
ABSTRACT
While the association between microbiomes and inflammatory bowel disease (IBD) is well known, establishing causal relationships between the two is difficult in humans. Germ-free (GF) mice genetically susceptible to IBD can address this question. Smad3-/- mice with defective transforming growth factor ß signaling are a model of IBD and colon cancer. They develop IBD upon colonization with Helicobacter under specific pathogen-free conditions, suggesting a role of the microbiome in IBD in this model. Thus, we rederived Smad3-/- mice GF to determine the potential of using these mice for testing the causative role of microbiomes in IBD. We found that fecal microbiomes from mice with IBD cause more severe gut inflammation in GF Smad3-/- and wild type mice compared to microbiomes from healthy mice and that Helicobacter induces gut inflammation within the context of other microbiomes but not by itself. Unexpectedly, GF Smad3+/+ and Smad3+/- mice given IBD microbiomes develop IBD despite their lack of disease in SPF conditions upon Helicobacter infection. This was not unique to the background strain of our Smad3 model (129); both wild type C57BL/6 and 129 strains developed IBD upon fecal transfer. However, wild type Swiss Webster stock was not susceptible, indicating that the genetic background of recipient mice influences the severity of IBD following fecal transfer. Our data suggest that the microbiome is an independent risk factor contributing to IBD development, and careful characterization of new GF models is needed to understand potential sources of confounding factors influencing microbiome studies in these mice.
Introduction
While the precise etiology of inflammatory bowel disease (IBD) is not known, multiple factors, including genetic susceptibility, immune responses, and intestinal microbiota, contribute to disease development.Citation1,Citation2 Various murine models have been employed to evaluate factors that can alter the risk of IBD and to elucidate mechanisms of disease development and progression.Citation3 Our laboratory has developedCitation4 and used Smad3-/- mice as a model for IBD and colitis-associated colon cancer (CAC). Smad3-/- mice are susceptible to IBD development in response to both chemicalCitation5 and bacterialCitation4 insults because they lack SMAD3, a transcription factor that is part of the transforming growth factor-ß (TGF-ß) signaling cascade. This signaling pathway is important in regulating inflammatory responses and tissue repair which can both contribute to IBD. Smad3-/- mice develop acute inflammation in the cecum and colon within a week of bacterially driven inflammation triggered by a commensal bacterium, Helicobacter bilis, in a specific pathogen free (SPF) facility.Citation4 Although H. bilis does not induce inflammation in wild type mice, it triggers acute inflammation in Smad3-/- mice and thus, could be considered a pathobiont (“resident microbe with pathogenic potential”Citation6). Most H. bilis-infected Smad3-/- mice recover and live for an extended asymptomatic period before a subset develops colon cancer.Citation4 Interestingly, mice that develop colon cancer have differences in their gut microbe composition during the early inflammatory phase compared to those that do not develop colon cancer (manuscript in preparation). While these data suggest to us that the gut microbiome plays a role in IBD/CAC, we have not been able to establish a causative relationship between microbiomes and IBD/CAC. Thus, we rederived Smad3-/- mice germ-free (GF) to enable future investigations to test the hypothesis that the gut microbiome plays a causative role in IBD/CAC. Our goals for the studies reported here were (1) to characterize IBD development and severity in the mice following the transfer of various microbiomes and (2) to determine the potential of using GF Smad3-/- mice as a model to assess the functional potential of particular microbiomes in IBD/CAC.
Results
Fecal microbiota of SPF Smad3-/- mice with IBD causes more severe gut inflammation in GF Smad3-/- mice than fecal microbiota of Smad3-/- mice without IBD
To determine the causative role of the gut microbiome in IBD, we performed a series of fecal transfer studies using feces collected from SPF Smad3-/- mice with and without IBD. The healthy microbiome was prepared from pooled fecal samples from random SPF Smad3-/- mice in our colony that are free of Helicobacter (Suppl. Table 1). IBD microbiome was prepared from SPF Smad3-/- mice infected with H. bilis (Suppl. Table 1). In the first study, we used GF Smad3-/- mice as recipients because Smad3+/+ or Smad3+/- mice are not susceptible to IBD development with H. bilis infection in SPF facilities. Fecal transfer from SPF Smad3-/- mice with IBD caused severe clinical signs of IBD in GF Smad3-/- mice within 2 days of transfer. In comparison, GF Smad3-/- mice colonized with microbiomes of SPF Smad3-/- mice without IBD (referred to as “healthy microbiome” or “no IBD” in subsequent sections) did not show any clinical signs of IBD at 2 days post transfer. Histopathology confirmed clinical findings; microbiomes from mice with IBD induced severe inflammation with extensive mucosal necrosis and ulceration throughout the colon and cecum of recipient mice while the healthy microbiome induced inflammation that was less ulcerative and limited mostly to the cecum ().
