in the environment, we examined the presence of this metabolic capability in hypoxic sediments (). Sediment samples were collected from various wetlands around Woods Hole, MA, USA. A subset of these samples was first incubated in the presence of Hyp or Hyp-containing supplements before inoculation into a defined medium containing Hyp as an electron acceptor and carbon source (–). Consumption of Hyp was confirmed in all enrichment cultures via High Performance Liquid Chromatography-Refractive Index Detector (HPLC-RID) (). Four enrichment cultures were selected for further passaging (). To identify the bacteria present in these enrichments, 16S rDNA sequences were amplified using universal primers ().Citation62 Phylogenetic analysis of these 16S rDNA sequences revealed that the enrichments were dominated by Firmicutes (). Very few 16S rDNA sequences yielded identical matches in the NCBI non-redundant nucleotide database,Citation63 indicating that most of these species have not been cultivated nor sequenced. In particular, two sequences are part of recently identified phyla (Candidatus Cloacimonete and Elusimicrobia) that have no or very few cultivated representatives.Citation64,Citation65 Overall, most of the species present in the Hyp enrichments are closely related to sequenced organisms that encode HypD, indicating they likely also possess this enzyme ().
Figure 6. (A) Workflow for enrichment culturing to obtain Hyp-degrading environmental microbes. Hyp-containing supplements were added to certain samples before inoculation into the Hyp enrichment medium. Turbidity and Hyp degradation were observed in all initial cultures before passaging into fresh medium. All cultures were incubated anaerobically at 30ºC in sealed vessels. (B) High Performance Liquid Chromatography-Refractive Index Detector (HPLC-RID) traces for the four enrichments further passaged for sequencing to show Hyp degradation. Samples were derivatized with nitrosonium, generated from potassium nitrite and hydrogen chloride, to obtain N-nitroso derivatives of Pro and Hyp for separation on an Aminex HPX-87P column.Citation69 The previously reported method was modified to include an isocratic elution at 60ºC for 40 minutes.
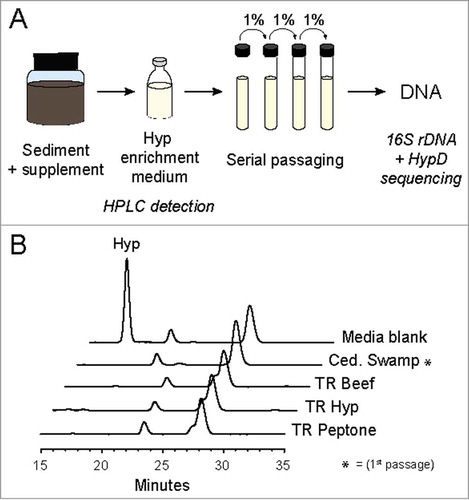
Table 1. All samples were collected at various locations around Woods Hole, MA, USA.
Table 2. Hyp enrichment medium composition.
Table 3. Primers and annealing temperatures used in this study.
Figure 7. 16S rDNA phylogenetic tree of enrichment culture isolates and reference species. 16S rRNA genes were amplified using primers 8F and 1510R () and sequenced from a pCR4-TOPO clone library (Invitrogen). Chimeric, low-quality, and redundant sequences were removed. 28 sequences from enrichment cultures and 12 reference sequences were aligned with MUSCLE.Citation70 The alignment was manually trimmed to yield a final length of 1371 bp and a maximum likelihood tree was constructed in MEGA767 using the Tamura–Nei method of substitution.Citation71 Bootstrap values from 50 to 100% are displayed. The highest similarity score calculated by RDP SeqMatch was used to assign the lowest classification level for each enrichment isolate.Citation72 The full-length sequences obtained for this analysis have been deposited in GenBank (accession: MG367094-MG367121).
