
ABSTRACT
The cellular degradative pathway of autophagy prevents unrestrained inflammatory signaling by removing intracellular microbes, damaged organelles, and other factors that trigger immune reactions. Consistent with this function, a common variant of the autophagy gene ATG16L1 is associated with susceptibility to inflammatory bowel disease (IBD), a disorder characterized by a chronic immune reaction directed against the gut microbiota. We recently contributed to our understanding of the link between autophagy and inflammatory signaling in the intestine by demonstrating that autophagy proteins including ATG16L1 are necessary in the epithelium to prevent a spontaneous type I interferon response to the gut microbiota. Enhanced innate immunity that occurs upon autophagy inhibition is protective in mouse models of infection by an enteric bacterial pathogen and acute epithelial injury. Although avoiding excess immune reactions towards the microbiota is necessary to prevent IBD, these observations indicate that autophagy hampers productive immunity at the intestinal epithelial barrier in certain contexts. Here, we discuss how this counterintuitive consequence of autophagy inhibition can be reconciled with the established beneficial role of the pathway.
Introduction
A single layer of epithelial cells lies between the rest of our body and a multitude of infectious agents in the gut, including members of the microbiota that establish long-term colonization and pathogens capable of causing life-threatening disease.Citation1 In addition to serving as a physical barrier, the intestinal epithelium relays signals back-and-forth between these microbes in the lumen and immune cells on the other side of the fence to achieve an immune response of appropriate magnitude and quality. This bidirectional communication is essential for the differentiation of immune cell subsets that promote host defense. However, maladaptive immune reactions directed towards the gut microbiota or other infectious agents can give rise to Crohn’s disease and ulcerative colitis, types of inflammatory bowel disease (IBD).Citation2 Therefore, innate immune signaling in the gut must be controlled, which poses a challenge at the level of the epithelial barrier. Microbes are a constant source of microbe-associated molecular patterns (MAMPs) that are ligands for pattern recognition receptors (PRRs), many of which are expressed in epithelial cells to allow a swift innate immune response to invasive infections.Citation1 What are the cell biological pathways that prevent spontaneous activation of these PRRs in the absence of a bona fide threat?
Autophagy is a cell-intrinsic mechanism by which cytosolic material is degraded. Through the coordinated action of numerous autophagy (ATG) proteins, cellular contents are engulfed in a double membrane vesicle and subsequent fusion with the lysosome leads to the breakdown and recycling of vesicle contents. In addition to countering nutritional stress, autophagy plays a critical role in cellular, tissue, and organismal homeostasis by removing protein aggregates, depolarized mitochondria, internalized microbes, and other unwanted material.Citation3 We and others have demonstrated that ATG proteins support the intestinal epithelial barrier, likely through this cellular homeostatic function of autophagy. Inhibiting ATG proteins in the intestinal epithelium leads to accumulation of intracellular bacteria in enterocytes (absorptive epithelial cells), dysfunction of Paneth cells and goblet cells (secretory epithelial lineages), and overall susceptibility to cell death.Citation4–Citation13 Additionally, ATG protein function in the intestinal epithelium is critical for preventing inflammation following a range of enteric infections.Citation14–Citation17 These observations may explain the genetic association between a common polymorphism in the autophagy gene ATG16L1 (T300A allele) and susceptibility to Crohn’s disease.Citation3,Citation18
To better understand the link between autophagy and mucosal immunity in the gut, we previously generated mice harboring a germ line gene-trap mutation that leads to decreased Atg16L1 expression and reduced autophagy.Citation4 These Atg16L1 hypomorph (Atg16L1HM) mice develop intestinal abnormalities upon infection with murine norovirus (MNV), which include lesions observed in Crohn’s disease patients such as morphological and functional defects in the antimicrobial Paneth cells that reside within the small intestinal epithelium.Citation4,Citation17 In a subsequent study, we showed that the immune response to the virus triggers necroptosis (programmed necrosis) in the ATG16L1-deficient epithelium, which was associated with defective autophagy-mediated removal of damaged mitochondria (mitophagy).Citation5 Of note, we found that MNV typically establishes an asymptomatic infection and can even be a beneficial virus in other animal models of disease.Citation19 Thus, Atg16L1 mutation leads to defects in the intestinal epithelium in response to an otherwise beneficial virus, indicating that autophagy is important for tolerating infectious threats in the gut.
