
ABSTRACT
The gastrointestinal microbiome plays a pivotal role in physiological homeostasis of the intestine as well as in the pathophysiology of diseases including inflammatory bowel diseases (IBD) and colorectal cancer (CRC). Emerging evidence suggests that gut microbiota signal to the mitochondria of mucosal cells, including epithelial cells and immune cells. Gut microbiota signaling to mitochondria has been shown to alter mitochondrial metabolism, activate immune cells, induce inflammasome signaling, and alter epithelial barrier function. Both dysbiosis of the gut microbiota and mitochondrial dysfunction are associated with chronic intestinal inflammation and CRC. This review discusses mitochondrial metabolism of gut mucosal cells, mitochondrial dysfunction, and known gut microbiota-mediated mitochondrial alterations during IBD and CRC.
Introduction
Mitochondria are vital organelles for energy production in the cell. Emerging evidence suggesting that altered functioning of the mitochondria of mucosal cells is associated with intestinal diseases, including inflammatory bowel diseases (IBD) and colorectal cancer (CRC), has led to a renewed interest in the role of mitochondria in intestinal health. The intestinal microbiome, consisting of trillions of microorganisms such as bacteria, viruses, and fungi, plays a crucial role in intestinal homeostasis and disease. It is widely accepted that mitochondria were evolutionarily derived from a proteobacterium, resulting in common characteristics between eukaryotic mitochondrial and prokaryotic genomes. Due to these similarities, mitochondria can play a central role in triggering immune responses using mechanisms similar to bacteria. Additionally, bacteria can target mitochondria in host cells. Such cross-talk between gut microbiota and host mitochondria during health and disease is emerging as a significant area of research. Studies are beginning to elucidate the intricate molecular signaling between gut microbiome and mitochondria, but further studies are necessary to identify the mechanistic link between microbiome and host mitochondria in the pathogenesis of IBD or CRC. This review aims to present current evidence demonstrating the role of gut microbiome in signaling to the mitochondria of intestinal cells and the implications of this signaling in driving IBD or CRC.
Mitochondria structure, dynamics, and energy metabolism
Mitochondrial structure
Mitochondria are double membrane-bound organelles found generally in large numbers in the cytoplasm of eukaryotic cells. Mitochondria are important organelles due to their primary function to generate energy for the cell in the form of adenosine 5′-triphosphate (ATP). In 1957, Phillip Skiekevitz coined the phrase “powerhouse of the cell” for the organelle. In addition to ATP production, mitochondria are an important site of intracellular calcium storage and the induction of apoptosis during cellular stress predominantly by the release of cytochrome c into the cytosol. The outer mitochondrial membrane (OMM) is composed of a phospholipid bilayer separating the mitochondria and its contents from the cytosol. Ions and proteins smaller than 5 kDa diffuse into and out of the mitochondria freely via porins, and therefore, the concentration of small proteins and ions in the mitochondrial intermembrane space matches the cytosol. Proteins larger than 5 kDa possessing a 5′ mitochondrial localization sequence translocate across the OMM via binding to translocase of the outer membrane (TOM) transporters. Proteins transferred into the mitochondria include nuclear DNA encoded proteins such as those for oxidative phosphorylation and heat shock proteins.Citation1 The inner mitochondrial membrane (IMM) is also a phospholipid bilayer and houses electron transport chain (ETC) complexes which drive ATP production. Transporters called translocase of the inner membrane (TIMs) are required for all ions and proteins to enter the mitochondrial matrixCitation2 allowing formation of the protein gradient in the space between the OMM and IMM, driving electron flow through the ETC.Citation3 Inward folds of the IMM are called cristae, which increase the surface area for ATP production to match cellular demand.Citation4 Within the IMM is the mitochondrial matrix which houses ribosomes, mitochondrial DNA (mtDNA), and ATP synthase proteins that facilitate the catalysis from adenosine diphosphate (ADP) to ATP.Citation1
Mitochondrial genome
Contrary to other eukaryotic organelles, mitochondria possess their own genome, mtDNA, located in the mitochondrial matrix.Citation5 mtDNA has been traced to a proteobacterial ancestor which is responsible for the first known case of ATP production in correlation to the ETC complex.Citation6 The bacterium-like protist Reclinomonas americana and the gram-negative, alphaproteobacteria Rickettsia prowazekii have been identified as potential ancestors of the eukaryotic mitochondrion.Citation6 Human mtDNA is ~16.6 kb long and contains 37 genes coding for two rRNAs, 22 tRNAs and 13 polypeptide mRNAs.Citation7 mtDNA is circular, double-stranded, and is exclusively transmitted from the female germline.Citation8 Circular molecules of DNA such as mtDNA are composed of two DNA strands consisting of the heavy, guanine-rich strand and the light strand. Majority of the mtDNA code is found in the heavy strand, including seven subunits of ETC complex I (NADH dehydrogenase), three subunits of ETC complex IV (cytochrome c oxidase), and two subunits of ETC complex V (ATP synthase) and ETC complex III.Citation9 Conversely, the light strand codes for eight tRNAs and a single polypeptide.Citation9 All the necessary transcription and translation machinery and protein folding chaperones are present in the matrix to facilitate production of functional proteins from the mitochondrial genome.Citation5 The majority of the proteins involved in this process are encoded by nuclear genes and not mtDNA and therefore, are transported from the cytosol into the mitochondrial matrix by TOMs/TIMs.Citation8 Unique features of mtDNA compared to nuclear DNA include circular structure, short introns, and lack of histone structures. The lack of histones and short intergenic regions contribute to mtDNA susceptibility to damage by oxidative stress and other noxious agents.Citation10 Mitochondria possess many of the same DNA repair pathways as found in the nucleus to repair DNA damage.Citation8
Mitochondrial dynamics
Mitochondria are dynamic organelles which readily respond to environmental stimuli and cellular demands for energy.Citation11 Quality control of mitochondria is critical in maintaining cell homeostasis and consists of fission, fusion, and removal of damaged mitochondria by autophagy, known as mitophagy. In the event that these quality control pathways cannot restore the mitochondrial population, apoptosis of the cell ensues. Mitochondria exhibit high plasticity of structure with constant fission, fusion, and relocation throughout the cytoplasm. The process of mitochondrial fission and fusion are mediated by guanosine triphosphatases (GTPases) of the dynamin family.Citation11 These GTPases coordinate to divide (fission) and fuse (fusion) the two lipid bilayers of the mitochondria. A balance between fusion and fission events maintains the overall morphology of the mitochondrial population. An increased fusion/fission ratio generates elongated, tubular mitochondria, whereas an increased fission/fusion ratio results in fragmented mitochondria.
Mitochondrial fusion is the merging of two or more damaged mitochondria, enabling mixing of contents within the mitochondrial population and subsequently diluting damaged components while preserving essential components.Citation11 Fusion proteins such as Mitofusin 1 (Mfn 1) and Mfn 2 as well as Optic Atrophy 1 (OPA1) are members of the dynamin GTPase family.Citation1,Citation11 Mfn1 and Mfn2 form homo- or heterodimers to fuse the outer membranes of adjacent targeted mitochondria together.Citation11 Subsequently, OPA1 that is located on the IMM coordinates with Mfn1 to facilitate IMM fusion.Citation11 Through these proteins, fusion facilitates the sharing of mtDNA between mitochondria thereby providing more support for critical functions such as oxidative phosphorylation, mitophagy, apoptosis, cell proliferation, and migration.Citation12
Mitochondrial fission is the process by which mitochondria divide into smaller daughter mitochondria.Citation1 Mitochondrial fission is essential to generate adequate numbers of mitochondria for growing cells and is mediated by Dynamin-Related Protein1 (Drp1), Fission 1 (Fis1), Mitochondria Fission Factor (Mff), Mitochondrial Dynamics Protein (MID49) and MID51. The OMM protein Mff recruits Drp1 from the cytosolCitation13 to the OMM of mitochondria where it assembles into oligomeric complexes that compress the mitochondrial tubule to mediate fission.Citation11 This action leads to OMM constriction and cleavage.Citation11 Fis1 is located on the OMM as the receptor protein for Drp1 during its translocation from the cytosol.Citation14 The interaction of Drp1 with Fis1 can lead to the release of cytochrome c which can induce apoptosis.Citation11 MID49/51 are also found on the OMM and facilitate randomized screening of uncharacterized proteins whose expression cause changes in mitochondrial morphology.Citation13
Autophagy is an evolutionarily conserved catabolic pathway that removes cytoplasmic components, including damaged organelles, through lysosomal degradation.Citation15 Autophagic flux is the dynamic process of autophagosome synthesis containing cargo to be degraded, fusion of the autophagosome with a lysosome, and degradation of autophagosome/cargo inside the lysosome. Selective autophagy of mitochondria, termed mitophagy, eliminates damaged mitochondria to prevent excessive mitochondrial reactive oxygen species (mtROS) generation and cell toxicity.Citation16 For detailed reviews, see refs.Citation17,Citation18 Mitochondrial dysfunction (depolarization, excessive misfolded proteins, mtROS accumulation)Citation19,Citation20 stimulates OMM-localized autophagy receptors to incorporate mitochondria into the autophagosome.Citation21 Some autophagy receptors are dependent on Parkin, which is recruited to dysfunctional mitochondria and targets OMM proteins for ubiquitination; polyubiquitinated chains are then recognized by the autophagy receptor, thereby incorporating the mitochondrion into the autophagosome via their LC3-interacting region.Citation21 Parkin-dependent autophagy receptors that target mitochondria include p62, NBR1, and AMBRA1. Other autophagy receptors that target mitochondria and act independently of Parkin include Bnip3, Nix/Bnip3L, FUNDC1, and BCl2L13. Parkin-independent autophagy receptors contain transmembrane domains and upon expression constitutively localize to the OMM.Citation16 Mitochondrial fission facilitates mitophagy by dividing mitochondria into fragments of small enough size to be engulfed by the autophagosome.Citation1
Energy metabolism
Mitochondria provide efficient mechanisms for eukaryotic cells to generate ATP and produce building blocks for biosynthesis required for cell proliferation and differentiation. A key function of mitochondria is to produce ATP through oxidative phosphorylation. In the presence of oxygen, this process begins with cytosolic glycolysis of glucose to produce pyruvate, which is the main source of mitochondrial energy production and intermediate metabolism by conversion into acetyl-CoA. Acetyl-CoA is the initial molecule powering the citric acid cycle, also known as the tricarboxylic acid (TCA) cycle, in the mitochondrial matrix.Citation3 In addition, fatty acid β-oxidation (FAO) produces acetyl-CoA from fatty acids to feed the TCA cycle. The TCA cycle provides the required precursors for oxidative phosphorylation: 2 pyruvate molecules, 6 NADH, and 2 FADH2 are produced from 1 glucose molecule. NADH and FADH2 provide electrons to complexes I or II of the ETC, respectively, which in turn utilize the energy of passing electrons to translocate protons from the mitochondrial matrix to the intermembrane space. This accumulation of protons generates an electrical and chemical gradient across the IMM, that in turn drives transport of protons through the ATP synthase complex. This process transforms ADP and an inorganic phosphate into ATP.Citation22 During oxidative phosphorylation, 38 ATP molecules are made from one glucose molecule (two from glycolysis, two from the TCA cycle, and approximately 34 from the ETC). When limited amounts of oxygen are available, anaerobic glycolysis occurs which produces lactate from glucose and generates two ATP molecules. Aerobic glycolysis is a well-documented phenomenon in cancer (called the Warburg effect). During aerobic glycolysis, cells ferment glucose to lactate even in the presence of oxygen. This produces only two ATP molecules but provides glycolytic intermediates to feed anabolic pathways necessary for rapidly dividing cells and is discussed further below. Importantly, through these metabolic pathways, mitochondria produce the majority of the acetyl-CoA and S-adenosylmethanione needed for protein acetylation and methylation, respectively.Citation23 Thus, alterations in mitochondrial biology and metabolism have profound outcomes on the epigenetic state of the genome.