We sought to directly identify hypD genes in the enrichment cultures by designing degenerate primers to target conserved regions present in all HypD sequences from the UniProt database (). Amplification conditions were optimized using genomic DNA from Clostridiales and Bacteroidales (, ). A fragment of the expected size for the hypD amplicon was amplified from DNA extracted from all passaged enrichment cultures (). These amplicon sequences do not have highly identical hits (<80% ID) in the NCBI nucleotide database, further demonstrating that Hyp-metabolizing species in these enrichments have not been sequenced. To confirm that the amplicons do represent hypD fragments, BLASTPCitation27 was performed using unique translated amino acid sequences of HypD sequences amplified from each enrichment against all GREs from our previously constructed sequence similarity network.Citation5 All top 50 hits mapped back to nodes within the HypD cluster of the sequence similarity network, with most of the enrichment sequences resembling HypD sequences from Firmicutes. This result is consistent with the high abundance of Firmicutes in these samples as determined from the 16S rDNA analysis. The two putative HypD active site residues (C. difficile HypD: F152, H160)Citation5 encoded within the amplified region are conserved in all enrichment amplicons (). Together, these results strongly support the presence of HypD in the four sediment enrichment cultures and suggest that this enzyme was responsible for the observed Hyp consumption. Lastly, the discovery of new, partial HypD sequences points to a much broader distribution of this activity across microbial diversity than is currently captured in sequenced genomes.
Figure 8. (A) A degenerate primer pair amplified an expected 357 bp product from genomic DNA of HypD-encoding isolates. No amplification product was observed using the genomic DNA of species lacking HypD. (B) Amplification of hypD from 100 ng of DNA from enrichment cultures. Amplicons were sequenced from a clone library generated using TOPO TA cloning kit (Invitrogen). 24 clones from each culture were sequenced by Beckman Coulter Genomics. High quality sequences that are non-redundant within each culture have been deposited on GenBank (accession: MG367122-MG367168). (C) Non-redundant translated amino acid sequences from each enrichment culture were aligned using Clustal OmegaCitation73 along with the biochemically characterized HypD from C. difficile 70-100-2010 (UniProt ID: A0A031WDE4). The two HypD putative active site residues encoded within this amplicon are present in all enrichment sequences and are indicated by asterisks. “Residue numbering is based on HypD sequence from C. difficile 70-100-2010.”

Conclusion
The discovery of HypD revealed a prominent metabolic pathway in the human gut microbiota that is responsible for anaerobic Hyp metabolism. Hyp is converted to P5C, which can then be processed in different ways to support microbial growth and survival. Given the strong evidence for Hyp utilization in the human gut microbiota and the wide distribution of HypD in strains from this habitat, we hypothesize that this activity has important roles in shaping gut microbiota composition and could affect host health and disease. Identification of the hypD gene will now enable the use of bacterial genetics and defined microbial consortia to elucidate the biological roles of this pathway in animal models. Further efforts to detect and quantify hypD in patient metagenomes and metatranscriptomes may reveal unanticipated correlations between this pathway and additional host phenotypes. Overall, the elucidation of a new enzymatic activity in the human gut microbiota has both expanded our knowledge of gut microbial metabolism and illuminated exciting directions for future research.
Disclosure of potential conflicts of interest
The authors report no potential conflicts of interest.
Acknowledgments
Enrichment culturing and HPLC measurements were carried out as a part of the Microbial Diversity course at Marine Biological Laboratory with Dianne Newman and Jared Leadbetter as course directors. Y.H. is grateful to Jared Leadbetter, Sebastian Kopf, and Kurt Hanselmann for helpful discussions about enrichment culturing during the Microbial Diversity course. Y.H. thanks Mikhail Tikhonov for help with sample collection.
Additional information
Funding
References
- Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 2016;14(8):e1002533. doi:10.1371/journal.pbio.1002533. PMID:27541692.
- Young VB. The role of the microbiome in human health and disease: An introduction for clinicians. BMJ. 2017;356:j831. doi:10.1136/bmj.j831. PMID:28298355.
- Joice R, Yasuda K, Shafquat A, Morgan Xochitl C, Huttenhower C. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metabolism. 2014;20(5):731–41. doi:10.1016/j.cmet.2014.10.003. PMID:25440055.
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLOS Comput Biol. 2009;5(12):e1000605. doi:10.1371/journal.pcbi.1000605. PMID:20011109.