Atg16L1HM mice display exacerbated disease in response to other inflammatory triggers such as Staphylococcus aureus infection and allogeneic bone marrow transplantation.Citation20,Citation21 However, Atg16L1HM mice are resistant to uropathogenic E. coli (UPEC) in a model of urinary tract infection and display decreased vertical transmission of Zika virus,Citation22,Citation23 demonstrating that reducing ATG16L1 function does not unilaterally compromise the immune system. Also, we found that Atg16L1HM mice are extraordinarily protected from disease following oral inoculation with Citrobacter rodentium.Citation24 This finding was unexpected because C. rodentium is a model of intestinal infection by enteropathogenic E. coli (EPEC), representing a striking contrast from our results with MNV. This protection from C. rodentium was associated with decreased colonic inflammation (which occurs due to binding of C. rodentium to the epithelial surface) and a striking reduction in bacterial burden. While CD4+ T cells and neutrophils were not required for enhanced C. rodentium resistance, we found monocytes play a crucial role in the mechanismof protection. Atg16L1 deficiency was associated with a hyperimmune transcriptional response characterized by increased expression of interferon-stimulated genes (ISGs) in the colon that precedes infection and becomes exacerbated in the presence of C. rodentium. These observations led us to question if ATG16L1 and autophagy were regulating inflammatory responses at the gut mucosal surface.
Autophagy proteins suppress protective type I interferon signaling
In our recent manuscript, we make progress in elucidating the mechanism by which Atg16L1 mutation enhances antimicrobial immunity in the gut.Citation25 Using cell type-specific knockout mice, we demonstrate that loss of ATG16L1 in the epithelium is sufficient to confer resistance to C. rodentium infection. Thus, ATG16L1 function in the intestinal epithelium prevents an adverse immune response to an enteric commensal-like virus (i.e., MNV), while suppressing a protective response to an enteric bacterial pathogen (i.e., C. rodentium). Autophagy can inhibit immune responses by mediating the degradation of PRRs (or their downstream signaling intermediates), MAMPs, and mitochondria that release reactive oxygen species (ROS). The two most established examples are autophagy-mediated inhibition of IL-1β production downstream of the NLRP3 inflammasome and IFN-I signaling following nucleic acid sensing.Citation3 We found that removing the IFN-I receptor (IFNAR1) in Atg16L1HM mice, but not NLRP3, abrogated the resistance to C. rodentium conferred by Atg16L1 mutation. Resistance to C. rodentium upon autophagy inhibition, therefore, is mediated by an enhanced IFN-I response, reflected by the increased ISG expression in the colon. These results are reminiscent of findings by other groups showing that certain viruses promote mitophagy to inhibit IFN-I signaling.Citation26–Citation28 Our results suggest that dampening the local IFN-I response through mitophagy may also increase susceptibility to extracellular bacterial pathogens of the gut.
We found that Atg16L1HM mice display an increase in MAVS, a mitochondria-associated signaling molecule involved in viral RNA sensing, which suggests that reduced ATG16L1 function leads to inhibition of mitophagy and associated proteins in the intestine. We further found that mice deficient in another ATG protein (ATG4B) were also resistant to C. rodentium and displayed a similar increase in ISG expression. Our observations are consistent with a model in which the enhanced resistance conferred by Atg16L1 mutation reflect suppression of IFN-I signaling through mitophagy. One point of caution, however, is that ATG proteins function in cellular processes other than autophagy that contribute to immunity.Citation29 An increase in MAVS can be secondary to other defects downstream of ATG inhibition, and thus, we cannot rule-out the contribution of autophagy-independent pathways. As discussed below, identifying the exact upstream trigger of IFN-I signaling may clarify this issue.
ATG16L1 inhibits spontaneous nucleic acid sensing in the presence of the gut microbiota
Chronic low levels of IFN-I are notoriously difficult to detect in situ, but we could infer its presence based on the genetic evidence of its activity (the observation that Atg16L1HM Ifnar1–/ – double mutant mice lose resistance to C. rodentium), the presence of phospho-STAT1 (p-STAT1) in the epithelium, and local ISG expression. p-STAT1 and ISG expression were observed in Atg16L1HM mice prior to C. rodentium infection. Germ-free Atg16L1HM mice did not display these markers of IFN-I activity, indicating that intestinal microbes were necessary for spontaneous signaling. We also found that resistance to C. rodentium infection required MAVS and STING, which mediate IFN-I production in response to cytosolic RNA and DNA, respectively. Hence, ATG16L1 suppresses a protective nucleic acid sensing response that is dependent on the gut microbiota ().