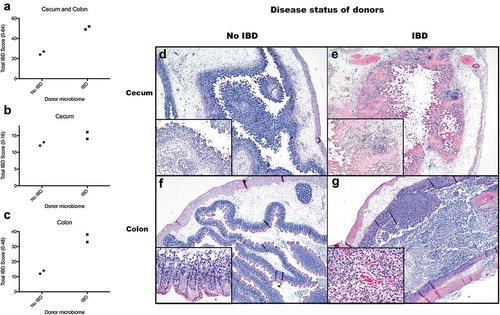
Figure 1. Microbiomes of mice with IBD induce more severe gut inflammation in recipient GF Smad3-/- mice compared to microbiomes of mice without IBD. GF Smad3-/- mice were colonized with microbiomes of mice with and without IBD. IBD was induced in donors by H. bilis infection, and the healthy microbiome was obtained from Smad3-/- SPF colony mice housed in a facility that excludes Helicobacter. Two days following the colonization, ceca and colons were scored for inflammation by histological analysis. (a) IBD scores of colon and cecum, (b) cecum, and (c) colon are shown. Possible score ranges for each tissue type are indicated on the Y-axis labels. (d) Cecum from a GF Smad3-/- mouse colonized with microbiomes from donors without IBD shows marked submucosal edema with diffuse inflammation and attenuation of the mucosa that, although pronounced, is less severe compared to cecum from a GF Smad3-/- mouse colonized with microbiome of donors with IBD (e), in which there is marked mucosal congestion and more extensive mucosal ulceration. In the proximal colon, a mouse colonized with microbiome from donors without IBD (f) shows only minimal to mild inflammation, with more extensive and severe inflammation in a mouse colonized with microbiome of donors with IBD (G).
Fecal microbiota from mice without IBD induces cecal inflammation in recipient GF mice independent of the recipient genotype
Because the healthy microbiome from SPF Smad3-/- mice also induced significant inflammation in the cecum of colonized recipient Smad3-/- mice, we next tested whether fecal transfer itself can cause cecal inflammation and if the development of inflammation was dependent upon recipient genotype. We transferred healthy microbiome from SPF Smad3-/- mice to GF Smad3+/+, Smad3+/- and Smad3-/- mice. All mice were euthanized at 6 days post colonization. Unexpectedly, gross observations of the ceca and colons of GF Smad3-/- mice showed less thickening compared to Smad3+/+ or Smad3+/- animals indicative of less inflammation, which was corroborated by histological analysis (Suppl. Figure 1). All recipient mice had substantial inflammation in the cecum with only mild inflammation in colons. Thus, it appears that fecal colonization, even from healthy donors, induces severe inflammation in the ceca of GF mice independent of recipient genotypes.
Microbiomes from mice with IBD (acute disease or remission) induced IBD in recipient GF mice
Under SPF conditions, following inoculation with H. bilis, Smad3-/- mice develop a transient period of IBD, which typically resolves clinically over a period of 2–4 weeks. Mice exhibit a period of disease remission for several months until the potential development of colon cancer. We therefore examined whether the disease activity (acute disease vs. remission) of donor mice is associated with the severity of IBD in recipient GF mice. Because GF Smad3-/- mice developed acute and severe inflammation when colonized with IBD-associated microbiomes, requiring early euthanasia, we addressed this question by colonizing GF Smad3+/- mice with microbiomes from SPF Smad3-/- mice without IBD, with acute IBD, and in remission from IBD and monitoring them for up to 6 days. GF Smad3+/- mice colonized with healthy microbiome did not show any clinical signs of IBD while those that received microbiomes from mice with IBD (acute or remission) had diarrhea and weight loss. In agreement with this observation, GF mice colonized with microbiomes of mice with IBD developed more severe inflammation throughout the colon (). However, the phase of H. bilis-induced IBD in the donor mice (acute vs. remission) did not influence the severity of inflammation in recipient mice. Though the cecal inflammation scores were not significantly different between recipients of healthy microbiomes vs. IBD-associated microbiomes ()), the ceca of mice treated with acute IBD feces had marked, extensive intraluminal necrotic debris not observed in the other groups ()). These data suggest that microbiomes from donors with IBD (either active or in remission) can induce more severe inflammation in GF recipients compared to those from healthy donors. Additionally, multiple studies comparing transfer of healthy vs. IBD microbiomes (from 2–3 pooled donor samples) into GF Smad3+/+, Smad3+/- and Smad3-/- mice consistently showed that IBD-associated microbiomes induce more severe gut inflammation in GF mice compared to healthy microbiomes (Suppl. Table 1, Suppl. Figure 2).