Mitochondria and innate and adaptive immune responses
Dysfunctional mitochondria are emerging as key activators of the nucleotide binding domain and leucine-rich repeat pyrin 3 domain (NLRP3) inflammasome. Inflammasomes are multiprotein complexes in the cytosol that promote inflammation in response to pathogens and cellular distress. The NLRP3 inflammasome is the best-characterized inflammasome to date with known function to activate Caspase-1. Caspase-1 in turn cleaves and activates interleukin-1β (IL-1β) and IL-18 cytokines, thereby promoting inflammation and self-defense pathways. The NLRP3 inflammasome is most prominently expressed in myeloid cells. The NLRP3 inflammasome is activated by mitochondrial signals including mtROS, oxidized mtDNA, extracellular ATP efflux, loss of mitochondrial membrane potential, and alterations in cell metabolism.Citation24 Other inflammasomes including NLRP1, NLRC4, and NLRP6 also function to activate Caspase-1 but in response to distinct stimuli compared to NLRP3. Mice deficient in NLRP3 exhibited less severe experimental colitis with decreased production of pro-inflammatory cytokines.Citation25 Furthermore, recent findings suggested that IL-10 deficiency led to the accumulation of damaged mitochondria and dysregulated activation of the NLRP3 inflammasome in macrophages of mouse models of colitis and in IBD patients.Citation26 These studies imply that activation of the NLRP3 inflammasome is involved in IBD pathogenesis.
Activation, cell differentiation, and polarization of immune cells are dependent on metabolic reprogramming during bacterial infection. Neutrophils, dendritic cells, and CD4+ T cells undergo a metabolic shift from oxidative phosphorylation to glycolysis.Citation27 Elimination of activated neutrophils by mitochondria-induced apoptosis is crucial to preventing extended and exaggerated inflammation.Citation28 M1 macrophage polarization in response to lipopolysaccharide is dependent on activation of glycolysis to acquire bactericidal activity, while M2 macrophage polarization requires increased FAO and oxidative phosphorylation.Citation29 Shifts in mitochondrial metabolism play a central role in memory T-cell quiescence and activation responses during bacterial infection.Citation30 Stimulation of B lymphocytes induces glycolysis and oxidative phosphorylation to facilitate the production of IgG or IgA antibodies.Citation30 The signaling pathways mediating metabolic reprogramming in immune cells are not fully understood, but hypoxia-inducible factor-1α (HIF-1α) has been implicated as a master regulator of mitochondrial responses during bacterial infection.Citation31,Citation32
Mitochondria and the intestinal epithelium
Mitochondrial metabolism is distinct in intestinal epithelial cell types
The epithelial lining of the small intestine (SI) is regenerated in its entirety at a rapid pace of every three to five days. In tissues of high turnover such as the intestinal epithelium, stem cells are crucial regulators of tissue homeostasis. Crypt base columnar (CBC) stem cells, a population of rapidly dividing cells at the crypt base expressing leucine-rich-repeat containing G-protein coupled receptor 5 (Lgr5), give rise to all terminally differentiated intestinal epithelial cell (IEC) types (enterocytes, Paneth, goblet, enteroendocrine, tuft, and M cells).Citation33 CBCs divide into progenitor cells which move upward within the crypt into the transit amplifying zone.Citation34 It is here that the cells differentiate further and travel to the villus where their functions are required. At the villus tip, senescent IECs slough off through anoikis, a specific type of programmed cell death for anchorage-dependent cells, and make room for newly formed cells to take their place.Citation34 Paneth cells are the exception as these cells are long-lived secretory cells that migrate to the crypt base and reside between Lgr5+ CBCs where they produce and secrete antimicrobial peptides and stem cell factors such as epidermal growth factor, Wnt3, and Notch ligand D114 that sustain the stem cell niche.Citation35–Citation38 Local conditions in the stem cell niche regulate cell proliferation, differentiation, and stem cell self-renewal.Citation34
Compared to other differentiated intestinal epithelial cells and relatively quiescent stem cell populations in the adult, such as hematopoietic stem cells and neural stem cells, Lgr5+ CBCs show higher mitochondrial oxidative phosphorylation activity.Citation37,Citation39 Rodriguez-Colman et al. 2017 revealed that in support of increased demand for mitochondrial oxidative phosphorylation for Lgr5+ CBCs to maintain the immense amount of cell turn over in the small intestine, lactate from Paneth cells is fed into neighboring Lgr5+ CBCs fueling their oxidative phosphorylation processes.Citation40 Inhibition of glycolysis, and subsequently lactate production, in Paneth cells affects Lgr5+ CBC function and hinders crypt maturation.Citation40 Therefore, the glycolytic phenotype in Paneth cells is crucial for lactate production, which drives increased oxidative phosphorylation in Lgr5+ CBCs. Mechanistically, Rodriguez-Colman et al. demonstrated that mtROS produced in Lgr5+ CBCs during oxidative phosphorylation stimulate activation of p38 MAP kinase, which in turn regulates Lgr5+ CBC self-renewal and differentiation.Citation40 These results show that the glycolytic phenotype in Paneth cells and increased oxidative phosphorylation in Lgr5+ CBCs are required in supporting both stem cell niche and function. Provision of lactate from Paneth cells is therefore another critical aspect of the interdependent relationship between Lgr5+ CBCs and Paneth cells, essential to the maintenance and differentiation of SI epithelium.
Mitochondrial dysfunction and intestinal stem cell homeostasis
Mitochondrial metabolism, function, and biogenesis are all strictly regulated within intestinal stem cells (ISCs) as well as epithelial cells along the crypt-villus axis and in turn, play critical roles in the regulation and maintenance of ISCs. ISCs possess a large number of mitochondria, and crypt formation was shown to be dependent on an abundance of mitochondria.Citation41 IEC-specific deletion of the mitochondrial chaperone protein heat-shock protein 60 (HSP60) led to mitochondrial dysfunction, impairment of cell proliferation and loss of stemness of ISCs.Citation41 Furthermore, mitochondrial dysfunction impaired the ability of the ISCs to produce ATP, leading to altered ISC self-renewal and differentiation.Citation41 Therefore, a functioning mitochondrial population in ISCs is crucially important for intestinal epithelial regeneration and homeostasis.
ROS are well-established primary signaling molecules for downstream signaling from mitochondria and excessive mtROS not eliminated by mitophagy can lead to multiple cellular dysfunctions.Citation42 Intestinal organoids treated with buthionine sulfoximine to generate excessive mtROS, or intestinal organoids generated from mice with ISCs deficient in autophagy with subsequent mtROS accumulation, exhibited reduced number and self-renewal of ISCs.Citation43 Treatment with the antioxidant N-acetyl-L-cysteine, which dampens excessive ROS levels and restores ATP synthesis in mitochondria, restored the number of ISCs in intestinal organoids deficient in autophagy.Citation43 The role of mitochondrial function and ROS generation on stem cell fate and self-renewal is also important in relatively quiescent stem cell populations including hematopoietic stem cellsCitation44 and neural stem cellsCitation45,Citation46 in addition to more active ISCs of frequent turnover tissues requiring more bioenergetics activity such as the intestinal epithelium.