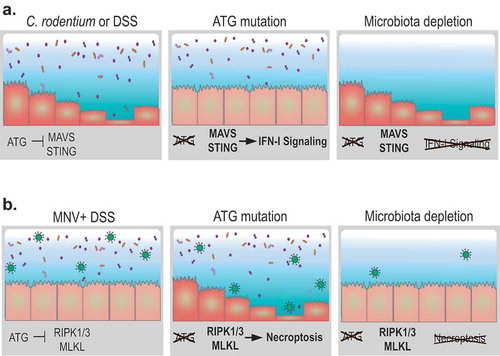
Figure 1. Autophagy in the intestinal epithelium can be harmful or protective during injury depending on the type of perturbation and the presence of the microbiota. (A) Autophagy (ATG) proteins prevent spontaneous triggering of the type I interferon (IFN-I) pathway by restraining the activity of cytosolic nucleic acid sensors MAVS and STING. In the absence of ATG proteins in the intestinal epithelium, enhanced IFN-I signaling increases resistance to infection by the bacterial pathogen Citrobacter rodentium or chemical injury by dextran sodium sulfate (DSS) treatment. These consequences of ATG protein inhibition are absent in germ-free mice lacking intestinal microbes. (B) Murine norovirus (MNV) infection is typically innocuous because ATG proteins promote epithelial homeostasis and viability. In the absence of ATG proteins, MNV infection triggers structural and functional abnormalities in Paneth cells. DSS treatment in this setting exacerbates epithelial defects by promoting necroptosis signaling through the RIPK1-RIPK3-MLKL complex, leading to loss of Paneth cells and mortality. Depletion of the microbiota reduces MNV infection and restores resistance to intestinal injury.
A simple model would be one in which direct sensing of bacterial nucleic acid in the cytosol of intestinal epithelial cells occurs upon inhibition of autophagy. MAVS and STING can act in parallel or regulate each other’s activity in this scenario. An alternative model would involve other MAMPs derived from the microbiota that indirectly trigger the release of RNA and DNA from damaged mitochondria or neighboring cells. While we were revising our manuscript, an important study was published demonstrating that ATG16L1 inhibits IFN-I production downstream of LPS recognition by TLR4 through autophagy-mediated degradation of TRIF.Citation30 Such a role for the TLR4-TRIF pathway is not mutually exclusive from the role of MAVS and STING that we identified, and may also contribute to the enhanced IFN-I response we observe in the Atg16L1 mutant conditions. An important future direction would be to identify the specific microbes in the gut that are responsible for the spontaneous IFN-I response observed in Atg16L1HM mice.
Atg16L1 mutation enhances monocyte competence for wound repair
In the above experiments, autophagy inhibition increased epithelial proliferation in a manner dependent on the microbiota and IFN-I signaling. Macrophages responding to enhanced IFN-I production increase epithelial turnover and improve wound repair,Citation31 which led us to examine this cell type in our model. Following local activation of the PRR NOD2, monocytes are recruited to the gut during C. rodentium infection and differentiate into macrophages to contribute to the ongoing immune response.Citation32 In our previous study, we found that removing NOD2 on the Atg16L1HM background reversed the protection independently of CD4+ T cells.Citation24 In our more recent manuscript, we found that removing the monocyte chemokine receptor CCR2 in Atg16L1HM mice led to a similar loss in resistance towards C. rodentium. RNAseq analysis, combined with additional functional analyses, indicated that monocytes isolated from the colon of C. rodentium-infected Atg16L1HM mice display increased expression of cell cycle genes and markers of activation and phagocytosis compared with their counterparts harvested from wild-type control mice. These isolated monocytes also displayed a gene expression signature indicative of increased fatty acid β-oxidation associated with wound-healing macrophages. These various properties of the recruited monocytes and macrophages are consistent with enhanced resolution of the infection and would repair.
