
ABSTRACT
Patients presenting with Inflammatory bowel disease have been shown to exhibit an altered microbiome in both Crohn’s disease and Ulcerative colitis. This shift in the microbial content led us to question whether several of these microbes are important in inflammatory processes present in these diseases and more specifically whether lipopolysaccharides from the gram-negative cell wall differentially stimulates resident cells. We, therefore, investigated the possible contribution of five major species of gram-negative bacteria found to be altered in presence during disease progression and evaluate their pathogenicity through LPS. We demonstrated that LPS from these different species had individual capacities to induce NF-κB and pro-inflammatory IL-8 production from HEK-TLR4 cells in a TLR4 dependent manner. Additional work using human intestinal colonic epithelial cell monolayers (Caco-2) demonstrated that the cells responded to the serotype specific LPS in a distinct manner, inducing many inflammatory mediators such as TNF-α and IL-10 in significantly altered proportions. Furthermore, the permeability of Caco-2 monolayers, as a test for their ability to alter intestinal permeability, was also differentially altered by the serotype specific LPS modulating trans-epithelial electrical resistance, small molecule movement, and tight junction integrity. Our results suggest that specific species of bacteria may be potentiating the pathogenesis of IBD and chronic inflammatory diseases through their serotype specific LPS responses.
Introduction
Inflammatory bowel disease (IBD) constitutes of two major phenotypes of gastrointestinal diseases, Ulcerative colitis (UC) and Crohn’s disease (CD). Both diseases contain an inflammatory component, which ultimately results in impaired nutrient absorption, increased cellular recruitment, and chronic inflammation.Citation1 Usually, onset in adolescence, and persistent throughout the life of the affected individual, the symptoms of IBD are not only limited to the GI tract but also can result in systemic complications such as fever, weight loss, delayed puberty and growth, to name but a few.Citation2–Citation4 Furthermore, peripheral inflammation can subsequently result in a high incidence of arthritis associated with the disease, issues within the cardiovascular system, and lymphatic remodeling.Citation5–Citation8 CD is characterized by a chronic and transmural inflammation that can be noted along the entire length of the GI tract, from mouth to anus, but primarily occurs within the colon and small intestine. The associated inflammation can promote the development of lesions at any point along the tract effecting all layers of the intestine, producing strictures and fistulae.Citation9 UC, primarily affects the superficial layer of the colon, with a continual chronic inflammation within the large intestine, a feature that persists, disrupts the barrier function of the intestinal lamina and can result in systemic infection.Citation10
There have been several different studies demonstrating the correlation between environmental and genetic factors that could induce dysfunction of the epithelial barrier. Concurrence of the disease in identical twins demonstrated the possible implications to genetics with an incidence of 50–55% being observed.Citation11,Citation12 These alterations can promote an inappropriate immunological response through the recognition of commensal gut microbiota or through an increased exposure to opportunistic pathogens such as Clostridium difficile co-infections, which in turn leads to an increase in-hospital mortalities.Citation13 However, the exact mechanisms of action are still to be discovered and delineated.