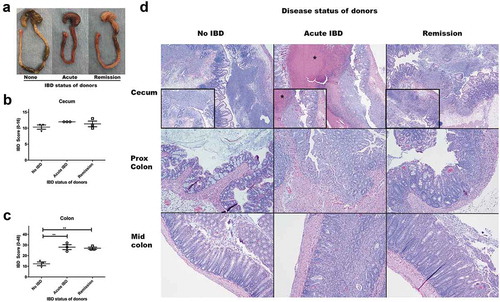
Figure 2. Microbiomes of mice with acute IBD or in remission from IBD induce more severe IBD in GF Smad3+/- mice compared to microbiomes of healthy mice. GF Smad3+/- mice were colonized with microbiomes of mice with acute IBD, in remission, and without IBD for 6 days. IBD was scored in the colon and cecum of individual mice by histological analysis. (a) Representative pictures of the cecum and colon of recipient mice that were colonized with microbiomes of mice without disease, with acute IBD or in remission. IBD scores of (b) cecum and (c) colon are shown. Possible score ranges for each tissue type are indicated on the Y-axis labels. (d) Representative histological images are shown of the cecum, proximal colon and mid colon for each colonization group. All mice have severe inflammation with regionally extensive severe mucosal ulceration in the cecum, although the mice colonized with acute IBD microbiomes have marked necrotic material expanding the lumen of the cecum (asterisk). In the proximal and mid colon, mice colonized with healthy microbiomes (no IBD) have minimal inflammation, whereas mice colonized with acute and remission IBD microbiomes have moderate diffuse inflammation. Horizontal bars represent mean ± SEM. One-way ANOVA test was performed followed by a post-hoc test of pair-wise comparisons with Bonferroni’s multiple comparison adjustment. **, p < 0.01.
H. bilis mono-colonization does not cause IBD in GF mice
Because severe IBD was induced in GF Smad3+/- mice by microbiomes collected from SPF Smad3-/- mice during acute inflammation or remission and H. bilis was used to induce IBD in donor mice, we investigated whether H. bilis alone can induce IBD in GF mice by conducting two studies. In one study, GF Smad3+/- and Smad3-/- were colonized with H. bilis and observed for clinical symptoms of IBD for up to 8 weeks. Three out of four mice gained weight steadily throughout the study without significant signs of disease. One mouse briefly experienced weight loss but recovered within a week. At necropsy, all mice had grossly normal ceca and colons although ceca were bigger than those in SPF mice or GF mice colonized with a fecal microbiome (Suppl. Figure 3(a)). Histological analysis showed minimal to mild inflammation in ceca and colons of H. bilis-mono-colonized mice (Suppl. Figure 3(b-d)). In another study, GF Smad3+/- mice were colonized with H. bilis and subsets of mice were euthanized 7, 10 and 21 days post colonization to evaluate the degree of gut inflammation at time points earlier than 8 weeks. Histological analysis showed that moderate inflammation occurred in the cecum and colon 7 days post colonization which subsided significantly by 3 weeks post colonization (-c)). These data suggest that H. bilis itself is not the cause of the severe inflammation seen in GF mice colonized with microbiomes of SPF Smad3-/- mice with IBD induced by H. bilis (). Instead, it acts in concert with other microbes to trigger IBD.
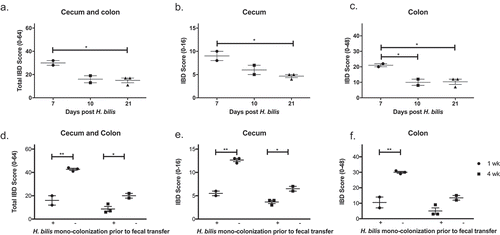
Figure 3. H. bilis is not a cause of IBD and prevents severe IBD in GF mice. GF Smad3+/- mice were mono-colonized with H. bilis, and the severity of inflammation was determined 7, 10 and 21 days post colonization by histological analysis of ceca and colons (a-c). IBD scores of cecum and colon (a), cecum (b), and colon (c) are shown. Possible score ranges for each tissue type are indicated on the Y-axis labels. GF Smad3+/- mice were colonized with microbiomes of mice with IBD, with or without prior H. bilis mono-colonization. The severity of IBD was determined 1 week and 4 weeks post fecal colonization by histological analysis of ceca and colons (d-f). IBD scores of cecum and colon (d), cecum (e), and colon (f) are shown. Horizontal bars represent mean ± SEM. One-way ANOVA test was performed followed by a post-hoc test of pair-wise comparisons with Bonferroni’s multiple comparison adjustment (a-c). Unpaired t-test was used for comparisons between the groups with and without mono-colonization prior to fecal transfer (D-F). *, p < 0.05; **, p < 0.01.