In these experiments, we did not separate specialized myeloid subsets to be examined individually, such as the recently characterized Ym1+Ly6c+ monocytes that plays a role in resolving colitis.Citation33 This limitation in our approach likely explains why we observe gene expression-level changes that are not representative of a single monocyte or macrophage population. Also, the exact relationship between IFN-I signaling and monocytes is unclear. Because ATG16L1-deficiency in the intestinal epithelium is sufficient to confer resistance to C. rodentium, and monocytes are not recruited until the infection is underway, we favor a model where higher basal IFN-I responses creates a local environment that is permissive for enhanced monocyte and macrophage function. However, monocytes and macrophages are not only targets of IFN-I, they can be the source of IFN-I during C. rodentium infection that can enhance resistance when amplified.Citation34 Also, in addition to directly producing effector molecules, IL-1β production following caspase-11 inflammasome activation in monocytes induces IL-22 production by innate lymphoid cells (ILCs) during C. rodentium infection.Citation35 We also observed a role for caspase-1 and caspase-11 in C. rodentium resistance in Atg16L1HM mice (in our study, we genetically deleted the caspases together). When our results are taken in the context of this literature, a complex feedback loop involving multiple cell types and cytokines appears to explain how an increase in IFN-I above the normal amount results in protection from an enteric bacterial pathogen.
Although we have not completely addressed these important questions surrounding how cell–cell interactions are rewired when ATG16L1 is inhibited, we were able to demonstrate that the enhanced IFN-I signaling and monocyte response improve intestinal injury. Atg16L1HM mice displayed increased survival in the dextran sodium sulfate (DSS) model of chemical injury to the gut, and this improved response was nullified upon inhibition of MAVS, STING, or CCR2. Germ-free Atg16L1HM mice were not protected from DSS, again implicating factors derived from the microbiota. This enhanced intestinal injury response likely contributes to the protection from C. rodentium that occurs upon Atg16L1 mutation.
Outlook: ATG16L1 function in intestinal disease susceptibility and treatment
The ATG16L1 T300A risk allele is present in 40–50% of individuals with 15% homozygosity in certain populations. It is unclear why this variant was maintained in the human population. The threonine to alanine coding change increases the cleavage of ATG16L1 by caspase-3 and leads to reduced autophagy in the presence of TNFα, internalized bacteria, or metabolic stress.Citation5,Citation36–Citation39 To determine the impact of this variant on C. rodentium infection, we used Atg16L1T316A knock-in (KI) mice that harbor the equivalent of the human T300A mutation in the endogenous Atg16L1 locus. We found that treating homozygous KI mice with PAC1, a chemical activator of caspase-3, reduced amounts of full-length ATG16L1 and increased ISG expression in the colon. Pre-treatment of Atg16L1T316A KI mice with PAC1 also boosted protection against C. rodentium infection. PAC1 treatment was necessary for this protection, and did not improve resistance to infection in wild-type mice. Similar, but mechanistically distinct, observations have been made in models of urinary tract infection and Salmonella dissemination.Citation23,Citation30 Given that diarrheal pathogens such as pathogenic E. coli remain a major contributor to early childhood mortality, a provocative implication of our observation and these other findings from the field is that ATG16L1T300A was maintained in the population because its presence leads to improved defense against certain types of bacterial pathogens, even if autophagy is generally beneficial in other circumstances.
In the mouse model we describe above, we artificially induced ATG16L1T300A cleavage with a chemical. What factors activate caspase-3 to process ATG16L1 in humans? As mentioned above, the presence of another infectious agent or malnutrition can lead to processing of ATG16L1T300A in vitro. Whether infection or nutritional stress have similar effects at the whole organism level remains to be determined. For modern humans, short-term treatment of NSAIDs induces caspase-3 activation.Citation40,Citation41 This observation is relevant to the link between this allele and disease because NSAIDs are associated with IBD flares in mice and humans.Citation42,Citation43 Also, cigarette smoke is associated with Paneth cell defects in Atg16L1T316A KI mice and Crohn’s disease patients harboring the ATG16L1T300A allele.Citation44 Thus, multiple environmental triggers may induce ATG16L1 processing in the epithelium, and depending on the surrounding circumstances, the outcome can be beneficial (infection by an enteric bacterial pathogen) or detrimental (sustained inflammation directed against the gut microbiota).