The intestinal epithelial barrier physically separates the microbially dense intestinal lumen from the relatively sterile environment of the sub-mucosa, which includes structures such as the lamina propria, lymphatic initial-lacteals, and resident immune cells.Citation14 Comprised of four major cell types, the intestinal epithelial barrier is designed to aid in nutrient absorption, immune defence, and structural integrity. Absorptive enterocytes have a digestive function and can also respond to infection through secretion of many antimicrobial peptides;Citation15 goblet cells secrete mucus; enteroendocrine cells secrete regulatory hormones;Citation16 and Paneth cells secrete antimicrobial peptides into the crypts of the small intestine.Citation17 Through the intestinal barrier, there are several important receptors that monitor the environment of the intestinal lumen and are present in these cells. These pattern-recognition receptors (PRRs) are expressed by many cell types in different ratios, based on the function of that cell, and can recognize a wide variety of pathogen associated molecular patterns (PAMPs), such as viral or bacterial components.Citation18,Citation19 PRRs include many super-families such as: Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs) and C-type lectin receptors (CLRs). RLRs and CLRs are primarily involved in the recognition of many viral and fungal components, respectively, whilst TLRs can recognize almost all microbial constituents including fungi, virus, bacteria, and protozoa. Usually expressed on the cell surface of cells (TLR1, TLR2, TLR4, TLR5, TLR6) or within the endosomes (TLR3, TLR7, TLR8, TLR9) TLRs are disproportionally distributed along the intestine and can be polarised in nature being expressed on the apical or basolateral side of the expressing cell.Citation20 This physiological characteristic allows for an initial physical distinction between ligands based on location, for example, endosomal TLRs such as TLR3 are present within the cytoplasm bound to the early endosome and recognize intracellular dsRNA during the replication of viruses.Citation21
TLR4, the earliest described Toll-like receptor, recognizes the gram-negative bacterial cell wall component lipopolysaccharide (LPS). TLR4, CD14, and co-receptor MD-2 interaction can trigger both a MyD88-dependent and MyD88-independent pathway leading to the nuclear translocation of NF-kB or IRF3, respectively, with subsequent production of inflammatory cytokines and other mediators.Citation22 Innate immune signaling such as this is important for the recruitment of immune cells to the site of inflammation promoting reparative mechanisms, however, many view the processes as a double-edged sword regulating wound healing and organ fibrosis as well as regenerative myogenesis.Citation23,Citation24 Due to the close proximity of the colonic microbiota to the intestinal epithelium, regulation of TLR4 signaling must be tightly controlled. Therefore, the expression is very low in various cell types of the intestine, including the epithelium where decreased expression correlates to protection against bacterial LPS, a phenomenon later attributed to be regulated by immune signals, primarily IFN-α.Citation25,Citation26 TLR4 expression was found to be greatly increased in IBD patient biopsies suggesting a role for TLR4-mediated signaling in the disease-associated inflammation.Citation25 Albeit a finding that is still poorly understood mechanistically, a window into the regulation of intestinal tight junction permeability has been described whereby enterocyte membrane expression and localization of TLR4 and CD14 are important factors.Citation27 Whilst the mucosa and superficial layers of the intestine provide a substantial barrier between the cells and microbiome, a more complete understanding of TLR4 with disease function, especially in promoting inflammation, is still required. An intimate relationship between TLRs, the microbiota and intestinal immunity has been shown to influence GI pathology.Citation28 However, due to an abundant microbiome, the exact mechanisms of action are still to be answered.
The main activator of TLR4, LPS, is well known in every laboratory environment for use as a stimulant or co-stimulant in a variety of in vitro and in vivo applications.Citation29,Citation30 However, the use of the term LPS seems indifferent to most, as a common misconception falls to many that LPS is identical between all bacteria. This is not the case. Due to many factors, species-specific, or “serotypes”, of LPS exist that are described as being distinctly different from one another in structure. Within a single species of bacteria, there can be thousands of different serotypes so it is perhaps unwise to assume that intra-species LPS would be the same, let alone unique.
Throughout the progression of IBD, in both CD and UC patients, the microbial content of the gut is substantially altered.Citation31 Whether this is a contributing factor of disease or a consequence of dysfunction is still debated. We, therefore, wanted to investigate whether the altered microbiota was influencing TLR4 activated inflammation through LPS produced by species of bacteria that dominated the newly established microbiome. By analyzing the activity of five species of gram-negative bacterial LPS through the use of TLR4 transfected HEK293 cells, we demonstrate an altered NF-κB and pro-inflammatory responses to species-specific LPS. Furthermore, utilizing a model the Caco-2 model of intestinal epithelial permeability, we showed distinctly individual impacts of specific LPS on monolayer integrity, which presents a novel concept to serotype specific epithelial injury and maintenance of epithelial barrier function in the “leaky gut”.
To our knowledge, this is the first study to suggest that LPS serotypes from individual species of gram-negative bacteria promote intestinal epithelial dysfunction through altered TLR4 signaling and provide novel insights into disease pathogenesis within a disease concurrent with an altered microbiome and opens up new avenues and targets for therapeutic intervention.