- Levin BJ, Huang YY, Peck SC, Wei Y, Martínez-del Campo A, Marks JA, Franzosa EA, Huttenhower C, Balskus EP. A prominent glycyl radical enzyme in human gut microbiomes metabolizes trans-4-hydroxy-l-proline. Science. 2017;355(6325):eaai8386. doi:10.1126/science.aai8386. PMID:28183913.
- Backman LRF, Funk MA, Dawson CD, Drennan CL. New tricks for the glycyl radical enzyme family. Crit Rev Biochem Mol Biol. 2017;54(6):674–95. doi:10.1080/10409238.2017.1373741.
- Selmer T, Pierik AJ, Heider J. New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol Chem. 2005;386(10):981–8. doi:10.1515/BC.2005.114. PMID:16218870.
- Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14(4):169–81. doi:10.1093/dnares/dsm018. PMID:17916580.
- Kolmeder CA, de Been M, Nikkilä J, Ritamo I, Mättö J, Valmu L, Salojärvi J, Palva A, Salonen A, de Vos WM. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLOS ONE. 2012;7(1):e29913. doi:10.1371/journal.pone.0029913. PMID:22279554.
- Wagner AF, Frey M, Neugebauer FA, Schäfer W, Knappe J. The free radical in pyruvate formate-lyase is located on glycine-734. Proc Natl Acad Sci USA. 1992;89(3):996–1000. PMCID:PMC48372. PMID:1310545.
- Reddy SG, Wong KK, Parast CV, Peisach J, Magliozzo RS, Kozarich JW. Dioxygen inactivation of pyruvate formate-lyase: EPR evidence for the formation of protein-based sulfinyl and peroxyl radicals. Biochemistry. 1998;37(2):558–63. doi:10.1021/bi972086n. PMID:9425077.
- Stickland LH. Studies in the metabolism of the strict anaerobes (genus Clostridium). I. The chemical reactions by which C. sporogenes obtains its energy. Biochem J. 1934;28(5):1746–59. PMCID:PMC1266487. PMID:16745572.
- Jackson S, Calos M, Myers A, Self WT. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol. 2006;188(24):8487–95. doi:10.1128/JB.01370-06. PMID:17041035.
- Kabisch UC, Gräntzdörffer A, Schierhorn A, Rücknagel KP, Andreesen JR, Pich A. Identification of D-proline reductase from Clostridium sticklandii as a selenoenzyme and indications for a catalytically active pyruvoyl group derived from a cysteine residue by cleavage of a proprotein. J Biol Chem. 1999;274(13):8445–54. doi:10.1074/jbc.274.13.8445. PMID:10085076.
- Neumann-Schaal M, Hofmann JD, Will SE, Schomburg D. Time-resolved amino acid uptake of Clostridium difficile 630Δerm and concomitant fermentation product and toxin formation. BMC Microbiology. 2015;15:281–93. doi:10.1186/s12866-015-0614-2. PMID:26680234.
- Barker HA, D'Ari L, Kahn J. Enzymatic reactions in the degradation of 5-aminovalerate by Clostridium aminovalericum. J Biol Chem. 1987;262(19):8994–9003. PMID:3597403.
- Hardman JK, Stadtman TC. Metabolism of ω-amino acidS: II. Fermentation of δ-aminovaleric acid by Clostridium aminovalericum. N. Sp. J Bacteriol. 1960;79(4):549–52. PMCID:PMC278729
- Medema MH, Takano E, Breitling R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol Biol Evol. 2013;30(5):1218–23. doi:10.1093/molbev/mst025. PMID:23412913.
- Dannheim H, Riedel T, Neumann-Schaal M, Bunk B, Schober I, Spröer C, Chibani CM, Gronow S, Liesegang H, Overmann J, et al. Manual curation and reannotation of the genomes of Clostridium difficile 630Δerm and C. difficile 630. J Med Microbiol. 2017;66(3):286–93. doi:10.1099/jmm.0.000427. PMID:28357980.