With this dichotomy in mind, it is interesting to note that MNV infection induces disease outcomes in the same Atg16L1T316A mice and other autophagy mutant models we discuss in this addendum. Notably, DSS treatment leads to severe histopathology in Atg16L1 mutant mice in a manner dependent on MNV infection, which is reversed by antibiotics.Citation17 There are several potential explanations for this opposing consequence of ATG protein dysfunction. First, our findings could reflect differences in the consequence of disrupting autophagy in the small intestine versus colon. MNV infection primarily leads to defects in Paneth cells, which are found in the small intestine, and ATG16L1T300A is predominantly associated with small intestinal Crohn’s disease in humans.Citation18 In contrast, the highest burden of C. rodentium during the course of infection is observed in the cecum and colon. Another difference in the two infection models is that the MNV strain we have been examining establishes a persistent infection while C. rodentium is transient. It stands to reason that an enhanced immune response may be beneficial during a self-resolving infection, but that chronic stimulation of the mucosal immune system may be undesirable. Consistent with this idea, a non-persistent strain of MNV fails to induce disease in Atg16L1HM mice.Citation17 The reason depletion of the bacterial microbiota has opposite results during DSS treatment of MNV-infected versus naïve Atg16L1 mutant mice is likely due to the impact of bacteria on the viral life cycle; intestinal bacteria promote MNV infection by facilitating attachment to host cells and dampening immune signaling.Citation19,Citation45,Citation46
It will be important to determine whether the enhanced IFN-I signaling that protects against C. rodentium contributes to MNV-induced disease in Atg16L1 mutant mice. A recent study showed that IL-22, which promotes epithelial wound repair and is essential for defense against C. rodentium, mediates necroptosis in mice harboring a loss of ATG16L1 in the intestinal epithelium.Citation12 The authors also found a spontaneous IFN-I signature that was dependent on STING. It is possible that excess IFN-I may have a pathologic role in our virally-triggered IBD model despite its protective effect during bacterial infection. Although counterintuitive because IFN-I is typically associated with inhibiting viral replication, we found that IFNAR1 is necessary for promoting the beneficial effects of persistent MNV infection with a relatively minor role in controlling viral burden.Citation19 Recent findings help explain this observation. In contrast to MNV strains that establish a self-limiting infection controlled by IFN-I, persistent MNV infection can be detected in the epithelium and is sensitive to type III IFN (IFN-λ) instead.Citation47–Citation49 Interestingly, the gut microbiota facilitates MNV persistence by inhibiting IFN-λ activity,Citation45 reinforcing the idea that IFN-I and IFN-λ have significantly divergent regulation and function in the gut despite activating similar transcription factors.
Clinical trials for IBD are examining the therapeutic efficacy of blocking JAK/STAT molecules, which function downstream of multiple cytokine receptors including IFNAR1. It is tempting to speculate that one mechanism by which this class of drugs can ameliorate intestinal disease is through inhibiting IFN-I signaling and other pathways that are problematic when the epithelium is autophagy-deficient. However, great care must be taken before considering therapy that targets autophagy, IFN-I signaling, or any of the players mentioned here. IBD patients may be vulnerable to enteric infections by inflammatory microbes such as adherent invasive E. coli (AIEC). It is unknown whether individuals who are positive for these pathogens should be treated differently from other patients during flares.Citation50 Segregating IBD patients based on their autophagy activity or their genetic status together with screening for enteric infections may help determine the conditions under which therapeutically targeting signaling pathways is safe and effective.
Supplemental Material
Download PDF (1.7 MB)Acknowledgments
K.C. is supported by US National Institute of Health (NIH) grants R01 HL123340, R01 DK093668, R01 DK103788, R01 AI121244; Faculty Scholar grant from the Howard Hughes Medical Institute, Rainin Foundation Innovator Award, Crohn’s & Colitis Senior Research Award, Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases Award, and Pfizer. P.K.M. is supported by the NIH grant F31 DK111139. K.C. has consulted for PureTech Health and AbbVie Inc. and is an inventor on U.S. patent application 62/608.404.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Ramanan D, Cadwell K. Intrinsic defense mechanisms of the intestinal epithelium. Cell Host Microbe. 2016;19:434–441. doi:10.1016/j.chom.2016.03.003.
- Wong SY, Cadwell K. There was collusion: microbes in inflammatory bowel disease. PLoS Pathog. 2018;14:e1007215. doi:10.1371/journal.ppat.1007215.
- Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and Inflammation. Annu Rev Immunol. 2018;36:73–101. doi:10.1146/annurev-immunol-042617-053253.
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi:10.1038/nature07416.
- Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, Dewan MZ, Lieberman SR, Lazrak A, Marinis JM, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. 2017;214:3687–3705. doi:10.1084/jem.20170558.
- Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, Guan J-L, Saitoh T, Akira S, Seglen PO, et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. Embo J. 2013;32:3130–3144. doi:10.1038/emboj.2013.233.
- Benjamin JL, Sumpter R Jr., Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. 2013;13:723–734. doi:10.1016/j.chom.2013.05.004.
- Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, Leal T, Winter SE, Xavier RJ, Hooper LV. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017;357:1047–1052. doi:10.1126/science.aal4677.
- Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–276. doi:10.1038/nature12599.
- Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, Bhattacharyya J, Flak MB, Shih DQ, Fuhler GM, et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J Exp Med. 2017;214:401–422. doi:10.1084/jem.20160791.
- Diamanti MA, Gupta J, Bennecke M, De Oliveira T, Ramakrishnan M, Braczynski AK, Richter B, Beli P, Hu Y, Saleh M, et al. IKKalpha controls ATG16L1 degradation to prevent ER stress during inflammation. J Exp Med. 2017;214:423–437. doi:10.1084/jem.20161867.
- Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW, Tschurtschenthaler M, Saveljeva S, Bhattacharyya J, Häsler R, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med. 2018;215:2868–2886. doi:10.1084/jem.20171029.
- Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, Billiar TR, Lotze MT, Boone DL, Chang EB. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125:1098–1110. doi:10.1172/JCI80323.
- Burger E, Araujo A, Lopez-Yglesias A, Rajala MW, Geng L, Levine B, Hooper LV, Burstein E, Yarovinsky F. Loss of paneth cell autophagy causes acute susceptibility to toxoplasma gondii-mediated inflammation. Cell Host Microbe. 2018;23:177–190 e4. doi:10.1016/j.chom.2018.01.001.
- Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145:1347–1357. doi:10.1053/j.gastro.2013.08.035.
- Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe. 2018;23:191–202 e4. doi:10.1016/j.chom.2017.12.017.
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi:10.1016/j.cell.2010.05.009.
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Philip Schumm L, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi:10.1038/nature11582.
- Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature. 2014;516:94–98.
- Hubbard-Lucey VM, Shono Y, Maurer K, West ML, Singer NV, Ziegler CG, Lezcano C, Motta A, Schmid K, Levi S, et al. Autophagy gene atg16l1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity. 2014;41:579–591. doi:10.1016/j.immuni.2014.09.011.
- Maurer K, Reyes-Robles T, Alonzo F 3rd, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to staphylococcus aureus alpha-toxin. Cell Host Microbe. 2015;17:429–440. doi:10.1016/j.chom.2015.03.001.
- Cao B, Parnell LA, Diamond MS, Mysorekar IU. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J Exp Med. 2017;214:2303–2313. doi:10.1084/jem.20170957.
- Wang C, Mendonsa GR, Symington JW, Zhang Q, Cadwell K, Virgin HW, Mysorekar IU. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc Natl Acad Sci U S A. 2012;109:11008–11013. doi:10.1073/pnas.1203952109.
- Marchiando AM, Ramanan D, Ding Y, Gomez LE, Hubbard-Lucey VM, Maurer K, Wang C, Ziel J, van Rooijen N, Nuñez G, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe. 2013;14:216–224. doi:10.1016/j.chom.2013.07.013.
- Martin PK, Marchiando A, Xu R, Rudensky E, Yeung F, Schuster SL, Kernbauer E, Cadwell K. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol. 2018;3:1131–1141. doi:10.1038/s41564-018-0229-0.
- Ding B, Zhang L, Li Z, Zhong Y, Tang Q, Qin Y, Chen M. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21:538–47 e4. doi:10.1016/j.chom.2017.03.004.
- Kuo SM, Chen CJ, Chang SC, Liu TJ, Chen YH, Huang SY, Shih SR. Inhibition of avian influenza a virus replication in human cells by host restriction factor TUFM is correlated with autophagy. mBio. 2017;88(3). pii: e00481-17. doi:10.1128/mBio.00481-17.
- Xia M, Gonzalez P, Li C, Meng G, Jiang A, Wang H, Gao Q, Debatin K-M, Beltinger C, Wei J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J Virol. 2014;88:5152–5164. doi:10.1128/JVI.03851-13.
- Cadwell K, Debnath J. Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J Cell Biol. 2018;217:813–822. doi:10.1083/jcb.201706157.