Results
Stimulation of HEK-TLR4 cells in vitro
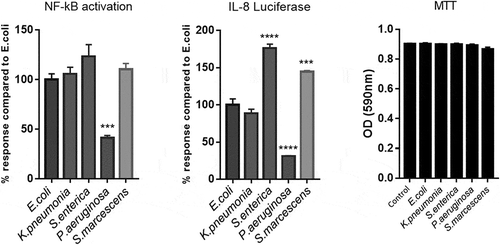
Cell stimulation by LPS is primarily achieved through the activation of TLR4.Citation32 Receptor dimerization then leads to activation of a series of downstream mediators, culminating in the phosphorylation and nuclear translocation of NF-κB and production of inflammatory cytokines.Citation33 Results illustrated in , demonstrate a significant difference in activation of the NF-κB SEAP reporter and IL-8 Luciferin production between the serotypes. We observe that LPS from P.aeurginosa, induced substantially less NF-κB and IL-8 production (P = .0006 and P = .0001, respectively). However, both S.enterica and S.marcescens induced a significantly higher amount of IL-8 production (P = .0001 and P = .0004, respectively). Cell viability was unaffected at this concentration. For the purpose of the assays values were normalized to E.coli in order to discern differences against a stimulation that lacks a positive control given that all LPS serotypes tested will stimulate TLR4.
Figure 1. Activation of HEK293-TLR4 cells in vitro with species-specific LPS.
LPS serotypes differentially affect pro- and anti-inflammatory cytokine production in Caco-2 monolayers
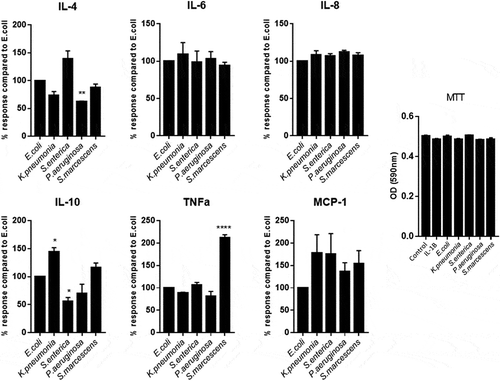
During chronic inflammation of the intestine, an imbalance of pro- and anti-inflammatory mediators is observed as well as increased bacterial translocation through proposed membrane permeability increase.Citation34 We, therefore, wanted to determine whether direct contact of epithelial cell monolayers to LPS would differentially alter inflammatory cytokine production from the cells thereby suggesting which bacterium could be more significantly contributing to disease through modulation of intestinal permeability. Stimulation of Caco-2 monolayers for 24 h with LPS demonstrated a significantly unique pattern of pro- and anti-inflammatory cytokine production between each serotype. Data was normalized to the induction of cytokines by the common lab strain of E.coli (O127:B8) LPS and demonstrated that S.macescens induced TNF-α at a significantly higher proportion than any other serotype (P < .0001) whilst K.pnemoniae induced substantially more IL-10 than its counterparts (P < .0001) (). It is also interesting to note that the induction of IL-6 and IL-8 were very stable between species, and it was primarily anti-inflammatory mediators, IL-4 and IL-10, that were differentially expressed suggesting an imbalance in pro- vs anti-inflammatory cytokines as a driver of inflammation. All serotypes induced all cytokines presented significantly over control ().
Figure 2. Comparing the response of Caco-2 cells to induce inflammatory cytokines dependent on species specific LPS.
Serotype specific toxicity
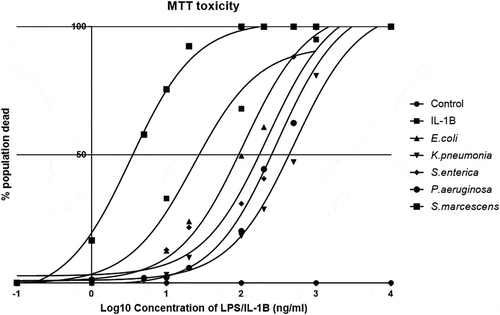
Many thousands of species of bacteria can colonize a human without any adverse pathogenic effect. However, some only need a few thousand cells to produce a pathogenic effect such as salmonella-induced diarrhea or the fatal build-up of Shiga toxin.Citation35,Citation36 We, therefore, hypothesized that some bacterial species that are usually low in prevalence may be repressed by the host in order to prevent a fatal cellular interaction. So far, we demonstrated that the inflammatory response to the LPS serotypes was different, could the toxicity to a cell of interest be unique? In order to determine cell viability, the MTT assay was utilized and demonstrated a stark difference in potency of individual serotypes. Using increasing doses of LPS upon cells allowed us to determine the LD50 of the serotypes over a 24-h stimulation (). We found that the LD50 of S.marcescens (LD50-3.33ng/ml) was more than 30 times greater than observed from each of the other species with Caco-2 cells being most resistant to K.pneumoniae (508.16ng/ml), S.enterica (222.33ng/ml), and P.aeruginosa (297.17ng/ml). Interestingly, the common strain of laboratory E.coli (O127:B8) was mild in toxicity (106.17ng/ml) and responded in the most controlled manner compounding its use within a laboratory environment. The importance of this data will have to be correlated to the bacterial load that corresponds to this concentration of LPS used; however, it does demonstrate that whilst “the same” LPS from specific species can activate cells in a manner that promotes cell death. Further analysis into the pathways associated with activation is ongoing.