Mitochondria–microbiota cross talk and implication in intestinal disease
Mitochondrial dysfunction and inflammatory bowel diseases
Inflammatory bowel diseases (IBD) are a group of idiopathic diseases of the gut characterized by recurring, chronic inflammation. The two predominant IBDs are Crohn’s disease (CD) and ulcerative colitis (UC). These multifactorial diseases exhibit dysfunction of the intestinal epithelial barrier and dysregulated immune cell responses to gut microorganisms due to unknown environmental triggers in genetically predisposed individuals.Citation47 IBD patients with colonic involvement exhibit an increased risk of developing colorectal cancer (CRC) dependent on disease severity and duration.Citation48
Emerging studies have begun to reveal the involvement of mitochondrial stress in the pathophysiology of IBD (). Over 30 years ago, UC was thought to be an “energy deficient” disease of the epithelium.Citation49 Electron microscopy studies demonstrated that ultrastructural alterations to epithelial cell mitochondria, such as dissolved/irregular cristae indicative of impaired function, were evident in CD patients as an early event during inflammation, prior to alterations to the tight junctions controlling barrier function.Citation50 Decreased activity of ETC complexes has been observed in mucosal biopsies of CD and UC patients, which was restored by anti-TNFα treatment, suggesting TNFα regulates mitochondrial function during intestinal inflammation.Citation51–Citation54 Induction of the mitochondrial-unfolded protein response (mtUPR) was evident in epithelial cells from CD and UC.Citation55 The mtUPR is activated upon an accumulation of unfolded or misfolded proteins in the mitochondrial matrix to upregulate protein folding chaperone proteins to return homeostasis.Citation41 In a recent study, it was demonstrated that mtDNA released into the serum in IBD patients was recognized as a damage-associated molecular pattern (DAMP) potentially by toll-like receptor 9 (TRL9), and could provide a biomarker of inflammation.Citation56 Paneth cell defects in CD patients, a phenotype shown to associate with early disease recurrence after resection,Citation57 were shown to correlate with dysbiosis of the microbiome and reduced expression of a large cluster of oxidative phosphorylation genes,Citation58 suggesting patients with Paneth cell defects may have altered mucosal mitochondrial function but this was not further elucidated. Recent reports on the pediatric RISK stratification study using RNA sequencing analysis on CD inflamed biopsies demonstrated that patients who were at risk of structuring, but upon follow-up through 36 months remained complication free, exhibited an enriched mitochondrial function gene signature compared to those patients who developed complications.Citation59 In a proteome study of pediatric CD patients with colonic involvement, impaired mitochondrial function was implicated and correlated with increased disease severity.Citation60 Importantly, mitochondrial-encoded DNA polymorphisms have been identified to be associated with UC in European cohorts.Citation61,Citation62 Furthermore, 5% of genes highlighted by IBD genome-wide association studies were functionally linked to the maintenance of mitochondrial health, further underpinning the role of deregulated mitochondrial function in IBD.Citation63
Figure 1. Mitochondrial dysfunction associated with IBD. Pro-inflammatory cytokines and bacterial toxins are implicated in mitochondrial alterations during IBD. Decreased activity of ETC complexes, decreased ATP levels, accumulation of mtROS, accumulation of misfolded or unfolded proteins in the matrix, and ultrastructural changes such as dissolved cristae have been reported in mitochondria of the epithelium of IBD patients. Subsequent loss of epithelial barrier integrity, epithelial cell apoptosis, and bacterial invasion have been demonstrated following mitochondrial dysfunction in the epithelium. mtDNA is released into the serum of IBD patients and serves as a DAMP for immune cell activation. Additionally, damaged mitochondria can signal inflammasome activation, leading to pro-inflammatory cytokine production.
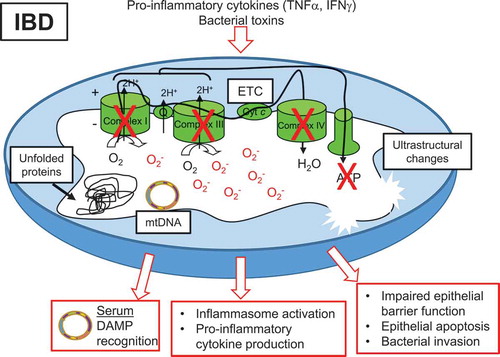
Multiple mouse models implicate mitochondrial dysfunction in the pathogenesis of intestinal inflammation. Mice treated with dextran sodium sulfate (DSS) to induce colitis exhibited swollen mitochondria, increased mtROS generation, decreased ETC activity and ATP level in the colonic mucosa that was ameliorated by anti-TNFα treatment.Citation54,Citation64 Mice with deletion of IRGM, a CD susceptibility gene that affects mitochondrial fission and induces autophagy, exhibited increased susceptibility to experimental colitis.Citation65,Citation66 Polymorphisms in SLC22A5, which encodes OCTN2 the carnitine transporter involved in mitochondrial FAO, are associated with CD risk.Citation67 Deletion of OCTN2 in mice caused spontaneous colitis.Citation68 Mice deficient in PGC1α, a master regulator of mitochondrial biogenesis, exhibited exacerbated intestinal inflammation associated with decreased mtDNA levels.Citation69 Deficiency of MDR1, which is an ATP-dependent efflux transporter, impaired mitochondrial homeostasis and promoted intestinal inflammation.Citation63 Bar et al. utilized conplastic strains of mice that have the same nuclear DNA but distinct mtDNA to demonstrate that strains of mice with increased ATP levels and increased mitochondrial oxidative phosphorylation were protected from experimental colitis. This protection was associated with enhanced IEC barrier function and epithelial proliferation.Citation70 Mice unable to mount a mtUPR due to deletion of PKR exhibited protection from experimental colitis, suggesting mtUPR is involved in the pathology of intestinal inflammation.Citation55 Genetic overexpression in IECs or therapeutic delivery (via adenovirus or nanoparticles) of the mitochondrial protein Prohibitin 1, which provides chaperone function for proteins of ETC complexes, protected mice from experimental colitis.Citation71,Citation72 Collectively, these mouse studies demonstrate the importance of mitochondrial health in protecting against intestinal inflammation.
IEC culture and enteroid models have provided important mechanistic insight suggesting that decreased mitochondrial function in epithelial cells drives a loss in barrier integrity and subsequent bacterial invasion of the underlying intestinal tissue. Loss of barrier function can manifest from epithelial cell death or leakiness of paracellular epithelial cell-cell junctions. DSS-induced colitis is associated with epithelial barrier dysfunction and mechanistic studies using Caco-2 cell monolayers demonstrated that mtROS plays a key role in loss of barrier integrity during DSS via stimulating the redistribution of Occludin and ZO-1 from intercellular junctions into intracellular compartments causing leakiness of the tight junctions without altering cell viability.Citation73 Many forms of ROS have been implicated in disrupting tight junctions through the rearrangement of the actin cytoskeleton to decrease its interaction with tight junction proteins Occludin and ZO-1 and interaction with myosin heavy chain.Citation74 Additionally, hydrogen peroxide alters phosphorylation of Occludin, disrupting the tight junction, and phosphorylation of β-catenin, disrupting the adherens junction due to the redistribution of E-cadherin preventing interaction with β-catenin.Citation74 The role of ROS specifically derived from mitochondria in inducing intestinal epithelial barrier dysfunction was demonstrated by rescue experiments using a mitochondrial-targeted ROS scavenger called mitoquinone (MitoQ).Citation75 Indeed, dysfunctional mitochondria and accumulation of mtROS during deficiency of the autophagy protein ATG16L1 induced epithelial barrier defects and the transcellular passage of bacteria that perpetuated intestinal inflammation.Citation75,Citation76
In addition to epithelial barrier integrity, intestinal epithelial mitochondria serve as both targets and facilitators of inflammatory pathways. Cultured colon epithelial cells with reduced mitochondrial activity (p0 cells) exhibited activation of pathways associated with inflammation through an AMPK-mediated mechanism.Citation54 Since colonic p0 cells are unable to produce mtROS but actively induce inflammatory pathways, decreased oxidative phosphorylation likely plays a role in triggering intestinal inflammation.Citation54 TNFα, a pro-inflammatory cytokine that plays a central role in IBD inflammation, inhibited mitochondrial function in cultured IECs, inducing mtROS accumulation and subsequently leading to impaired barrier function.Citation54,Citation77 Infliximab, an anti-TNFα biologic therapy for IBD, was shown to be beneficial to reduce mitochondrial dysfunction in cultured IECs and IBD patients.Citation54,Citation78 The role of microbiome in mitochondrial alterations driving epithelial barrier dysfunction will be discussed in the following section.
Gut microbiome signaling to mitochondria – IBD
The interface between the IECs and intestinal luminal contents stimulates a variety of immune and metabolic activities, with the mucosa armed with mechanisms able to sense nutrients, microbes, and microbial metabolites.Citation79 The gut microbiome actively interacts and shapes the host mucosal immune system and regulates intestinal homeostasis. Bacteria lead the microbial ecosystem in terms of abundance inside the GI tract, at over 90% of the healthy gut microbiome belonging to the Bacteriodetes and the Firmicutes phyla. However, inter-individual variation can be more than an order of magnitude in their Fimicutes/Bacteriodetes ratios. Typical gut bacterial families found in healthy individuals includes Bacteroidaceae, Clostridiaceae, Prevotellaceae, Eubacteriaceae, Ruminococcaceae, Bifidobacteriaceae, Lactobacillaceae, Enterobacteriaceae, Saccharomycetaceae, and Methanobacteriaceae.Citation80–Citation82
Bacterial metabolites, including short-chain fatty acids (SCFAs) and hydrogen sulfide (H2S), serve as messengers to colonic epithelial and immune cells, impacting their metabolism, epigenetic modifications, and gene expression. SCFAs are currently the most studied bacterial metabolites and are beneficial to colon homeostasis. The three major SCFAs, acetate, propionate, and butyrate, are produced in the colon by bacterial fermentation of carbohydrates and are an important source of energy for colon epithelial cells. SCFAs are ligands for free fatty acid receptors 2 and 3, which modulate glucose metabolism and FAO. Additionally, SCFAs regulate PGC1α, a master regulator of mitochondrial biogenesis.Citation83 These responses to SCFAs result at the organelle level in increased glucose uptake, FAO, oxidative phosphorylation, and mitochondrial biogenesis. In terms of intestinal homeostasis, these responses to SCFAs in colon epithelial cells facilitate the development of a tolerant mucosal immune system, promote epithelial barrier integrity, promote “physiologic hypoxia”, and suppress colitis.Citation83–Citation87 In addition, steady-state NLRP3 inflammasome activation in the colon is mediated by SCFAs, which produces basal IL-18 levels, regulates the microbiome composition, and dampens overt inflammatory responses.Citation88
H2S is largely produced by colonic enteric bacteria such as Escherichia coli (E. coli) and Salmonella through degradation of sulfur amino acids. Some studies report anti-inflammatory activity of H2S in the intestine, while others demonstrate detrimental effects, suggesting that these results are likely context dependent.Citation89 In IECs, H2S exhibits detrimental responses. Relative levels of H2S in the colon directly regulate oxidative phosphorylation in epithelial cells, with elevated H2S levels inhibiting complex IV of the ETC.Citation90 Additionally, H2S induces genotoxic damage to the epithelium, inhibits metabolism of SCFAs, and induces breaks in the mucus barrier allowing exposure of luminal contents to the underlying tissue.Citation91
IBD is associated with disruption of commensal bacteria and their downstream genes, which has been extensively reviewed elsewhere.Citation92 IBD patients exhibit decreased overall diversity of the microbiome and alterations in the gut microbiota (), but it is not understood if these changes to the microbiota initiate disease pathogenesis or are secondary to inflammation. Commensal bacteria, such as mucosa-associated E. coli and Mycobacterium avium paratuberculosis, are thought to serve a pathogenic role in the development of IBD. Mucosal-associated microbiota is 50% less diverse in the colon of patients with active CD and 30% less diverse in UC patients.Citation93 A large-scale study describing the microbiome in 447 newly diagnosed CD pediatric patients prior to anti-inflammatory treatment reported dysbiosis of the microbiome with increased abundance of Enterobacteriaceae, Pasteurellaceae, Fusobacteriaceae, Neisseriaceae, Veillonellaceae, and Gemellacee, and decreased abundances of Bifidobacteriaceae, Erysipelotrichaceae, Clostridiales, and Bacteroidales.Citation94 Some reports suggest that increased amounts of sulfate-reducing bacteria, enhancing the capacity to elevate H2S levels, are present in the gut microbiome of IBD patients.Citation91 Additionally, IBD patients exhibit decreased levels of SCFA-producing bacteria (Clostridium and Faecalibacterium prausnitzzi) and increased levels of mucolytic bacteria that degrade the mucus layer (Ruminococcus gnavas and Ruminococcus torques).Citation95 The central involvement of gut microbiota in the development of intestinal inflammation is further identified in studies demonstrating that genetically altered mice that manifest spontaneous intestinal inflammation fail to do so in a germ-free environment.Citation96
Table 1. Alterations to gut microbiome during IBD and CRC.