- Samie M, Lim J, Verschueren E, Baughman JM, Peng I, Wong A, Kwon Y, Senbabaoglu Y, Hackney JA, Keir M, et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol. 2018;19:246–254. doi:10.1038/s41590-017-0042-6.
- Sun L, Miyoshi H, Origanti S, Nice TJ, Barger AC, Manieri NA, Fogel L, French A, Piwnica-Worms D, Piwnica-Worms H, et al. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe. 2015;17:85–97. doi:10.1016/j.chom.2014.11.004.
- Kim YG, Kamada N, Shaw MH, Warner N, Chen GY, Franchi L, Núñez G. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity. 2011;34:769–780. doi:10.1016/j.immuni.2011.04.013.
- Ikeda N, Asano K, Kikuchi K, Uchida Y, Ikegami H, Takagi R, Yotsumoto S, Shibuya T, Makino-Okamura C, Fukuyama H, et al. Emergence of immunoregulatory Ym1(+)Ly6C(hi) monocytes during recovery phase of tissue injury. Sci Immunol. 2018;33(28). pii: eaat0207. doi:10.1126/sciimmunol.aat0207.
- Normand S, Waldschmitt N, Neerincx A, Martinez-Torres RJ, Chauvin C, Couturier-Maillard A, Boulard O, Cobret L, Awad F, Huot L, et al. Proteasomal degradation of NOD2 by NLRP12 in monocytes promotes bacterial tolerance and colonization by enteropathogens. Nat Commun. 2018;9:5338. doi:10.1038/s41467-018-07750-5.
- Seo SU, Kuffa P, Kitamoto S, Nagao-Kitamoto H, Rousseau J, Kim YG, Núñez G, Kamada N. Intestinal macrophages arising from CCR2(+) monocytes control pathogen infection by activating innate lymphoid cells. Nat Commun. 2015;6:8010. doi:10.1038/ncomms9010.
- Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–+.
- Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu T-C, Heath RJ, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111:7741–7746. doi:10.1073/pnas.1407001111.
- Gao P, Liu H, Huang H, Zhang Q, Strober W, Zhang F. The inflammatory bowel disease-associated autophagy gene Atg16L1T300A acts as a dominant negative variant in mice. J Immunol. 2017;198:2457–2467. doi:10.4049/jimmunol.1502652.
- Zhang H, Zheng L, McGovern DP, Hamill AM, Ichikawa R, Kanazawa Y, Luu J, Kumagai K, Cilluffo M, Fukata M, et al. Myeloid ATG16L1 facilitates host-bacteria interactions in maintaining intestinal homeostasis. J Immunol. 2017;198:2133–2146. doi:10.4049/jimmunol.1601293.
- Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004;11:1009–1016. doi:10.1038/sj.cdd.4401436.
- Utzeri E, Usai P. Role of non-steroidal anti-inflammatory drugs on intestinal permeability and nonalcoholic fatty liver disease. World J Gastroentero. 2017;23:3954–3963. doi:10.3748/wjg.v23.i22.3954.
- Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. 2014;41:311–324. doi:10.1016/j.immuni.2014.06.015.
- Feagins LA, Cryer BL. Do non-steroidal anti-inflammatory drugs cause exacerbations of inflammatory bowel disease? Dig Dis Sci. 2010;55:226–232. doi:10.1007/s10620-009-1042-7.
- Liu TC, Kern JT, VanDussen KL, Xiong S, Kaiko GE, Wilen CB, Rajala MW, Caruso R, Holtzman MJ, Gao F, et al. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. J Clin Invest. 2018;128:5110–5122. doi:10.1172/JCI120453.
- Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science. 2015;347:266–269. doi:10.1126/science.1258025.
- Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinjé J, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 2014;346:755–759. doi:10.1126/science.1255826.
- Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347:269–273. doi:10.1126/science.1258100.
- Baldridge MT, Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, Virgin HW. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J Virol. 2017;91(7). pii: e02079-16. doi:10.1128/JVI.02079-16.
- Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL Jr., McAllaster MR, Balce DR, Feehley T, Brestoff JR, et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science. 2018;360:204–208. doi:10.1126/science.aar3799.
- Axelrad JE, Joelson A, Green PHR, Lawlor G, Lichtiger S, Cadwell K, Lebwohl B. Enteric infections are common in patients with flares of inflammatory bowel disease. Am J Gastroenterol. 2018;113:1530–1539. doi:10.1038/s41395-018-0120-x.