Figure 3. Species-specific LPS toxicity in Caco-2 monolayers.
Serotype specific alterations in trans-epithelial flux of FITC in caco-2
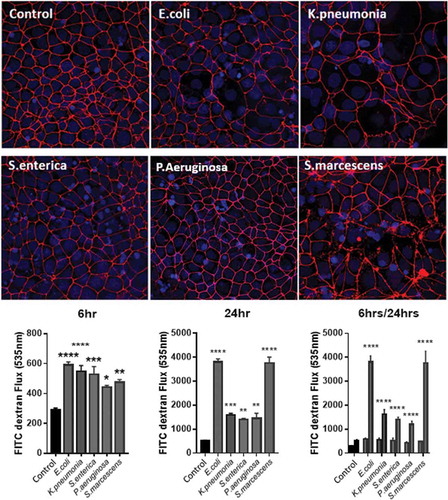
Epithelial barrier function has been described earlier as being important in maintaining the divide between the microbe-rich lumen and the intestinal lamina. During inflammatory insults to the gut, bacterial translocation to the mesenteric lymph node and even to the periphery is a notable occurrence.Citation37,Citation38 With implications such as fever and sepsis to consider, the so-called “leaky gut” is a method to cause both localized and widespread inflammation.Citation39 We hypothesized that specific LPS could increase intestinal permeability and therefore allow many different bacteria to spread, not only those who produce the potent immune activator. In order to assess this, we stimulated Caco-2 monolayers seeded on transwell membrane inserts and measured their permeability with a FITC-dextran flux assay. Tight-junction formation was assessed using ZO-1 staining and was quantified prior-to and post-treatment.
As illustrated in , all LPS stimulations induced a significant increase in monolayer permeability evident by an increased FITC-dextran flux across the membrane. Interestingly, this effect could be seen at 6 h where there was a significance of LPS induced flux present in all treatments in relation to control flux. Additionally, ZO-1 staining was already notably disturbed (). Measuring flux after 24 h of LPS stimulation demonstrated a larger disruption of monolayer stability evident by severe ZO-1 disruption and increased permeability. It is at this time point however that species specific differences become evident whereby S.marcescens and E.coli have the greatest effect on monolayer permeability, at more than double the flux of the other stimuli. The flux correlates to severely ablated ZO-1 leading to complete monolayer disruption. Conversely, P.aeruginosa, K.pnemoniae, and S.enterica have significantly less increases in permeability at 24 h appearing to have their largest response within 6 h.
Figure 4. LPS stimulation reduces ZO-1 tight junctions in Caco-2 cells in a species-specific manner altering monolayer permeability.
Trans-epithelial permeability is modulated by species specific LPS in a TLR4 dependent manner
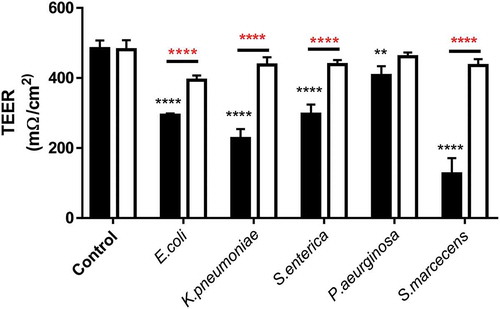
In addition to observing changes in transcellular permeability of Caco-2 monolayers, the trans-epithelial electrical resistance (TEER) was measured as a marker of tight-junction disruption and permeability changes. Caco-2 monolayers exhibited a significant drop in TEER after 24-h treatment with all LPS tested (). Mirroring the FITC flux and ZO-1 staining S.marcescens had the most pronounced effect on reducing TEER and P.aeruginosa the least. Referring back to our previous observations that each LPS had a differential ability to promote TLR4 activation, Caco-2 monolayers were co-treated with the specific TLR4 blocking small compound C34 (10ng/ml) which significantly attenuated LPS-induced TEER reduction, furthermore outlining the importance of TLR4 as a driver of this phenomenon ().