- Watanabe S, Morimoto D, Fukumori F, Shinomiya H, Nishiwaki H, Kawano-Kawada M, Sasai Y, Tozawa Y, Watanabe Y. Identification and characterization of D-hydroxyproline dehydrogenase and Δ1-pyrroline-4-hydroxy-2-carboxylate deaminase involved in novel L-hydroxyproline metabolism of bacteria. J Biol Chem. 2012;287(39):32674–88. doi:10.1074/jbc.M112.374272. PMID:22833679.
- Adams E, Frank L. Metabolism of proline and the hydroxyprolines. Annu Rev Biochem. 1980;49(1):1005–61. doi:10.1146/annurev.bi.49.070180.005041. PMID:6250440.
- Brown JR, Doolittle WF. Archaea and the prokaryote-to-eukaryote transition. Microbiol Mol Biol Rev. 1997;61(4):456–502. PMCID:PMC232621 PMID:9409149.
- Falb M, Müller K, Königsmaier L, Oberwinkler T, Horn P, von Gronau S, Gonzalez O, Pfeiffer F, Bornberg-Bauer E, Oesterhelt D. Metabolism of halophilic archaea. Extremophiles. 2008;12(2):177–96. doi:10.1007/s00792-008-0138-x. PMID:18278431.
- Adams E. Hydroxyproline metabolism: I. Conversion to α-ketoglutarate by extracts of Pseudomonas. J Biol Chem. 1959;234(8):2073–84. PMID:13673016.
- Chen S, White CE, diCenzo GC, Zhang Y, Stogios PJ, Savchenko A, Finan TM. l-Hydroxyproline and d-proline catabolism in Sinorhizobium meliloti. J Bacteriol. 2016;198(7):1171–81. doi:10.1128/JB.00961-15. PMID:26833407.
- Blumer C, Haas D. Mechanism, regulation, and ecological role of bacterial cyanide biosynthesis. Arch Microbiol. 2000;173(3):170–7. doi:10.1007/s002039900127. PMID:10763748.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi:10.1016/S0022-2836(05)80360-2. PMID:2231712.
- Smith EA, Macfarlane GT. Dissimilatory amino acid metabolism in human colonic bacteria. Anaerobe. 1997;3(5):327–37. doi:10.1006/anae.1997.0121. PMID:16887608.
- Mergenhagen SE, Scherp HW. Lysis of reconstituted collagen and catabolism of products of collagenolysis by the oral microbiota. Arch Oral Biol. 1960;1:333–8. doi:10.1016/0003-9969(60)90094-7.
- Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45(2):106–24. doi:10.3109/10409231003627991. PMID:20199358.
- Hijazi M, Velasquez SM, Jamet E, Estevez JM, Albenne C. An update on post-translational modifications of hydroxyproline-rich glycoproteins: toward a model highlighting their contribution to plant cell wall architecture. Front Plant Sci. 2014;5(395). doi:10.3389/fpls.2014.00395. PMID:25177325.
- Nguema-Ona E, Vicré-Gibouin M, Gotté M, Plancot B, Lerouge P, Bardor M, Driouich A. Cell wall O-glycoproteins and N-glycoproteins: aspects of biosynthesis and function. Front Plant Sci. 2014;5(499). doi:10.3389/fpls.2014.00499.
- Kim KH, Jia B, Jeon CO. Identification of trans-4-hydroxy- l-proline as a compatible solute and its biosynthesis and molecular characterization in Halobacillus Halophilus. Front Microbiol. 2017;8(2054). doi:10.3389/fmicb.2017.02054.
- Katz E, Prockop DJ, Udenfriend S. Precursors of the hydroxyproline and ketoproline in actinomycin. J Biol Chem. 1962;237(5):1585–8. PMID:14454373.
- Lawrence CC, Sobey WJ, Field RA, Baldwin JE, Schofield CJ. Purification and initial characterization of proline 4-hydroxylase from Streptomyces griseoviridus P8648: a 2-oxoacid, ferrous-dependent dioxygenase involved in etamycin biosynthesis. Biochem J. 1996;313(Pt 1):185–91. PMCID:PMC1216881. PMID:8546682.