The gut microbiome has emerged as a dynamic central regulator of mitochondrial function in intestinal cells, including immune and epithelial cells.Citation97 Studies have reported mitochondria to be the most responsive organelle to microbiotic signaling.Citation98 mtROS produced in immune cells plays an important role in the eradication of invading pathogens via direct bactericidal effects or via indirect effects to activate the NLRP3 inflammasome and produce pro-inflammatory cytokines. Invading bacteria as well as fermentation products of the gut microbiome such as SCFAs induce mtROS production in immune cells via upregulation of mitochondrial respiration and enhanced oxidative phosphorylation. Activation of Toll-like receptors (TLRs) 1, 2, or 4 at the plasma membrane or endoplasmic reticulum membrane occurs upon bacterial or viral infection and subsequently induces mtROS accumulation in macrophages and neutrophils.Citation99 This was shown to be dependent on TRAF6-induced accumulation of evolutionarily conserved signaling intermediate in Toll pathways (ECSIT), a protein important for ETC complex assembly and activity.Citation99 Immune-Responsive Gene 1 (Irg1) is a mitochondrial enzyme induced in macrophages during inflammation that regulates FAO and is required for enhanced oxidative phosphorylation and mtROS production during infection.Citation100 Such increased mtROS via enhanced oxidative phosphorylation is crucial in macrophage- and neutrophil-mediated eradication of invading pathogens.
Microbial signaling to mitochondria is also emerging in key regulation of intestinal epithelial function. Studies suggest that commensal bacterial-induced mitochondrial signaling potentiates epithelial homeostasis, while pathogenic bacterial-induced mitochondrial responses are detrimental to the function of gut epithelial cells. Colon epithelial metabolism of the SCFA butyrate promoted “physiologic hypoxia” via mitochondrial O2 consumption to stabilize HIF and induce HIF-targeted barrier-protective genes, linking microbe, epithelial metabolism, and mucosal homeostasis.Citation87 Generation of ROS in epithelial cells induced by commensal bacteria promoted restitution of damaged epithelium and wound healing.Citation101 Furthermore, mtROS produced by IECs was shown to protect mice from experimental colitis dependent on the recruitment and polarization of intestinal macrophages to the M2 anti-inflammatory phenotype.Citation102 The ability of the epithelium to be tolerant of commensal bacteria was lost when epithelial cells exhibited mitochondrial dysfunction and was dependent on the expression of TNFα and IFNγ.Citation103 When treated with E. coli to mimic bacterial invasion during IBD, human colonic epithelial cells exhibiting mitochondrial dysfunction were more susceptible to impaired barrier function, suggesting that during mitochondrial stress, epithelial cells sense commensal bacteria as a pro-inflammatory threat.Citation103 Mitochondria are important targets of bacterial pathogens to manipulate eukaryotic cellular machinery and exploit apoptotic pathways. Pathogenic bacteria produce toxins that directly damage IEC mitochondria, including the suppression of critical mitochondrial ATP-sensitive potassium channels, initiating mitochondrial membrane hyperpolarization and apoptosis and subsequently impairing epithelial barrier function.Citation104 Epithelial barrier defects mediated by pathogenic E. coli involve relocalizing Occludin from the peri-junctional region to the cytosol, thereby disrupting tight junctions.Citation105 This was mechanistically shown to be mediated by two effector proteins, called EspF and Mitochondrial-Associated Protein (MAP), released into the cell by pathogenic E. coli. MAP is targeted to the mitochondria where it interferes with mitochondrial function and induces loss of mitochondrial membrane potential via pore-forming activity.Citation106 Mitochondrial depolarization leads to mtROS accumulation, which in turn, disrupts tight and adherens junctions.Citation74 In a mouse model of IBD induced by murine norovirus, accumulation of damaged mitochondria during loss of the autophagy protein ATG16L1 induced epithelial cell death via necroptosis, driving barrier dysfunction and inflammation,Citation76 suggesting that disruption of autophagy and mitochondrial function can confer susceptibility to epithelial cell death and subsequent inflammation. These mechanisms whereby the microbiota elicits alterations to epithelial cell mitochondria play crucial roles in intestinal homeostasis and disease pathogenesis.
Gut microbiome signaling to mitochondria – CRC
CRC is the third most common cause of cancer-related deaths worldwide.Citation107,Citation108 90% of CRCs are adenocarcinomas originating in the epithelium via sporadic DNA mutations or as a consequence of chronic inflammation.Citation109 Patients with chronic IBD affecting the colon have increased risk for developing CRC, accounting for nearly 10–15% of deaths in IBD patients.Citation110,Citation111 It is well established that mitochondrial alterations are associated with all stages of tumorigenesis, including oncogenic transformation, tumor progression, and metastasis (). However, the contribution of mitochondria to cell transformation and tumor progression are not completely understood. The Warburg effect, first postulated in the 1950s, suggested that mitochondrial dysfunction causes cells to undergo aerobic glycolysis, a characteristic of cancer cells.Citation112 However, more recent studies convey that cancer metabolism is not the consequence of mitochondrial dysfunction, but the result of mitochondrial reprogramming by loss-of-function mutations in tumor suppressor genes or proto-oncogenic-directed gene modulation.Citation113 Mitochondrial reprogramming alters cellular metabolism to enhance anabolic pathways critical in supplying the increased requirements of tumor growth.Citation114 Alteration of cellular metabolism is fundamental to transformation, selected for during tumorigenesis, and does not occur passively as a response to damaged mitochondria or reduction in ATP concentration.Citation113 The assumption that cancer cells do not utilize mitochondria to derive ATP through oxidative phosphorylation is a misconception; oxidative phosphorylation yields a significant amount of ATP in cancer cells. However, these cells concurrently alter use of mitochondrial enzymes to create anabolic precursors such as reduced carbon, reduced nitrogen, and cytosolic NADPH for macromolecular synthesis of nucleotides, proteins, and lipids necessary for cell proliferation.Citation114 It has recently been proposed that cancer cells exhibit a hybrid metabolic state, utilizing both oxidative phosphorylation and glycolysis allowing adaptation to changing microenvironments.Citation115 In a dynamic process, the function of tumor mitochondria is complemented to the metabolic needs necessary to achieve rapid cell growth and enhance metastatic potential.
Figure 2. Mitochondrial alterations play a central role in CRC tumorigenesis. Cancer cells exhibit hybrid metabolism, utilizing both glycolysis and oxidative phosphorylation to drive ATP production and adapt to ever-changing microenvironments. Acetyl-CoA produced by glycolysis feeds the TCA cycle to produce cofactors NADH and FADH2 that donate electrons to the ETC and drive oxidative phosphorylation. Altered use of mitochondrial enzymes generate anabolic precursors necessary for rapid cell proliferation. Hypoxia is implicated as a driver of progression to CRC, with Hif-1α providing protection via enhanced D2HGDH production to inhibit the oncometabolite D2HG produced by the TCA cycle. Hif-2α directly targets STEAP4 which modulates mitochondrial iron balance and function. Although mtDNA mutations are demonstrated early in neoplasias, progression to malignancy selects for non-pathogenic mtDNA mutations and functioning mitochondria. These mitochondrial alterations enhance proliferation and metastatic potential and decrease apoptosis, driving CRC tumorigenesis.
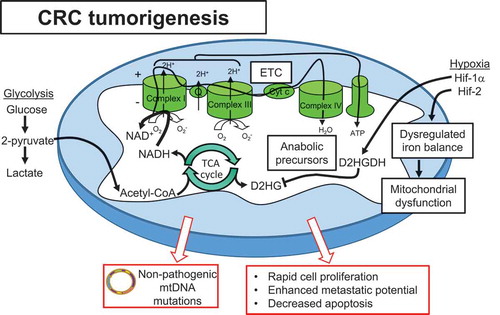
Mitochondrial alterations are evident early in cancer progression in both sporadic and IBD-related tumorigenesis. Emerging evidence suggests an early loss of mitochondrial function during pre-neoplasia and restoration after malignant transformation. UC-associated progression to tumorigenesis demonstrated early loss of mitochondria and increased glycolysis followed by restoration of oxidative phosphorylation and positive-selection of mitochondria in cancer cells to further growth.Citation116 This was further corroborated by a recent study demonstrating frequent mtDNA mutations contributing to early UC tumorigenesis that were no longer evident in cancer, suggesting functional mitochondria are required for transformation.Citation117 The same phenomenon has been observed in sporadic CRC tumorigenesis with increased mtDNA mutations demonstrated in adenomas that decrease in CRC.Citation118 This supports a model in which mtDNA mutations clonally expand in the colon epithelium during pre-neoplasia, exacerbated by increased epithelial cell proliferation as an injury response during inflammation. Progression to malignancy involves selection of clones carrying fewer and/or non-pathogenic mtDNA mutations.Citation117 Interestingly, methylation and silencing of DNA polymerase γ (POLG), which is the only mtDNA polymerase tasked with mtDNA repair and replication and maintenance of mitochondrial numbers, occurs early during a mouse model of colitis-associated CRC. This was associated with reduced mitochondrial levels, altered mitochondrial metabolism, and resistance to oxidative stress.Citation119 Studies using p0 cells, which are deficient in mitochondrial energy function, suggest that multiple established regulators of CRC depend on reduced mitochondrial function for their expression and are linked to lower patient survival.Citation120 CRC cells exhibit remodeling of intracellular calcium homeostasis characterized by increased mitochondrial calcium storage.Citation121 This increased mitochondrial calcium storage was dependent on aerobic glycolysis metabolism and contributed to enhanced cell proliferation, invasion, and resistance to cell death. Further studies are needed to gain an understanding of the role of mitochondrial metabolic reprogramming, mtROS accumulation, and mitochondrial biogenesis in this progression of mitochondrial alterations during tumorigenesis.