Figure 5. Trans-epithelial electrical resistance reduction in response to LPS is modulated by TLR4 activation in Caco-2 monolayers.
Discussion
Taken for granted, the microbiome of the gastrointestinal tract provides the host with a symbiotic relationship of untold proportion. Co-evolved to exist in harmony, the relentless onslaught of bacterial antigens, held at bay by the mucosa and epithelial barrier. Any disruption in this key barrier can lead to many complications through extravasation of commensal microbes, leading to systemic infections such as sepsis amongst other pathologies.Citation40 During IBD, a poignant shift in the microbial content of the gut is evident.Citation41 Whether the shift in bacteria is a causal effect of the disease or just a surreptitious consequence is still debated, yet the contribution of these newly dominated bacteria could lead to a plethora of issues. Our hypothesis alluded to the fact that many of these bacteria, being gram negative, could have drastically different efficacies in activating TLR4, altering epithelial barrier function, activity of resident cells, and perpetuating disease through localized chronic inflammation.
The integrity of the intestinal epithelial monolayer is sustained by tight cell-cell junctions, preventing the uncontrolled flux of material as well as structurally stabilizing the tissue.Citation42 Many bacterial pathogens including endotoxins can target these tight-junctions in order to relax the structure, allowing for successful transmigration.Citation43 We report that treatment with LPS from different species differentially regulate tight-junctions within Caco-2 monolayers effectively increasing permeability to varying extents. This translated into S.marcescens LPS being identified to vastly down-regulate ZO-1 and increase epithelial barrier permeability within Caco-2 monolayer trans-well cultures suggesting the role for the aforementioned bacteria’s-LPS in modulating tight junctions in host cell barriers. A previous report has demonstrated the dangers presented by the hospital acquired infection of living S.marcescens highlighting its adherence and invasive capabilities on human epithelial cells. Our work supports their findings and presents S.marcescens-derived LPS as a causal target.Citation44 Indeed, we present information suggesting it is the LPS derived from this species that is directly toxic to cells in culture, affecting both pro-inflammatory cytokine production and epithelial permeability and toxicity. These results lead us to ponder that shifts in the microbiota during infection, disease, and trauma, may alter the inflammatory balance, cell death, and intestinal permeability through compositional changes in the resident LPS population.
Acting as tissue resident watchmen, intestinal epithelial cells are important in their role to activate an early innate immune response against foreign, and potentially dangerous particulates through the production of pro-inflammatory and chemotactic cytokines. TNF-α modulates many factors within the epithelium including mucus secretion, paracellular flow via tight junction control and is a master regulator of intracellular signaling cascades involved in cell survival, apoptosis, and proliferation, positioning it at the forefront of many inflammatory diseases and cancer studies.Citation45–Citation48 TNF-α is therefore notably important in the maintenance and healing of the intestinal mucosa but is also important in guiding cell recruitment and function with anti-TNF trials of Adalimumab (Humira™) evidencing TNF-α as a target for mucosal healing in Crohn’s patients.Citation49 We have demonstrated that LPS from specific species of bacteria can differentially modulate the production of TNF-α through individual profiles of activation of an NF-kB promoter. What effect this could have on a global level within a tissue is still to be investigated; however, we could hypothesize that intestinal permeability, cellular recruitment, mucus coverage, and wound healing could all be drastically affected.