- Scotti JS, Leung IKH, Ge W, Bentley MA, Paps J, Kramer HB, Lee J, Aik W, Choi H, Paulsen SM, et al. Human oxygen sensing may have origins in prokaryotic elongation factor Tu prolyl-hydroxylation. Proc Natl Acad Sci USA. 2014;111(37):13331–6. doi:10.1073/pnas.1409916111. PMID:25197067.
- Schnicker NJ, Dey M. Bacillus anthracis prolyl 4-hydroxylase modifies collagen-like substrates in asymmetric patterns. J Biol Chem. 2016;291(25):13360–74. doi:10.1074/jbc.M116.725432. PMID:27129244.
- Wold JP, Lundby F, Egelandsdal B. Quantification of connective tissue (hydroxyproline) in ground beef by autofluorescence spectroscopy. J Food Sci. 1999;64(3):377–83. doi:10.1111/j.1365-2621.1999.tb15045.x.
- Vázquez‐Ortíz FA, Morón‐Fuenmayor OE, González‐Méndez NF. Hydroxyproline measurement by HPLC: Improved method of total collagen determination in meat samples. J Liq Chrom Relat Tech. 2004;27(17):2771–80. doi:10.1081/JLC-200029339.
- Valiente C, Arrigoni E, Esteban RM, Amado R. Grape pomace as a potential food fiber. J Food Sci. 1995;60(4):818–20. doi:10.1111/j.1365-2621.1995.tb06237.x.
- Verbeken D, Dierckx S, Dewettinck K. Exudate gums: occurrence, production, and applications. Appl Microbiol Biotechnol. 2003;63(1):10–21. doi:10.1007/s00253-003-1354-z. PMID:12802529.
- Kivirikko KI. Urinary excretion of hydroxyproline in health and disease. Int Rev Connect Tissue Res. 1970;5:93–163. PMID:5533228.
- Wu GD, Compher C, Chen EZ, Smith SA, Shah RD, Bittinger K, Chehoud C, Albenberg LG, Nessel L, Gilroy E, et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut. 2016;65(1):63–72. doi:10.1136/gutjnl-2014-308209. PMID:25431456.
- Knight J, Jiang J, Assimos DG, Holmes RP. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney Int. 2006;70(11):1929–34. doi:10.1038/sj.ki.5001906. PMID:17021603.
- Weiss PH, Klein L. The quantitative relationship of urinary peptide hydroxyproline excretion to collagen degradation. J Clin Invest. 1969;48(1):1–10. doi:10.1172/JCI105957. PMID:5765022.
- Liu D, Nikoo M, Boran G, Zhou P, Regenstein JM. Collagen and gelatin. Annu Rev Food Sci Technol. 2015;6(1):527–57. doi:10.1146/annurev-food-031414-111800. PMID:25884286.
- Smith EA, Macfarlane GT. Enumeration of amino acid fermenting bacteria in the human large intestine: effects of pH and starch on peptide metabolism and dissimilation of amino acids. FEMS Microbiol Ecol. 1998;25(4):355–68. doi:10.1016/S0168-6496(98)00004-X.
- Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NHL, Zhou Z, Bao Y, Jia W, Nicholson JK, et al. The footprints of gut microbial–mammalian co-metabolism. J Proteome Res. 2011;10(12):5512–22. doi:10.1021/pr2007945. PMID:21970572.
- Jump RLP, Polinkovsky A, Hurless K, Sitzlar B, Eckart K, Tomas M, Deshpande A, Nerandzic MM, Donskey CJ. Metabolomics analysis identifies intestinal microbiota-derived biomarkers of colonization resistance in clindamycin-treated mice. PLOS ONE. 2014;9(7):e101267. doi:10.1371/journal.pone.0101267. PMID:24988418.
- Matsumoto M, Kibe R, Ooga T, Aiba Y, Kurihara S, Sawaki E, Koga Y, Benno Y. Impact of intestinal microbiota on intestinal luminal metabolome. Sci Rep. 2012;2(233):doi:10.1038/srep00233.
- Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: The Bacteroidetes Sus-like paradigm. J Biol Chem. 2009;284(37):24673–7. doi:10.1074/jbc.R109.022848. PMID:19553672.