H. bilis infection prior to fecal microbiome transfer prevents severe IBD in GF Smad3+/- and Smad3-/- mice
Due to the severe inflammation induced in GF Smad3-/- mice upon fecal transplantation of IBD-associated microbiomes necessitating euthanasia, we were unable to study the causality of specific microbiomes in CAC in this model. Because H. bilis mono-colonization induced mild to moderate inflammation that quickly resolved, we tested if pre-colonization with H. bilis would suppress IBD in GF mice colonized with IBD-associated microbiomes. To test this, GF Smad3+/- mice were gavaged with H. bilis or broth 3 weeks prior to being colonized with IBD-associated microbiomes. Recipient mice mono-colonized with H. bilis prior to fecal colonization had significantly less inflammation in the cecum and colon at 1 and 4 weeks after the fecal microbiome transfer compared to the mice without prior H. bilis mono-colonization (-f)). The differences in IBD severity in H. bilis-precolonized and -uncolonized mice were greater at 1 week compared to 4 weeks following the fecal transfer. Similarly, H. bilis mono-colonization protected Smad3-/- mice from developing severe IBD upon colonization with IBD-associated microbiomes. All Smad3-/- mice that were mono-colonized with H. bilis prior to fecal transfer survived up to 1 week post fecal transfer with IBD-associated microbiomes (pre-determined time point) without showing symptoms of severe inflammation seen in the first study (). In agreement with this observation, IBD scores of Smad3-/- mice pre-colonized with H. bilis were significantly lower (19.3 ± 2.2, mean ± SEM) compared to those in the first study (50.5 ± 1.5).
The genetic background of mice significantly influences susceptibility to IBD upon fecal transfer
The genetic background of mice influences susceptibility to gut inflammation in various models of IBD.Citation7,Citation8 We have also found that Smad3-/- mice on a 129 background are more susceptible to H. bilis-triggered IBD than those on a C57BL/6 background in an SPF facility (unpublished observation). We were surprised to observe that Smad3+/+ and Smad3+/- recipients of IBD microbiomes developed severe inflammation because these genotypes do not develop IBD upon H. bilis infection under SPF conditions. In light of these observations, we wondered if the severe gut inflammation in response to IBD microbiomes seen in GF Smad3+/+ and Smad3+/- recipients was due to their genetic background, 129. To determine whether background genetics influenced susceptibility to IBD in GF mice following fecal transfer, we colonized GF wild type mice of two different strains (C57BL/6 and 129) and one stock (Swiss Webster) with fecal microbiomes from SPF Smad3-/- mice with or without IBD. Recipients were euthanized 1 week following colonization to determine the incidence and severity of IBD. All mice had acute cecal inflammation in response to fecal transfer, regardless of the IBD status of the donor. However, microbiomes of donors with IBD caused more severe gut inflammation compared to those of donors without IBD in both 129 and C57BL/6 mice but not in Swiss Webster mice (), suggesting that background genetics plays a role in susceptibility to IBD in GF mice.
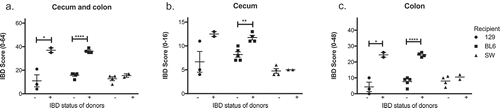
Figure 4. Microbiomes of mice with IBD induce inflammation in GF recipients in a manner dependent on the genetic background of recipient GF mice. Microbiomes of mice with or without IBD were transferred to GF C57BL/6, 129 and Swiss Webster mice, and the severity of IBD was determined 6 days post fecal transfer by histological analysis. IBD scores of cecum and colon (a), cecum (b), and colon (c) are shown. One-way ANOVA was performed followed by a post-hoc test of pair-wise comparisons with Bonferroni’s multiple comparison adjustment. *, p < 0.05; **, p < 0.01.
Discussion
Both animal and human studies suggest that gut bacteria can contribute to the risk for IBD. Incidence and severity of gut inflammation is significantly decreased in murine models of IBD, such as Il10-/-, TCRα-/-, and Mdr1a-/- mice, when they are treated with antibiotics or raised germ-free.Citation4,Citation9–Citation12 Antibiotics are also used to treat patients with IBD, and fecal transplantation is being studied as a treatment for IBD in humans.Citation13,Citation14 In addition, compositional changes in commensal bacteria (dysbiosis) occur in mouse and human IBDCitation15-Citation17 although it is not known whether the microbiome changes in IBD are a cause or consequence of the disease. To address this question, GF mice with genetic susceptibility to IBD can be colonized with specific microbiomes and followed for incidence and severity of the disease. Our laboratory uses Smad3-/- mice to study factors influencing IBD and CAC development as these mice are susceptible to gut inflammation due to alterations in the TGFß signaling pathwayCitation4,Citation5,Citation18,Citation19 that is frequently disturbed in human IBD/CAC.Citation20,Citation21 In addition, single nucleotide polymorphisms found in the SMAD3 gene in humans have been associated with increased recurring surgery risk in CD patientsCitation22 and with increased risk of UC.Citation23 Thus, we rederived Smad3-/- mice GF to determine whether microbiomes associated with IBD directly contribute to disease development. GF Smad3-/- mice were extremely susceptible to intestinal inflammation upon fecal transfer from SPF Smad3-/- mice with IBD, resulting in severe inflammation throughout the cecum and colon very early after colonization. Unexpectedly, healthy microbiomes from SPF mice without IBD also induced inflammation in the cecum of these mice. Therefore, we carried out additional studies to characterize the newly rederived GF model and to determine the role of the microbiome in IBD through experimenting with different donor microbiomes and genotypes of recipient mice (Smad3-/-, Smad3+/-, Smad3+/+). We found that severe gut inflammation could be triggered in any of the genotypes upon fecal transfer from mice with IBD even though Smad3+/+ and Smad3+/- mice are not susceptible to bacterially induced IBD/CAC under SPF conditions. In addition, IBD-associated microbiomes induced more severe inflammation throughout the colon and cecum compared to a healthy microbiome in GF recipients. Healthy microbiome-induced inflammation was limited mostly to the cecum of GF recipients. We do not know the exact mechanisms for why the onset of IBD is faster and more severe in the GF Smad3−/- model compared to SPF Smad3-/- mice. We postulate that perhaps in GF Smad3-/-, the severe rapid disease results from a completely uncontrolled nascent immune response driven by Smad3-/- immune cells with the proclivity to mount inflammatory Th1-type immune responses.Citation4 However, this needs to be tested in the future. These data suggest that microbiomes associated with IBD play a causative role in IBD in GF recipients.