As all of the LPS used in this study are derived from common hospital acquired infection pathogens that can colonize the GI tract, the translation to real-world implications could suggest involvement for these bacteria to modulate intestinal permeability, the cause of a myriad of inflammatory conditions including IBD. We would suggest that opportunistic infections such as these take holds in niches created through antibiotic ablation of the resident microbiome allowing growth in population, a phenomenon already observed in humans.Citation50 It is then possible that through bacterial lysis, either via complement induced opsonization or another cause, vast amounts of bacterially derived endotoxins are released into the luminal space potentially inducing a large TLR4 driven inflammatory response and increasing intestinal permeability. The disrupted epithelium can then facilitate bacterial translocation into the peritoneal cavity and the periphery or into intestinal mesenteric lymphatics to then be shuttled to the mesenteric lymph node where a larger immune response can be generated. A disruption of the mesenteric lymphatic system is a well-documented sign of intestinal inflammation in two animal models of human IBD.Citation51,Citation52 Whether the disruption in effective lymph flow is driven by the translocated bacteria is still to be validated. However, evidence points to it as being a key component.
In this study, using human colon-derived polarised epithelial (Caco-2) cell monolayers, we have demonstrated that LPS from five different species of gram-negative bacteria have significantly different activities upon cytokine release, toxicity, and permeability through tight-junction inhibition. We also demonstrate that different species of gram-negative bacteria produce LPS that are unique in their ability to activate an in vitro reporter HEK293 system, potentially linking direct involvement of TLR4 and altered affinity and/or downstream activation. Together, our data indicate that species specific LPS can distinctly modulate TLR4-induced inflammation and permeability of the gut epithelium, suggesting a contributing mechanism of action in inflammatory diseases, such as IBD, which could open the door to new biological therapeutic development.
Materials and methods
Lipopolysaccharide stocks
Lipopolysaccharides purchased from Sigma Aldrich were resuspended in the sterile condition in endotoxin free water and thoroughly mixed by vortex for 5 min. Samples were then aliquoted into glass vials at 10µg/ml as a stock solution for working concentrations and stored at 4°C until use ().
Table 1. Lipopolysaccharide stocks and species.
Immunofluorescent staining
Caco-2 monolayers were differentiated for 72 h in glass bottomed chamber slides. Samples were stimulated as described for 24 h with LPS or SHAM before fixation with 4% paraformaldehyde for 20 min. Once fixed cells were stained with ZO-1 (Invitrogen Cat No 33–9100).
Caco-2 cell maintenance
Human colonic epithelial cells (Caco-2) cells were seeded into 75 cm2 tissue culture flasks (T75) and maintained in Dulbecco’s Modified Eagle Medium (high glucose) (DMEM) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 1% L-glutamine (REF) 50 u/ml penicillin and streptomycin with 1% non-essential amino acids. Cells were kept at 37°C in 5% CO2, and media was changed every 2–3 d. Cells were passaged once they had reached 80% confluence by trypsin-EDTA dissociation and were split 1:8 every 7 d.
Trans-well assay
Cells were seeded onto 12 mm polycarbonate Costar tissue culture trans-well permeable insert filters with a 0.4 µm pore size (Corning Inc) and grown for 7 d until the monolayer and tight junctions had formed (confirmed by ZO-1 immuno-staining). Cells were stimulated for 24 h with 1ng/ml of serotype specific LPS before permeability increase was measured using FITC-dextran Flux assay. In brief, 200 µl of 10µM FITC-dextran was added to the apical side of the chamber and sample of supernatant from the basal side was collected every 15 min. Samples were assayed for FITC-Dextran presence using a fluorescent plate-reader (ex-485 em-535). A physical wound with a pipette tip was used as a positive control for permeability increase and monolayer disruption.
Trans-epithelial electrical resistance (TEER)
Cells were seeded onto 12 mm polycarbonate Costar tissue culture trans-well permeable insert filters with a 0.4 µm pore size (Corning Inc) and grown for 7 d until the monolayer and tight junctions had formed and TEER had reached 500mΩ. Cells were then stimulated for 24 h with 1ng/ml of serotype specific LPS, in the presence or absence of 10ng/ml C34 (Tocris, USA) Measurements of TEER were obtained at three different points per transwell using a Millicell ERS-2 Epithelial Volt-Ohm Meter (Millipore, USA) before and after stimulations. A physical wound with a pipette tip was used as a positive control for permeability increase and monolayer disruption.