Recent studies suggest that hypoxia plays an important role in mediating early mitochondrial alterations during the progression of colitis to cancer. Hypoxia is present during both inflammation and cancer and there are similar metabolic reprogramming pathways affected in both states.Citation64 HIF-1α and HIF-2α are transcription factors responsive to hypoxia (). HIF-1α is beneficial to reduce intestinal inflammation and regulate epithelial barrier function, in contrast to HIF-2α that has been shown to promote colitis.Citation122 A recent quantitative proteomics study demonstrated that mitochondrial iron dysregulation occurred early during colitis via upregulation of the mitochondrial iron reductase, six-transmembrane epithelial antigen of prostate 4 (STEAP4).Citation64 Mechanistically, STEAP4 was shown to be a direct target gene of HIF-2α in a hypoxia-dependent manner, that led to a dysregulation in mitochondrial iron balance, mitochondrial dysfunction, tissue injury, and potentiated colon cancer.Citation64 Another recent study implicated HIF-1α in the elimination of the mitochondrial oncometabolite D-2-hydroxyglutarate (D-2-HG) in colonic epithelial cells, thereby protecting against colitis-associated cancer.Citation123 D-2-HG, which is normally produced in the TCA cycle and quickly converted to α-ketoglutarate by D-2-HG dehydrogenase (D-2-HGDH), was highly elevated during colitis and directly correlated with the severity of tumorigenesis in a mouse model of colitis-associated cancer. Elevated D-2-HG enhanced the progression to cancer and conferred a hyper-proliferative, non-apoptotic phenotype in cultured IECs, which are prone to neoplastic progression in the setting of chronic inflammation. The colonic expression of D-2-HGDH was decreased in UC patients at baseline who progressed to cancer. HIF-1α was shown to directly induce transcription of D-2-HGDH, suggesting hypoxia associated with inflammation regulates mitochondrial TCA metabolites that can drive progression of colitis to cancer.Citation123
Similar to IBD, CRC increased risk has been associated with dysbiosis of the gut microbiome and decreased production of SCFAs (). Several studies have demonstrated CRC patients exhibit increased abundance of F. nucleatum, E. coli, Bacteriodes fragiles, and Enterococcus faecalis, and decreased Clostridiales, Faecalibacterium, Blautia, and Bifidobacterium.Citation124 It has been proposed that abundance of Butyricicoccus, E. coli, and Fusobacterium, may provide potential biomarkers for normal, adenomatous, and cancerous colon mucosa, respectively.Citation125 Bacterial targeting of mitochondria is considered a risk factor for carcinogenesis but the mechanism underpinning this association has not been elucidated.Citation126 Computational prediction models suggest that 87 E. coli proteins target host cell mitochondria that contribute to the etiology of CRC.Citation127 SCFAs exhibit anti-tumorigenic properties in colon epithelial cells through homeostasis of mitochondrial metabolism and cell proliferation.Citation128 Gut bacteria have been shown to generate mutations of mtDNA and to modulate DNA repair pathways of the mitochondria.Citation126 Additionally, bacteria-to-mitochondrial signaling can inhibit the inherent apoptotic pathway in the gut epithelium.Citation129 Future studies are needed to gain further understanding of whether direct bacterial toxins or bacterial metabolites are involved in the mechanism whereby bacterial-to-mitochondrial signaling enhances colon tumorigenesis.
Mitochondrial targeting by potential therapeutics
Mitochondrial-targeted therapeutics for intestinal inflammation have thus far targeted elimination of mtROS via antioxidant function. The mitochondrial-targeted antioxidant mitoTEMPO is a key example of experimental drugs being used in preclinical research moving towards the elucidation of mitochondrial-based therapeutic approaches for intestinal disorders. Prophylactic mitoTEMPO treatment in DSS-induced colitis demonstrated less severe inflammation and barrier dysfunction.Citation130 Furthermore, mitoTEMPO decreased metabolic stress in human CD biopsies.Citation130 TEMPO not targeted to the mitochondria has been shown to affect microbiota, but this has not been investigated for mitoTEMPO.Citation131 The mitochondrial-targeted antioxidant MitoQ protected against murine ileocolitis in glutathione peroxidase (GPx) −1 and −2 knockouts.Citation132 Moreover, DSS-induced colitis was ameliorated by MitoQ in mice.Citation133 These accounts suggest that the targeting of mitochondrial mechanisms, namely mtROS production, assist in quelling inflammation as it relates to IBD. However, future translational studies using mitochondrial-targeted antioxidants in IBD biopsies or patients are necessary. Use of mitochondrial-targeted antioxidants during inflammation in pancreas, arterial blood vessels, liver, and brain also demonstrate beneficial results.Citation134–Citation137
With mitochondria as inducers of apoptotic activity and providers of ATP and intracellular ROS, they have been heavily implicated in affecting cancer cell activity.Citation138 Mitochondria targeting therapeutics for CRC stimulate mitochondrial-induced apoptosis or inhibit ETC complexes, curbing necessary ATP production for cancer cells. Pro-apoptotic factors such as Bcl-2 homology domain 3 (BH3) mimetics, Bcl-2-associated X protein (BAX) and Bcl-2 antagonist/killer protein (BAK) are potential therapeutics for cancer – namely ABT-263, ABT-737 and GX15-070 (obatoclax) for example.Citation139–Citation141 In addition, voltage-dependent anion channel blockers for the triggering of apoptosis and ROS regulators which are geared towards increasing oxidative stress, and thereby cytotoxicity, are also being used in the clinical setting to decrease tumor progression via mitochondrial targeting.Citation141 Inhibition of glycolysis, thereby decreasing ATP sources of energy for progressing tumors, can also be initiated via 2-deoxy-D-glucose (2DG).Citation142 Additionally, numerous studies suggest the mechanism by which the diabetic drug metformin inhibits cancer cells might correlate with its mitochondrial-targeting activity to inhibit the ETC.Citation143 Population studies such as that of Seo et al. have shown that diabetic patients treated with metformin, which inhibits ETC Complex I, present an approximately 30% reduced overall cancer incidence and mortality rate.Citation144 Therefore, mitochondrial-targeted therapeutics are emerging as an exciting opportunity for cancer therapy.
The usage of nutraceuticals such as prebiotic molecules and probiotic bacterial species, as well as an alternative diet, can help maintain intestinal homeostasis, thereby providing therapeutics for intestinal diseases. Although controversial in terms of effectiveness, the usage of prebiotics and probiotics in some studies have been shown to ameliorate dysbiotic characteristics of the gut microbiome.Citation145 Human CRC cell lines treated with butyrate, an SCFA, exhibited decreased cell proliferation and increased apoptotic cell death, suggesting that SCFAs may be capable of CRC inhibition.Citation146 Westernized diets consisting of increased ingestion of saturated fats, red meat, refined carbohydrates, and decreased dietary fibers not only potentiate dysbiosis of the microbiota but have also been implicated in increased CRC incidence.Citation145 Evidence of invoking a modified diet and nutritional management has been presented in some cancers as beneficial. Ketogenic diets (KD), in which 90% of the calories source is from fat coupled with sufficient protein and low carbohydrates, produces ketone bodies by metabolic changes that imitate those during starvation.Citation147 This mechanism forces cells to rely on fat oxidation and mitochondrial respiration in lieu of glycolysis that is a major source of ATP for cancer cells.Citation147,Citation148 Given these findings, implementing KD based nutritional adjustments could potentially reduce tumor size and progression rates that are dependent upon glucose as an energy source during anaerobic glycolysis.Citation147 In this way, KD can provide a rationale for therapeutic strategies via tumor growth inhibition.
Conclusions/perspectives
As the powerhouse of the eukaryotic cell, mitochondria play an imperative role in intestinal homeostasis. Mitochondria invoke innate and adaptive immune responses, thereby promoting inflammation and self-defense pathways. Mitochondrial function is crucial to proper epithelial barrier integrity during inflammation and plays a central role in tumorigenesis. The gut microbiome plays an important role in modulating mitochondrial signaling in intestinal mucosal cells, but the involvement of microbiota-mitochondrial cross-talk in driving intestinal diseases such IBD and CRC is not fully understood. Mitochondrial-targeted therapies provide an exciting novel strategy for intestinal diseases. Additionally, therapies aimed at altering the intestinal microbiome may in turn drive mitochondrial responses in intestinal cells. Future studies elucidating the molecular mechanisms linking the gut microbiome and host mitochondria in the pathogenesis of IBD and CRC will be fundamental in advancing mitochondrial-targeted therapies for these intestinal diseases.