While a large number of bacteria reside in the mammalian gut, they do not elicit pathogenic reactions from hosts as long as the host maintains an intact gut barrier and balanced immune function.Citation24 In addition, commensals in the gut also protect hosts from pathogenic bacteria by occupying various niches in the gut environment and competing for nutrients.Citation25 GF mice exhibit abnormalities in many functional systems, including intestinal immunity and gut barrier function, in comparison to their counterparts raised in SPF facilities.Citation24 Since their immune system does not encounter “normal commensals” at birth, resulting in abnormal immune and gut barrier function, it is possible that “healthy” microbiomes may cause a severe immune reaction in adult GF mice acutely. In support of this notion, GF Il10-/- mice developed more severe cecal inflammation when they were transferred to an SPF facility at 20–24 weeks of age versus 3 weeks of age.Citation12 In addition, GF ICR (Institute of Cancer Research) mice (outbred stock) colonized with fecal bacteria developed significant inflammation in the cecum and colon that resolved by 2 weeks post colonization.Citation26 We observed that all GF recipient mice developed severe acute cecal inflammation independent of the recipient genotype and the type of microbiome used to colonize the mice (IBD-associated or not). Together, data from our and others’ studies suggest that GF mice are prone to acute and severe cecal inflammation in response to microbial colonization especially if the mice were colonized at adult stages, regardless of the donor microbiome composition.
Though our studies employ H. bilis as a trigger of IBD in the SPF donor mice, our mono-colonization experiments suggested that H. bilis on its own is not likely the cause of IBD in GF recipient mice; H. bilis alone only induced mild to moderate inflammation in the cecum that quickly resolved. Our results are similar to those reported by Dieleman et al. in that mono-colonization with another Helicobacter, H. hepaticus, does not cause IBD in GF Il10-/- mice at 9 and 12 weeks following colonization.Citation27 In addition, H. bilis was reported to promote more severe gut inflammation in wild type mice (C3H/HeN and C57BL/6) colonized with Altered Schaedler Flora compared to H. bilis mono-colonized mice when they were treated with a low dose of dextran sodium sulfate, a chemical trigger of IBD.Citation28 Our data and these previous studies demonstrate that other enteric bacteria are needed to induce severe intestinal inflammation upon Helicobacter infection.Citation4,Citation12,Citation29
In future studies, we want to identify causative microbiomes responsible for IBD and CAC. Because Smad3-/- mice develop severe inflammation in cecum and colon upon colonization with microbiota associated with H. bilis-induced IBD, requiring euthanasia, long-term studies needed for understanding the role of the microbiome in CAC development are not possible using fecal transfer of mouse IBD microbiomes in these mice. Previous studies have reported that an enteric virus, murine norovirus, can replace some of the functions of commensal bacteria in proper immune cell development in the intestines of GF mice.Citation30 Interestingly, mono-colonization with H. bilis, a commensal bacterium in immune-competent mice, prior to fecal transfer from mice with IBD significantly reduced inflammation in both cecum and colon compared to mice that were not mono-colonized prior to fecal transfer in both Smad3+/- and Smad3-/- recipients. Currently, we do not know whether other commensal bacteria can elicit the same suppressive effects on gut inflammation induced by microbiome-transfer into GF mice. Recent studies suggest that another Helicobacter species, Helicobacter hepaticus can induce regulatory T cells in wild type mice in a c-MAF-dependent manner while it induces expansion of Th17 cells in Il10-/- mice,Citation31 suggesting that interactions between microbiota and a host genetic susceptibility promote gut inflammation. Further studies will be needed to determine whether H. bilis mono-colonization alters cancer development at later time points and whether H. bilis mono-colonization suppresses inflammation by changing H. bilis-specific immune responses or if immune responses are generally more attenuated due to previous exposure to this commensal bacterium.