HEK293-TLR4 cell line and maintenance
HEK-TLR4 NF-kB/IL-8 reporter sells were purchased from Invivogen and maintained as per manufacturer instruction. Cells were maintained in DMEM containing 10% FCS, 10µg/ml Blasticidin, 100µg/ml Zeocin, and 100µg/ml Normocin. Cells were incubated at 37°C with 5% CO2 and passaged as needed when reaching 80–90% confluence. For use in assays, cells were stimulated without selection antibiotics and were trypsinized and seeded as indicated above.
Quantiblue assay
Supernatant removed from stimulated HEK-TLR4 dual reporter cells were analyzed for NF-κB activity through analysis of SEAP induction as per manufacturer guidelines. Simply, 10 µl of conditioned supernatant was incubated with 90 µl of QUANTIBlue substrate for 1 h at 37°C. Measurement of O.D. at 605nm quantified the induction of the reporter plasmid and was displayed in relation to control.
Luciferase assay
In a similar manner to the QuantiBlue reporter assay, 10 µl of conditioned supernatant was added to 90 µl of pre-warmed (37°C) QuantiLuc substrate solution and mixed briefly on a horizontal plate shaker protected from light. Within 15 min, fluorescence was quantified using a Victor X5 fluorescent plate reader and presented in relation to control wells.
Cell viability assay (MTT)
Stimulated cells were cleared of supernatant and 50 µl of MTT (5mg/ml in Complete media) was added for 4 h at 37°C. Media was then aspirated and intra-cellular formazan crystals, formed by the substrate conversion, were lysed in 50 µl DMSO. The plate was then read at 590 nm in order to determine cell viability.
Multiplex analysis
Samples for multiplex analysis were collected, filtered, and sent to Eve Technologies (Calgary, Canada) for quantification.
Additional information
Funding
References
- Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517.
- Rosen DS. Pubertal growth and sexual maturation for adolescents with chronic illness or disability. Pediatrician. 1991;18:105–120.
- Brain CE, Savage MO. Growth and puberty in chronic inflammatory bowel disease. Baillieres Clin Gastroenterol. 1994;8:83–100.
- Rigaud D, Angel LA, Cerf M, Carduner MJ, Melchior JC, Sautier C, René E, Apfelbaum M, Mignon M. Mechanisms of decreased food intake during weight loss in adult Crohn’s disease patients without obvious malabsorption. Am J Clin Nutr. 1994;60(5):775–781. doi:10.1093/ajcn/60.5.775.
- Andersen NN, Jess T. Risk of cardiovascular disease in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2014;5(3):359–365. doi:10.4291/wjgp.v5.i3.359.
- von der Weid PY, Rehal S, Ferraz JG. Role of the lymphatic system in the pathogenesis of Crohn’s disease. Curr Opin Gastroenterol. 2011;27(4):335–341. doi:10.1097/MOG.0b013e3283476e8f.
- Ge Y, Li Y, Gong J, Zhu W. Mesenteric organ lymphatics and inflammatory bowel disease. Ann Anat. 2018;218:199–204. doi:10.1016/j.aanat.2018.03.006.
- Li Y, Zhu W, Zuo L, Shen B. The Role of the Mesentery in Crohn’s Disease: the Contributions of Nerves, Vessels, Lymphatics, and Fat to the Pathogenesis and Disease Course. Inflamm Bowel Dis. 2016;22(6):1483–1495. doi:10.1097/MIB.0000000000000791.
- Lissner D, Sonnenberg E, Siegmund B. [Inflammatory bowel disease: cardinal signs and their diagnostics]. Dtsch Med Wochenschr. 2018;113(13):937–944. doi:10.1055/a-0612-3564.
- Adams SM, Bornemann PH. Ulcerative colitis. Am Fam Physician. 2013;87:699–705.
- Halfvarson J, Bodin L, Tysk C, Lindberg E, Järnerot G. Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology. 2003;124:1767–1773.
- Orholm M, Binder V, Sørensen TIA, Rasmussen LP, Kyvik KO. Concordance of Inflammatory Bowel Disease among Danish Twins: results of a Nationwide Study. Scand J Gastroenterol. 2000;35:1075–1081.
- Rezapour M, Galoosian A, Liu B, Bhuket T, Wong RJ. Clostridium difficile co-infection in inflammatory bowel disease is associated with significantly increased in-hospital mortality. Eur J Gastroenterol Hepatol. 2018;30(9):1041–1046. doi:10.1097/MEG.0000000000001185.