Abbreviations
| CD | = | Crohn’s Disease |
| CRC | = | Colorectal cancer |
| CBC | = | crypt base columnar |
| DAMP | = | damage associated molecular pattern |
| ETC | = | electron transport chain |
| FAO | = | fatty acid oxidation |
| HIF | = | hypoxia inducible factor |
| IBD | = | inflammatory bowel disease |
| IMM | = | inner mitochondrial membrane |
| IECs | = | intestinal epithelial cells |
| ISCs | = | intestinal stem cells |
| KD | = | ketogenic diet |
| mtDNA | = | mitochondrial DNA |
| mtROS | = | mitochondrial reactive oxygen species |
| NLRP | = | nucleotide binding domain and leucine rich repeat pyrin |
| OMM | = | outer mitochondrial membrane |
| SCFAs | = | short-chain fatty acids |
| SI | = | small intestine |
| TCA | = | tricarboxylic acid |
| UC | = | ulcerative colitis |
Disclosure of Potential Conflicts of Interest
The authors have declared that no conflict of interest exists.
Acknowledgments
We would like to thank Dr. Sheré L. Paris for her gracious comments and input.
Additional information
Funding
Related Research Data
References
- Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi:10.1016/j.redox.2014.11.006.
- Li J, Sha B. The structure of tim50(164-361) suggests the mechanism by which tim50 receives mitochondrial presequences. Acta Crystallogr F Struct Biol Commun. 2015;71(Pt 9):1146–1151. doi:10.1107/S2053230X15013102.
- Engelking LR. 2015. Tricarboxylic acid (tca) cycle in textbook of veterinary physiological chemistry. 3rd. Oxford: Elsevier.
- Salisbury-Ruf CT, Bertram CC, Vergeade A, Lark DS, Shi Q, Heberling ML, Fortune NL, Okoye GD, Jerome WG, Wells QS, et al. Bid maintains mitochondrial cristae structure and protects against cardiac disease in an integrative genomics study. Elife. 2018;7. doi:10.7554/eLife.40907.
- Lee SR, Han J. Mitochondrial nucleoid: shield and switch of the mitochondrial genome. Oxid Med Cell Longev. 2017;2017:8060949. doi:10.1155/2017/8060949.
- Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2:REVIEWS1018.
- Falkenberg M. Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem. 2018;62(3):287–296. doi:10.1042/EBC20170100.
- Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103–123.
- D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62(3):309–320. doi:10.1042/EBC20170102.
- Sifuentes-Franco S, Padilla-Tejeda DE, Carrillo-Ibarra S, Miranda-Diaz AG. Oxidative stress, apoptosis, and mitochondrial function in diabetic nephropathy. Int J Endocrinol. 2018;2018:1875870. doi:10.1155/2018/1875870.
- Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83(4):568–581. doi:10.1038/ki.2012.441.
- Naghdi S, Slovinsky WS, Madesh M, Rubin E, Hajnóczky G. Mitochondrial fusion and bid-mediated mitochondrial apoptosis are perturbed by alcohol with distinct dependence on its metabolism. Cell Death Dis. 2018;9(10):1028. doi:10.1038/s41419-018-1070-3.
- Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833(5):1256–1268. doi:10.1016/j.bbamcr.2013.02.002.
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hfis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein dlp1. Mol Cell Biol. 2003;23:5409–5420.
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi:10.1038/nature06639.
- Hamacher-Brady A, Brady NR. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016;73(4):775–795. doi:10.1007/s00018-015-2087-8.
- Bingol B, Sheng M. Mechanisms of mitophagy: pink1, parkin, usp30 and beyond. Free Radic Biol Med. 2016;100:210–222. doi:10.1016/j.freeradbiomed.2016.04.015.
- El-Hattab AW, Suleiman J, Almannai M, Scaglia F. Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab. 2018. doi:10.1016/j.ymgme.2018.10.003.
- Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by pink1 to induce park2/parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9(11):1750–1757. doi:10.4161/auto.26122.
- Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. Ros-induced mitochondrial depolarization initiates park2/parkin-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8(10):1462–1476. doi:10.4161/auto.21211.
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547–564. doi:10.1515/hsz-2012-0119.
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. Iii. The steady state. J Biol Chem. 1955;217:409–427.
- Minocherhomji S, Tollefsbol TO, Singh KK. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics. 2012;7(4):326–334. doi:10.4161/epi.19547.
- Prochnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26(1):71–93. doi:10.1016/j.cmet.2017.06.018.
- Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M. Colitis induced in mice with dextran sulfate sodium (dss) is mediated by the nlrp3 inflammasome. Gut. 2010;59(9):1192–1199. doi:10.1136/gut.2009.197822.
- Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of il-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–519. doi:10.1126/science.aal3535.
- Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol. 2014;32:609–634. doi:10.1146/annurev-immunol-032713-120236.
- McGrath EE, Marriott HM, Lawrie A, Francis SE, Sabroe I, Renshaw SA, Dockrell DH, Whyte MK. Tnf-related apoptosis-inducing ligand (trail) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol. 2011;90(5):855–865. doi:10.1189/jlb.0211062.
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and pgc-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi:10.1016/j.cmet.2006.05.011.
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing cd8 t-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–107. doi:10.1038/nature08097.
- Wang D, Malo D, Hekimi S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived mclk1± mouse mutants. J Immunol. 2010;184(2):582–590. doi:10.4049/jimmunol.0902352.
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces il-1beta through hif-1alpha. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986.
- Barker N, Clevers H. Leucine-rich repeat-containing g-protein-coupled receptors as markers of adult stem cells. Gastroenterology. 2010;138(5):1681–1696. doi:10.1053/j.gastro.2010.03.002.
- Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75:289–311. doi:10.1146/annurev-physiol-030212-183744.
- Henning SJ, von Furstenberg RJ. Gi stem cells - new insights into roles in physiology and pathophysiology. J Physiol. 2016;594(17):4769–4779. doi:10.1113/JP271663.
- Elphick DA, Mahida YR. Paneth cells: their role in innate immunity and inflammatory disease. Gut. 2005;54(12):1802–1809. doi:10.1136/gut.2005.068601.
- Umar S. Intestinal stem cells. Curr Gastroenterol Rep. 2010;12(5):340–348. doi:10.1007/s11894-010-0130-3.
- Srinivasan T, Than EB, Bu P, Tung KL, Chen KY, Augenlicht L, Lipkin SM, Shen X. Notch signalling regulates asymmetric division and inter-conversion between lgr5 and bmi1 expressing intestinal stem cells. Sci Rep. 2016;6:26069. doi:10.1038/srep26069.
- Chandel NS, Jasper H, Ho TT, Passegue E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol. 2016;18(8):823–832. doi:10.1038/ncb3385.
- Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, Sacchetti A, Hornsveld M, Oost KC, Snippert HJ, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature. 2017;543(7645):424–427. doi:10.1038/nature21673.
- Berger E, Rath E, Yuan D, Waldschmitt N, Khaloian S, Allgauer M, Staszewski O, Lobner EM, Schottl T, Giesbertz P, et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat Commun. 2016;7:13171. doi:10.1038/ncomms13171.
- Shadel GS, Horvath TL. Mitochondrial ros signaling in organismal homeostasis. Cell. 2015;163(3):560–569. doi:10.1016/j.cell.2015.10.001.
- Asano J, Sato T, Ichinose S, Kajita M, Onai N, Shimizu S, Ohteki T. Intrinsic autophagy is required for the maintenance of intestinal stem cells and for irradiation-induced intestinal regeneration. Cell Rep. 2017;20(5):1050–1060. doi:10.1016/j.celrep.2017.07.019.
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, et al. Reactive oxygen species act through p38 mapk to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–451. doi:10.1038/nm1388.
- Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: A relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082.
- Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19(2):232–247. doi:10.1016/j.stem.2016.04.015.
- Kim DH, Cheon JH. Pathogenesis of inflammatory bowel disease and recent advances in biologic therapies. Immune Netw. 2017;17(1):25–40. doi:10.4110/in.2017.17.1.25.
- Stidham RW, Higgins PDR. Colorectal cancer in inflammatory bowel disease. Clin Colon Rectal Surg. 2018;31(3):168–178. doi:10.1055/s-0037-1602237.
- Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet. 1980;2:712–715.
- Söderholm JD, Olaison G, Peterson KH, Franzén LE, Lindmark T, Wirén M, Tagesson C, Sjödahl R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of crohn’s disease. Gut. 2002;50:307–313.
- Schürmann G, Brüwer M, Klotz A, Schmid KW, Senninger N, Zimmer KP. Transepithelial transport processes at the intestinal mucosa in inflammatory bowel disease. Int J Colorectal Dis. 1999;14:41–46.
- Santhanam S, Rajamanickam S, Motamarry A, Ramakrishna BS, Amirtharaj JG, Ramachandran A, Pulimood A, Venkatraman A. Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflamm Bowel Dis. 2012;18(11):2158–2168. doi:10.1002/ibd.22926.
- Sifroni KG, Damiani CR, Stoffel C, Cardoso MR, Ferreira GK, Jeremias IC, Rezin GT, Scaini G, Schuck PF, Dal-Pizzol F, et al. Mitochondrial respiratory chain in the colonic mucosal of patients with ulcerative colitis. Mol Cell Biochem. 2010;342(1–2):111–115. doi:10.1007/s11010-010-0474-x.
- Heller S, Penrose HM, Cable C, Biswas D, Nakhoul H, Baddoo M, Flemington E, Crawford SE, Savkovic SD. Reduced mitochondrial activity in colonocytes facilitates ampkalpha2-dependent inflammation. FASEB J. 2017;31(5):2013–2025. doi:10.1096/fj.201600976R.
- Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D. Induction of dsrna-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61(9):1269–1278. doi:10.1136/gutjnl-2011-300767.
- Boyapati RK, Dorward DA, Tamborska A, Kalla R, Ventham NT, Doherty MK, Whitfield PD, Gray M, Loane J, Rossi AG, et al. Mitochondrial dna is a pro-inflammatory damage-associated molecular pattern released during active ibd. Inflamm Bowel Dis. 2018;24(10):2113–2122. doi:10.1093/ibd/izy095.
- VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of crohn’s disease. Gastroenterology. 2014;146(1):200–209. doi:10.1053/j.gastro.2013.09.048.
- Liu TC, Gurram B, Baldridge MT, Head R, Lam V, Luo C, Cao Y, Simpson P, Hayward M, Holtz ML, et al. Paneth cell defects in crohn’s disease patients promote dysbiosis. JCI Insight. 2016;1(8):e86907. doi:10.1172/jci.insight.86907.