The severity of intestinal inflammation in murine models of IBD may be dependent on genetic background: Il10-/- mice develop more severe inflammation when they are on a 129/SvEv background compared to a C57/BL6 background;Citation32 Mdr1a-/- mice on C57BL/6 background do not develop IBD upon H. bilis infection while these mice on FVB/N background do;Citation8 and various inbred strains of mice show differential degrees of gut inflammation upon treatment with dextran sodium sulfate.Citation7 Because we were surprised at the severity of IBD following fecal transfer from mice with IBD observed in GF Smad3+/- and Smad3+/+ recipients that are not susceptible to bacterially induced IBD in SPF conditions, we tested whether the 129 background is more susceptible to IBD development upon fecal transfer into GF mice. Fecal transfer into three GF wild type mice of different genetic backgrounds demonstrated that C57BL/6 wild type mice showed similar responses to those of 129 wild type mice. In contrast, GF Swiss Webster mice developed only mild inflammation regardless of the IBD status of the donors, suggesting that genetics play a significant role in IBD development induced by fecal transfer from mice with IBD and that acute cecal inflammation may occur in GF mice of various genetic backgrounds in response to fecal transfer. However, it is possible that our observation is due to reduced colonization in Swiss Webster mice compared to the other two strains rather than their ability to better control the inflammatory process in response to the same microbiomes. Future studies with larger numbers of donors and recipients will be needed to address the underlying cause of the differential responses to the same microbiome in mice with different genetic backgrounds.
In summary, we have shown that GF Smad3-deficient mice may be useful to determine the potential of a particular microbiome to induce IBD. Although the small sample sizes employed in our studies limit our ability to determine causative bacteria associated with IBD, consistent phenotypes across multiple studies using 2–3 different pooled microbiome samples suggest that IBD-associated microbiomes cause more severe gut inflammation in GF recipient mice. We demonstrated that H. bilis does not cause IBD without the presence of other enteric bacteria in GF Smad3-/- and Smad3+/- mice. Though mono-colonization with H. bilis prior to fecal transfer can protect these mice from developing severe gut inflammation upon fecal transfer from mice with IBD, more studies are needed to determine how this is being achieved and whether this impacts CAC development at later time points. We also showed that even healthy microbiomes can elicit acute cecal inflammation in GF mice, highlighting the need for detailed model validation before using new GF mice to study IBD.
Materials and methods
Mice and diet
Smad3-/- (129-Smad3tmCitation1Par/J) mice were originally obtained from The Jackson Laboratory and maintained in our facility by both homozygous and heterozygous breeding.Citation5,Citation19 Smad3-/- mice were rederived GF in the Gnotobiotic Animal Core (GNAC) facility at University of Washington by caesarian section. The first rederivation resulted in three male and two female Smad3-/- mice. Although SPF Smad3-/- mice are fertile and produce viable pups, the GF Smad3-/- mice produced a single non-viable pup after ~3.5 months of attempted breeding in several different combinations (as duos and trios). Thus, we attempted a second rederivation to generate GF Smad3+/- mice. Heterozygous breedings produced progeny with the expected Mendelian ratios. We also attempted a breeding scheme where homozygous male or female mice were paired with heterozygous mice for ~2 months. These pairs or trios did not produce any progeny. Because of unsuccessful breeding using germ-free Smad3-/- mice, heterozygous mice were used as breeders for our studies. Genotypes were confirmed by PCR of DNA extracted from ear punch or tail. A breeding colony was maintained in a flexible film isolator in open-top cages with Enrich-n’Pure bedding (The Andersons), fed an autoclaved rodent chow (Laboratory autoclavable rodent diet 5010 or 5021, Lab Diet) ad lib and provided autoclaved water.Citation33 GF status was confirmed as previously described.Citation33 GF 129 wild type mice were obtained from the cross-breeding of GF Smad3+/- mice. GF C57BL/6 and Swiss Webster mice were obtained from the GNAC. C57BL6 mice were originally obtained from Dr. Mazmanian (Caltech, CA) and Swiss-Webster mice were from Taconic. For fecal transfer studies, mice were transferred into ISOcage P cages (Tecniplast) prior to colonization, and the GF status of individual animals was determined by 16S rRNA PCR.Citation33 Handling of mice in ISOcage P cages was performed in a biosafety cabinet following the previously established protocol.Citation33 In all experiments, recipient mice were distributed among groups to evenly match age and sex. In most studies, recipient mice were 5–15 weeks of age at the start of the study except one study (Smad3-/- mice transferred with healthy vs. IBD fecal samples) where 25-week-old mice were used.