- Bouillaut L, Dubois T, Sonenshein AL, Dupuy B. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol. 2015;166(4):375–83. doi:10.1016/j.resmic.2014.10.002. PMID:25445566.
- Jenior ML, Leslie JL, Young VB, Schloss PD. Clostridium difficile colonizes alternative nutrient niches during infection across distinct murine gut microbiomes. mSystems. 2017;2(4):e00063–17. doi: 10.1128/mSystems.00063-17. PMID:28761936.
- Janoir C, Denève C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapetón-Montes D, Pereira FC, Henriques AO, Collignon A, et al. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun. 2013;81(10):3757–69. doi:10.1128/IAI.00515-13. PMID:23897605.
- Britton RA, Young VB. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology. 2014;146(6):1547–53. doi:10.1053/j.gastro.2014.01.059. PMID:24503131.
- Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem. 2009;78(1):929–58. doi:10.1146/annurev.biochem.77.032207.120833. PMID:19344236.
- Biancheri P, Di Sabatino A, Corazza GR, MacDonald TT. Proteases and the gut barrier. Cell Tissue Res. 2013;351(2):269–80. doi:10.1007/s00441-012-1390-z. PMID:22427120.
- Duarte AS, Correia A, Esteves AC. Bacterial collagenases – A review. Crit Rev Microbiol. 2016;42(1):106–26. doi:10.3109/1040841X.2014.904270. PMID:24754251.
- Martin F-PJ, Sprenger N, Montoliu I, Rezzi S, Kochhar S, Nicholson JK. Dietary modulation of gut functional ecology studied by fecal metabonomics. J Proteome Res. 2010;9(10):5284–95. doi:10.1021/pr100554m. PMID:20806900.
- `Muhyaddin M, Roberts PJ, Woodruff GN. Presynaptic γ-aminobutyric acid receptors in the rat anococcygeus muscle and their antagonism by 5-aminovaleric acid. Br J Pharmacol. 1982;77(1):163–8. doi:10.1111/j.1476-5381.1982.tb09282.x. PMID:6289954.
- Auteri M, Zizzo MG, Serio R. GABA and GABA receptors in the gastrointestinal tract: from motility to inflammation. Pharmacol Res. 2015;93:11–21. doi:10.1016/j.phrs.2014.12.001. PMID:25526825.
- Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. Chichester, United Kingdom: John Wiley and Sons;1991. p. 115–75.
- Coordinators NR. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44:D7–D19. doi:10.1093/nar/gkv1290. PMID:26615191.
- Pelletier E, Kreimeyer A, Bocs S, Rouy Z, Gyapay G, Chouari R, Rivière D, Ganesan A, Daegelen P, Sghir A, et al. “Candidatus Cloacamonas Acidaminovorans”: Genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol. 2008;190(7):2572–9. doi:10.1128/jb.01248-07. PMID:18245282.
- Zheng H, Dietrich C, Radek R, Brune A. Endomicrobium proavitum, the first isolate of Endomicrobia class. nov. (phylum Elusimicrobia) – an ultramicrobacterium with an unusual cell cycle that fixes nitrogen with a Group IV nitrogenase. Environ Microbiol. 2016;18(1):191–204. doi:10.1111/1462-2920.12960. PMID:26119974.
- Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017; bbx108. doi:10.1093/bib/bbx108. PMID:28968734.
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4. doi:10.1093/molbev/msw054. PMID:27004904.
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992;8(3):275–82. doi:10.1093/bioinformatics/8.3.275.
- Pleissner D, Wimmer R, Eriksen NT. Quantification of amino acids in fermentation media by isocratic HPLC analysis of their α-hydroxy acid derivatives. Analytical Chemistry. 2011;83(1):175–81. doi:10.1021/ac1021908. PMID:21121687.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi:10.1093/nar/gkh340. PMID:15034147.
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26. doi:10.1093/oxfordjournals.molbev.a040023. PMID:8336541.
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(D1):D633–D42. doi:10.1093/nar/gkt1244. PMID:24288368.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7(539). doi:10.1038/msb.2011.75. PMID:21988835.