- Kugathasan S, Denson LA, Walters TD, Kim MO, Marigorta UM, Schirmer M, Mondal K, Liu C, Griffiths A, Noe JD, et al. Prediction of complicated disease course for children newly diagnosed with crohn’s disease: A multicentre inception cohort study. Lancet. 2017;389(10080):1710–1718. doi:10.1016/S0140-6736(17)30317-3.
- Mottawea W, Chiang CK, Muhlbauer M, Starr AE, Butcher J, Abujamel T, Deeke SA, Brandel A, Zhou H, Shokralla S, et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset crohn’s disease. Nat Commun. 2016;7:13419. doi:10.1038/ncomms13419.
- Rosa A, Abrantes P, Sousa I, Francisco V, Santos P, Francisco D, Xavier JM, Oliveira SA. Ulcerative colitis is under dual (mitochondrial and nuclear) genetic control. Inflamm Bowel Dis. 2016;22(4):774–781. doi:10.1097/MIB.0000000000000694.
- Dankowski T, Schroder T, Moller S, Yu X, Ellinghaus D, Bar F, Fellermann K, Lehnert H, Schreiber S, Franke A, et al. Male-specific association between mt-nd4 11719 a/g polymorphism and ulcerative colitis: a mitochondria-wide genetic association study. BMC Gastroenterol. 2016;16(1):118. doi:10.1186/s12876-016-0509-1.
- Ho GT, Aird RE, Liu B, Boyapati RK, Kennedy NA, Dorward DA, Noble CL, Shimizu T, Carter RN, Chew ETS, et al. Mdr1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. 2018;11(1):120–130. doi:10.1038/mi.2017.31.
- Xue X, Bredell BX, Anderson ER, Martin A, Mays C, Nagao-Kitamoto H, Huang S, Gyorffy B, Greenson JK, Hardiman K, et al. Quantitative proteomics identifies steap4 as a critical regulator of mitochondrial dysfunction linking inflammation and colon cancer. Proc Natl Acad Sci U S A. 2017;114(45):E9608–e9617. doi:10.1073/pnas.1712946114.
- Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, Grossniklaus E, Schoenborn AA, Sartor RB, Taylor GA. Irgm1-deficient mice exhibit paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2013;305(8):G573–584. doi:10.1152/ajpgi.00071.2013.
- Rogala AR, Schoenborn AA, Fee BE, Cantillana VA, Joyce MJ, Gharaibeh RZ, Roy S, Fodor AA, Sartor RB, Taylor GA, et al. Environmental factors regulate paneth cell phenotype and host susceptibility to intestinal inflammation in irgm1-deficient mice. Dis Model Mech. 2018;11(2). doi:10.1242/dmm.031070
- Lin Z, Nelson L, Franke A, Poritz L, Li TY, Wu R, Wang Y, MacNeill C, Thomas NJ, Schreiber S, et al. Octn1 variant l503f is associated with familial and sporadic inflammatory bowel disease. J Crohns Colitis. 2010;4(2):132–138. doi:10.1016/j.crohns.2009.09.003.
- Shekhawat PS, Srinivas SR, Matern D, Bennett MJ, Boriack R, George V, Xu H, Prasad PD, Roon P, Ganapathy V. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (octn2(-/-)) mice. Mol Genet Metab. 2007;92(4):315–324. doi:10.1016/j.ymgme.2007.08.002.
- Cunningham KE, Vincent G, Sodhi CP, Novak EA, Ranganathan S, Egan CE, Stolz DB, Rogers MB, Firek B, Morowitz MJ, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (pgc1alpha) protects against experimental murine colitis. J Biol Chem. 2016;291(19):10184–10200. doi:10.1074/jbc.M115.688812.
- Bar F, Bochmann W, Widok A, von Medem K, Pagel R, Hirose M, Yu X, Kalies K, Konig P, Bohm R, et al. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology. 2013;145(5):1055–1063.e1053. doi:10.1053/j.gastro.2013.07.015.
- Theiss AL, Vijay-Kumar M, Obertone TS, Jones DP, Hansen JM, Gewirtz AT, Merlin D, Sitaraman SV. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology. 2009;137(1):199–208, 208.e191-196. doi:10.1053/j.gastro.2009.03.033.
- Theiss AL, Laroui H, Obertone TS, Chowdhury I, Thompson WE, Merlin D, Sitaraman SV. Nanoparticle-based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm Bowel Dis. 2011;17(5):1163–1176. doi:10.1002/ibd.21469.
- Gangwar R, Meena AS, Shukla PK, Nagaraja AS, Dorniak PL, Pallikuth S, Waters CM, Sood A, Rao R. Calcium-mediated oxidative stress: A common mechanism in tight junction disruption by different types of cellular stress. Biochem J. 2017;474(5):731–749. doi:10.1042/BCJ20160679.
- Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci. 2008;13:7210–7226.
- Lopes F, Keita AV, Saxena A, Reyes JL, Mancini NL, Al Rajabi A, Wang A, Baggio CH, Dicay M, van Dalen R, et al. Er-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J Biol Chem. 2018;293(9):3073–3087. doi:10.1074/jbc.RA117.000809.
- Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, Dewan MZ, Lieberman SR, Lazrak A, Marinis JM, et al. Autophagy protein atg16l1 prevents necroptosis in the intestinal epithelium. J Exp Med. 2017;214(12):3687–3705. doi:10.1084/jem.20170558.
- Han J, Yu C, Souza RF, Theiss AL. Prohibitin 1 modulates mitochondrial function of stat3. Cell Signal. 2014;26(10):2086–2095. doi:10.1016/j.cellsig.2014.06.006.
- Fratila OC, Craciun C. Ultrastructural evidence of mucosal healing after infliximab in patients with ulcerative colitis. J Gastrointestin Liver Dis. 2010;19:147–153.
- Coleman OI, Haller D. Bacterial signaling at the intestinal epithelial interface in inflammation and cancer. Front Immunol. 2017;8:1927. doi:10.3389/fimmu.2017.01927.
- Turroni F, Ribbera A, Foroni E, van Sinderen D, Ventura M. Human gut microbiota and bifidobacteria: from composition to functionality. Antonie Van Leeuwenhoek. 2008;94(1):35–50. doi:10.1007/s10482-008-9232-4.
- Zoetendal EG, Vaughan EE, de Vos WM. A microbial world within us. Mol Microbiol. 2006;59(6):1639–1650. doi:10.1111/j.1365-2958.2006.05056.x.
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307(5717):1915–1920. doi:10.1126/science.1104816.
- Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119. doi:10.1016/B978-0-12-800100-4.00003-9.
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory t-cell generation. Nature. 2013;504(7480):451–455. doi:10.1038/nature12726.
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science. 2013;341(6145):569–573. doi:10.1126/science.1241165.
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor gpr43. Nature. 2009;461(7268):1282–1286. doi:10.1038/nature08530.
- Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial hif augments tissue barrier function. Cell Host Microbe. 2015;17(5):662–671. doi:10.1016/j.chom.2015.03.005.
- Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, Maruya M, Ian McKenzie C, Hijikata A, Wong C, et al. Metabolite-sensing receptors gpr43 and gpr109a facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6:6734. doi:10.1038/ncomms7734.
- Wallace JL, Blackler RW, Chan MV, Da Silva GJ, Elsheikh W, Flannigan KL, Gamaniek I, Manko A, Wang L, Motta JP, et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: translation to therapeutics. Antioxid Redox Signal. 2015;22(5):398–410. doi:10.1089/ars.2014.5901.
- Saint-Georges-Chaumet Y, Edeas M. Microbiota-mitochondria inter-talk: consequence for microbiota-host interaction. Pathog Dis. 2016;74(1):ftv096. doi:10.1093/femspd/ftv096.
- Ijssennagger N, van der Meer R, van Mil SWC. Sulfide as a mucus barrier-breaker in inflammatory bowel disease?. Trends Mol Med. 2016;22(3):190–199. doi:10.1016/j.molmed.2016.01.002.
- Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11(1):1–10. doi:10.1007/s12328-017-0813-5.
- Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693.
- Hofer U. Microbiome: bacterial imbalance in crohn’s disease. Nat Rev Microbiol. 2014;12(5):312. doi:10.1038/nrmicro3255.
- Forbes JD, Van Domselaar G, Bernstein CN. The gut microbiota in immune-mediated inflammatory diseases. Front Microbiol. 2016;7:1081. doi:10.3389/fmicb.2016.01081.
- Butto LF, Schaubeck M, Haller D. Mechanisms of microbe-host interaction in crohn’s disease: dysbiosis vs. Pathobiont selection. Front Immunol. 2015;6:555. doi:10.3389/fimmu.2015.00555.
- Hirose M, Kunstner A, Schilf P, Sunderhauf A, Rupp J, Johren O, Schwaninger M, Sina C, Baines JF, Ibrahim SM. Mitochondrial gene polymorphism is associated with gut microbial communities in mice. Sci Rep. 2017;7(1):15293. doi:10.1038/s41598-017-15377-7.
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UC, Podowski RM, Näslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396(6707):133–140. doi:10.1038/24094.
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. Tlr signalling augments macrophage bactericidal activity through mitochondrial ros. Nature. 2011;472(7344):476–480. doi:10.1038/nature09973.
- Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, Ellett F, Lieschke GJ, Crosier KE, Crosier PS. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating beta-oxidation-dependent mitochondrial ros production. Cell Metab. 2013;18(2):265–278. doi:10.1016/j.cmet.2013.06.018.
- Swanson PA 2nd, Kumar A, Samarin S, Vijay-Kumar M, Kundu K, Murthy N, Hansen J, Nusrat A, Neish AS. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc Natl Acad Sci U S A. 2011;108(21):8803–8808. doi:10.1073/pnas.1010042108.
- Formentini L, Santacatterina F, Nunez de Arenas C, Stamatakis K, Lopez-Martinez D, Logan A, Fresno M, Smits R, Murphy MP, Cuezva JM. Mitochondrial ros production protects the intestine from inflammation through functional m2 macrophage polarization. Cell Rep. 2017;19(6):1202–1213. doi:10.1016/j.celrep.2017.04.036.
- Saxena A, Lopes F, Poon KKH, McKay DM. Absence of the nod2 protein renders epithelia more susceptible to barrier dysfunction due to mitochondrial dysfunction. Am J Physiol Gastrointest Liver Physiol. 2017;313(1):G26–G38. doi:10.1152/ajpgi.00070.2017.