Microbiome transfer studies
GF mice were colonized with fecal slurriesCitation33 prepared from feces of SPF Smad3-/- mice with or without IBD. IBD in the donor SPF mice was induced by H. bilis inoculation,Citation19 and fecal samples were collected during either the acute inflammatory phase or the remission phase (based on clinical symptoms of diarrhea and weight loss that were previously validated in other disease progression studies in this modelCitation4,Citation19). Control fecal samples (healthy microbiomes, no IBD) were collected from the same SPF Smad3-/- mice before H. bilis infection or from random colony SPF Smad3-/- mice (Helicobacter-free). All donor fecal samples were stored at −80°C until the fecal slurry preparation on the day of the experiment. For each experiment, a pooled sample from multiple donors was prepared and used to colonize multiple recipients. IBD-associated microbiomes were collected from two cohorts. Both cohorts were composed of 5-male and 5-female SPF Smad3-/- mice, and their fecal samples were collected at ~1 wk (6–8 days) following H. bilis infection (Suppl. Table 1). For H. bilis mono-colonization or pre-colonization experiments, H. bilis was cultured and inoculated as previously reportedCitation19 and infection status was confirmed by PCRCitation34 with DNA extracted from fecal samples at 1–2 wks post infection and/or at the end of the study. For H. bilis pre-colonization experiments, fecal transfer was carried out 3 weeks following H. bilis inoculation. Colonized GF mice were monitored daily for IBD development by observing diarrhea and body condition for predetermined periods of time post colonization (mostly ~1 week and in some cases, up to 8 weeks).
Tissue collection and histopathology
At the end of each study, mice were euthanized by CO2 asphyxiation, blood was collected by heart puncture to obtain sera, and cecum, colon, and mesenteric lymph nodes were fixed in 10% phosphate-buffered formalin and processed for histological analysis (IDEXX Laboratories). Tissue sections were stained with hematoxylin and eosin (H&E) and graded by a board-certified veterinary pathologist blinded to treatments, following a published protocol.Citation35
Statistical analyses
All statistical analyses were performed using Prism software (version 5, Graph Pad). Student’s t-test was employed for comparisons between two groups, and one-way ANOVA followed by a post-hoc test (Bonferroni) was used for comparisons among three groups. Non-parametric Mann–Whitney test was performed on data from combined studies (Suppl. Figure 2). Statistical significance was defined as p-values less than 0.05.
Abbreviations and acronyms
| IBD | = | inflammatory bowel disease |
| GF | = | germ-free |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (1.2 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Jones-Hall YL, Kozik A, Nakatsu C. Ablation of tumor necrosis factor is associated with decreased inflammation and alterations of the microbiota in a mouse model of inflammatory bowel disease. PloS One. 2015;10:e0119441. doi:10.1371/journal.pone.0119441.
- Yang Y, Jobin C. Microbial imbalance and intestinal pathologies: connections and contributions. Dis Model Mech. 2014;7:1131–1142. doi:10.1242/dmm.016428.
- Jiminez JA, Uwiera TC, Douglas Inglis G, Uwiera RR. Animal models to study acute and chronic intestinal inflammation in mammals. Gut Pathog. 2015;7:29. doi:10.1186/s13099-015-0076-y.
- Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006;66:828–838. doi:10.1158/0008-5472.CAN-05-2448.
- Seamons A, Treuting PM, Brabb T, Maggio-Price L. Characterization of dextran sodium sulfate-induced inflammation and colonic tumorigenesis in Smad3(-/-) mice with dysregulated TGFbeta. PloS one. 2013;8:e79182. doi:10.1371/journal.pone.0079182.
- Chow J, Tang H, Mazmanian SK. Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr Opin Immunol. 2011;23:473–480. doi:10.1016/j.coi.2011.07.010.
- Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–551. doi:10.1152/ajpgi.1998.274.3.G544.
- Staley EM, Schoeb TR, Lorenz RG. Differential susceptibility of P-glycoprotein deficient mice to colitis induction by environmental insults. Inflamm Bowel Dis. 2009;15:684–696. doi:10.1002/ibd.20824.
- Dianda L, Hanby AM, Wright NA, Sebesteny A, Hayday AC, Owen MJ. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol. 1997;150:91–97.
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274.
- Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744.
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231.
- Khan, KJ, Ullman T, Ford A, Abreu M, Abadir A, Marshall J, Talley N, Moayyedi P. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011;106:661–673. doi:10.1038/ajg.2011.72.
- Suskind DL, Brittnacher MJ, Wahbeh G, Shaffer ML, Hayden HS, Qin X, Singh N, Damman CJ, Hager KR, Nielson H, et al. Fecal microbial transplant effect on clinical outcomes and fecal microbiome in active Crohn‘s disease. Inflamm Bowel Dis. 2015;21:556–563. doi:10.1097/MIB.0000000000000307.
- Lepage P, Häsler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Doré J, Raedler A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011;141:227–236. doi:10.1053/j.gastro.2011.04.011.
- Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9:599–608. doi:10.1038/nrgastro.2012.152.
- Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. . The gut microbiome modulates colon tumorigenesis. MBio. 2013;4:e00692–00613. doi:10.1128/mBio.00692-13.
- Maggio-Price L, Shows D, Waggie K, Burich A, Zeng W, Escobar S, Morrissey P, Viney JL. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a-/-) mice. The American Journal of Pathology. 2002;160:739–751. doi:10.1016/S0002-9440(10)64894-8.
- Meeker S, Seamons A, Paik J, Treuting PM, Brabb T, Grady WM, Maggio-Price L. Increased dietary vitamin D suppresses MAPK signaling, colitis, and colon cancer. Cancer Res. 2014;74:4398–4408. doi:10.1158/0008-5472.CAN-13-2820.
- Daniel SG, Ball CL, Besselsen DG, Doetschman T, Hurwitz BL. Functional changes in the gut microbiome contribute to transforming growth factor beta-deficient colon cancer. mSystems. 2017:2. doi:10.1128/mSystems.00065-17.
- Troncone E, Marafini I, Stolfi C, Monteleone G. Transforming growth factor-beta1/Smad7 in intestinal immunity, inflammation, and cancer. Front Immunol. 2018;9:1407. doi:10.3389/fimmu.2018.01407.
- Fowler SA, Ananthakrishnan AN, Gardet A, Stevens CR, Korzenik JR, Sands BE, Daly MJ, Xavier RJ, Yajnik V. SMAD3 gene variant is a risk factor for recurrent surgery in patients with Crohn‘s disease. J Crohns Colitis. 2014;8:845–851. doi:10.1016/j.crohns.2014.01.003.
- Yamashita A, Inamine T, Suzuki S, Fukuda S, Unoike M, Kawafuchi Y, Machida H, Isomoto H, Nakao K, Tsukamoto K. Genetic variants of SMAD2/3/4/7 are associated with susceptibility to ulcerative colitis in a Japanese genetic background. Immunol Lett. 2019;207:64–72. doi:10.1016/j.imlet.2019.01.007.
- Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi:10.1016/j.smim.2006.10.002.
- Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–519. doi:10.1016/S0140-6736(03)12489-0.
- Ogawa H, Fukushima K, Sasaki I, Matsuno S. Identification of genes involved in mucosal defense and inflammation associated with normal enteric bacteria. Am J Physiol Gastrointest Liver Physiol. 2000;279:G492–499. doi:10.1152/ajpgi.2000.279.3.G492.
- Dieleman LA, Arends A, Tonkonogy SL, Goerres MS, Craft DW, Grenther W, Sellon RK, Balish E, Sartor RB. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect Immun. 2000;68:5107–5113.
- Gomes-Neto JC, Kittana H, Mantz S, Segura Munoz RR, Schmaltz RJ, Bindels LB, Clarke J, Hostetter JM, Benson AK, Walter J, et al. A gut pathobiont synergizes with the microbiota to instigate inflammatory disease marked by immunoreactivity against other symbionts but not itself. Sci Rep. 2017;7:17707. doi:10.1038/s41598-017-18014-5.
- Jergens AE, Wilson-Welder JH, Dorn A, Henderson A, Liu Z, Evans RB, Hostetter J, Wannemuehler MJ. Helicobacter bilis triggers persistent immune reactivity to antigens derived from the commensal bacteria in gnotobiotic C3H/HeN mice. Gut. 2007;56:934–940. doi:10.1136/gut.2006.099242.
- Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature. 2014;516:94–98. doi:10.1038/nature13960.
- Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, Littman DR. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature. 2018;554:373–377. doi:10.1038/nature25500.
- Berg DJ, Davidson N, Kühn R, Müller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi:10.1172/JCI118861.
- Paik J, Pershutkina O, Meeker S, Yi JJ, Dowling S, Hsu C, Hajjar AM, Maggio-Price L, Beck DAC. Potential for using a hermetically-sealed, positive-pressured isocage system for studies involving germ-free mice outside a flexible-film isolator. Gut Microbes. 2015;6:255–265. doi:10.1080/19490976.2015.1064576.
- Maggio-Price L, Bielefeldt-Ohmann H, Treuting P, Iritani BM, Zeng W, Nicks A, Tsang M, Shows D, Morrissey P, Viney JL. Dual infection with helicobacter bilis and helicobacter hepaticus in p-glycoprotein-deficient mdr1a-/- mice results in colitis that progresses to dysplasia. Am J Pathol. 2005;166:1793–1806. doi:10.1016/S0002-9440(10)62489-3.
- Torrence AE, Brabb T, Viney JL, Bielefeldt-Ohmann H, Treuting P, Seamons A, Drivdahl R, Zeng W, Maggio-Price L. Serum biomarkers in a mouse model of bacterial-induced inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:480–490. doi:10.1002/ibd.20347.