- Matarrese P, Falzano L, Fabbri A, Gambardella L, Frank C, Geny B, Popoff MR, Malorni W, Fiorentini C. Clostridium difficile toxin b causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of atp-sensitive mitochondrial potassium channels. J Biol Chem. 2007;282(12):9029–9041. doi:10.1074/jbc.M607614200.
- Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol Microbiol. 2004;54(3):665–675. doi:10.1016/j.bbrc.2005.09.130.
- Kenny B, Jepson M. Targeting of an enteropathogenic escherichia coli (epec) effector protein to host mitochondria. Cell Microbiol. 2000;2:579–590.
- Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3(4):464–471. doi:10.1001/jamaoncol.2016.5194.
- Wen J, Xu Q, Yuan Y. Single nucleotide polymorphisms and sporadic colorectal cancer susceptibility: A field synopsis and meta-analysis. Cancer Cell Int. 2018;18:155. doi:10.1186/s12935-018-0656-2.
- Munro MJ, Wickremesekera SK, Peng L, Tan ST, Itinteang T. Cancer stem cells in colorectal cancer: A review. J Clin Pathol. 2018;71(2):110–116. doi:10.1136/jclinpath-2017-204739.
- Mattar MC, Lough D, Pishvaian MJ, Charabaty A. Current management of inflammatory bowel disease and colorectal cancer. Gastrointest Cancer Res. 2011;4:53–61.
- Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in ibd: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6(5):297–305. doi:10.1038/nrgastro.2009.44.
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270.
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi:10.1016/j.ccr.2012.02.014.
- Ward PS, Thompson CB. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol. 2012;4(7):a006783. doi:10.1101/cshperspect.a006783.
- Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, Kaipparettu BA, Onuchic JN. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017;77(7):1564–1574. doi:10.1158/0008-5472.CAN-16-2074.
- Smolkova K, Plecita-Hlavata L, Bellance N, Benard G, Rossignol R, Jezek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43(7):950–968. doi:10.1016/j.biocel.2010.05.003.
- Baker KT, Nachmanson D, Kumar S, Emond MJ, Ussakli C, Brentnall TA, Kennedy SR, Risques RA. Mitochondrial dna mutations are associated with ulcerative colitis preneoplasia but tend to be negatively selected in cancer. Mol Cancer Res. 2018. doi:10.1158/1541-7786.MCR-18-0520.
- Ericson NG, Kulawiec M, Vermulst M, Sheahan K, O’Sullivan J, Salk JJ, Bielas JH. Decreased mitochondrial dna mutagenesis in human colorectal cancer. PLoS Genet. 2012;8(6):e1002689. doi:10.1371/journal.pgen.1002689.
- Maiuri AR, Li H, Stein BD, Tennessen JM, O’Hagan HM. Inflammation-induced dna methylation of dna polymerase gamma alters the metabolic profile of colon tumors. Cancer Metab. 2018;6:9. doi:10.1186/s40170-018-0182-7.
- Penrose HM, Heller S, Cable C, Nakhoul H, Ungerleider N, Baddoo M, Pursell ZF, Flemington EK, Crawford SE, Savkovic SD. In colonic rho(0) (rho0) cells reduced mitochondrial function mediates transcriptomic alterations associated with cancer. Oncoscience. 2017;4(11–12):189–198. doi:10.18632/oncoscience.386.
- Hernandez-Morales M, Sobradillo D, Valero RA, Munoz E, Ubierna D, Moyer MP, Nunez L, Villalobos C. Mitochondria sustain store-operated currents in colon cancer cells but not in normal colonic cells: reversal by non-steroidal anti-inflammatory drugs. Oncotarget. 2017;8(33):55332–55352. doi:10.18632/oncotarget.19430.
- Ramakrishnan SK, Shah YM. Role of intestinal hif-2alpha in health and disease. Annu Rev Physiol. 2016;78:301–325. doi:10.1146/annurev-physiol-021115-105202.
- Han J, Jackson D, Holm J, Turner K, Ashcraft P, Wang X, Cook B, Arning E, Genta RM, Venuprasad K, et al. Elevated d-2-hydroxyglutarate during colitis drives progression to colorectal cancer. Proc Natl Acad Sci U S A. 2018;115(5):1057–1062. doi:10.1073/pnas.1712625115.
- Zou S, Fang L, Lee MH. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol Rep (Oxf). 2018;6(1):1–12. doi:10.1093/gastro/gox031.
- Xu K, Jiang B. Analysis of mucosa-associated microbiota in colorectal cancer. Med Sci Monit. 2017;23:4422–4430.
- Strickertsson JAB, Desler C, Rasmussen LJ. Bacterial infection increases risk of carcinogenesis by targeting mitochondria. Semin Cancer Biol. 2017;47:95–100. doi:10.1016/j.semcancer.2017.07.003.
- Khan AA, Khan Z, Malik A, Shrivastava A, Jain SK, Alshamsan A. Computational prediction of escherichia coli proteins host subcellular targeting and their implications in colorectal cancer etiology. Cancer Lett. 2015;364(1):25–32. doi:10.1016/j.canlet.2015.04.024.
- Meng C, Bai C, Brown TD, Hood LE, Tian Q. Human gut microbiota and gastrointestinal cancer. Genom Proteom Bioinf. 2018;16(1):33–49. doi:10.1016/j.gpb.2017.06.002.
- Clark A, Mach N. The crosstalk between the gut microbiota and mitochondria during exercise. Front Physiol. 2017;8:319. doi:10.3389/fphys.2017.00319.
- Wang A, Keita AV, Phan V, McKay CM, Schoultz I, Lee J, Murphy MP, Fernando M, Ronaghan N, Balce D, et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am J Pathol. 2014;184(9):2516–2527. doi:10.1016/j.ajpath.2014.05.019.
- Aytekin FO, Teke Z, Aydin C, Kabay B, Yenisey C, Sacar S, Demir EM, Tekin K. Effects of a membrane-permeable radical scavenger, tempol, on healing of colonic anastomoses in the cecal ligation and puncture model of polymicrobial sepsis in rats. Am J Surg. 2007;193(6):723–729. doi:10.1016/j.amjsurg.2006.06.035.
- Chu FF, Esworthy RS, Doroshow JH, Grasberger H, Donko A, Leto TL, Gao Q, Shen B. Deficiency in duox2 activity alleviates ileitis in gpx1- and gpx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 2017;11:144–156. doi:10.1016/j.redox.2016.11.001.
- Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, Yoon KS, Kim HJ, Park JH, Murphy MP, et al. Mitochondria-targeted antioxidant mitoq ameliorates experimental mouse colitis by suppressing nlrp3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11:178. doi:10.1186/1741-7015-11-178.134.
- Huang W, Cash N, Wen L, Szatmary P, Mukherjee R, Armstrong J, Chvanov M, Tepikin AV, Murphy MP, Sutton R, et al. Effects of the mitochondria-targeted antioxidant mitoquinone in murine acute pancreatitis. Mediators Inflamm. 2015;2015:901780. doi:10.1155/2015/901780.
- Mao P, Manczak M, Shirendeb UP, Reddy PH. Mitoq, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim Biophys Acta. 2013;1832(12):2322–2331. doi:10.1016/j.bbadis.2013.09.005.
- Maiti AK, Saha NC, More SS, Panigrahi AK, Paul G. Neuroprotective efficacy of mitochondrial antioxidant mitoq in suppressing peroxynitrite-mediated mitochondrial dysfunction inflicted by lead toxicity in the rat brain. Neurotox Res. 2017;31(3):358–372. doi:10.1007/s12640-016-9692-7.
- Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M, Gioscia-Ryan RA, Murphy MP, et al. Chronic supplementation with a mitochondrial antioxidant (mitoq) improves vascular function in healthy older adults. Hypertension. 2018;71(6):1056–1063. doi:10.1161/HYPERTENSIONAHA.117.10787.
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi:10.1038/nature07733.
- Zhang E, Zhang C, Su Y, Cheng T, Shi C. Newly developed strategies for multifunctional mitochondria-targeted agents in cancer therapy. Drug Discov Today. 2011;16(3–4):140–146. doi:10.1016/j.drudis.2010.12.006.
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi:10.1038/nature03579.
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi:10.1038/nrd3137.
- Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-deoxy-d-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67(7):3364–3370. doi:10.1158/0008-5472.CAN-06-3717.
- Wang SQ, Cui SX, Qu XJ. Metformin inhibited colitis and colitis-associated cancer (cac) through protecting mitochondrial structures of colorectal epithelial cells in mice. Cancer Biol Ther. 2018:1–11. doi:10.1080/15384047.2018.1529108.
- Seo YS, Kim YJ, Kim MS, Suh KS, Kim SB, Han CJ, Jang WI, Kang SH, Tchoe HJ, Park CM, et al. Association of metformin use with cancer-specific mortality in hepatocellular carcinoma after curative resection: A nationwide population-based study. Medicine (Baltimore). 2016;95(17):e3527. doi:10.1097/MD.0000000000003527.
- Rossi M, Mirbagheri S, Keshavarzian A, Bishehsari F. Nutraceuticals in colorectal cancer: A mechanistic approach. Eur J Pharmacol. 2018;833:396–402. doi:10.1016/j.ejphar.2018.06.027.
- Zhang J, Yi M, Zha L, Chen S, Li Z, Li C, Gong M, Deng H, Chu X, Chen J, et al. Sodium butyrate induces endoplasmic reticulum stress and autophagy in colorectal cells: implications for apoptosis. PLoS One. 2016;11(1):e0147218. doi:10.1371/journal.pone.0147218.
- Khodadadi S, Sobhani N, Mirshekar S, Ghiasvand R, Pourmasoumi M, Miraghajani M, Dehsoukhteh SS. Tumor cells growth and survival time with the ketogenic diet in animal models: a systematic review. Int J Prev Med. 2017;8:35. doi:10.4103/2008-7802.207035.
- Warburg OH. The classic: the chemical constitution of respiration ferment. Clin Orthop Relat Res. 2010;468(11):2833–2839. doi:10.1007/s11999-010-1534-y.