- McKay DM, Perdue MH. Intestinal epithelial function: the case for immunophysiological regulation. Implications for disease (2). Dig Dis Sci. 1993;38:1735–1745.
- Gassler N, Newrzella D, Böhm C, Lyer S, Li L, Sorgenfrei O, van Laer L, Sido B, Mollenhauer J, Poustka A, et al. Molecular characterisation of non‐absorptive and absorptive enterocytes in human small intestine. Gut. 2006;55(8):1084–1089. doi:10.1136/gut.2005.073262.
- Latorre R, Sternini C, De Giorgio R, Greenwood-Van Meerveld B. Enteroendocrine Cells: A Review of Their Role In Brain-Gut Communication. Neurogastroenterol Motil. 2016;28(5):620–630. doi:10.1111/nmo.12754.
- Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9(5):356–368. doi:10.1038/nrmicro2546.
- Metzger RN, Krug AB, Eisenächer K. Enteric Virome Sensing—its Role in Intestinal Homeostasis and Immunity. Viruses. 2018;10(4):146. doi:10.3390/v10040146.
- Rhee SH. Basic and Translational Understandings of Microbial Recognition by Toll-Like Receptors in the Intestine. J Neurogastroenterol Motil. 2011;17(1):28–34. doi:10.5056/jnm.2011.17.1.28.
- Yu S, Gao N. Compartmentalizing Intestinal Epithelial Cell Toll-like Receptors for Immune Surveillance. Cell Mol Life Sci. 2015;72(17):3343–3353. doi:10.1007/s00018-015-1931-1.
- Chaturvedi A, Pierce SK. How location governs Toll like receptor signaling. Traffic. 2009;10(6):621–628. doi:10.1111/j.1600-0854.2009.00899.x.
- Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol. 2014;5:461. doi:10.3389/fimmu.2014.00461.
- Hindi SM, Kumar A. Toll-like receptor signalling in regenerative myogenesis: friend and foe. J Pathol. 2016;239(2):125–128. doi:10.1002/path.4714.
- Huebener P, Schwabe RF. Regulation of Wound Healing and Organ Fibrosis by Toll-like Receptors. Biochim Biophys Acta. 2013;1832(7). doi:10.1016/j.bbadis.2012.11.017.
- Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68(12):7010–7017. doi:10.1128/iai.68.12.7010-7017.2000.
- Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased Expression of Toll-Like Receptor-4 and MD-2 Correlates with Intestinal Epithelial Cell Protection Against Dysregulated Proinflammatory Gene Expression in Response to Bacterial Lipopolysaccharide. J Immunol. 2001;167(3):1609. doi:10.4049/jimmunol.167.3.1609.
- Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am J Pathol. 2013;182(2):375–387. doi:10.1016/j.ajpath.2012.10.014.
- Frosali S, Pagliari D, Gambassi G, Landolfi R, Pandolfi F, Cianci R. How the Intricate Interaction among Toll-Like Receptors, Microbiota, and Intestinal Immunity Can Influence Gastrointestinal Pathology. J Immunol Res. 2015;2015:489821. doi:10.1155/2015/489821.
- Ngkelo A, Lewkowich IP, Zhou P, Ledford JR, Page K. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3kinase signalling. J Inflamm. 2012;9(1):1. doi:10.1186/1476-9255-9-32.
- Tough DF, Sun S, Sprent J. T Cell Stimulation In Vivo by Lipopolysaccharide (LPS). J Exp Med. 1997;185(12):2089–2094. doi:10.1084/jem.185.12.2089.
- Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146(6):1489–1499. doi:10.1053/j.gastro.2014.02.009.
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. doi:10.1016/j.cyto.2008.01.006.
- Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23.
- Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53(1):1. doi:10.1136/gut.53.1.1.
- Terajima J, Iyoda S, Ohnishi M, Watanabe H. Shiga Toxin (Verotoxin)-Producing Escherichia coli in Japan. Microbiol Spectr. 2014;2(5). doi:10.1128/microbiolspec.EHEC-0011-2013.
- Zhang S, Kingsley RA, Santos RL, Andrews-Polymenis H, Raffatellu M, Figueiredo J, Nunes J, Tsolis RM, Adams LG, Bäumler AJ. Molecular Pathogenesis of Salmonella enterica Serotype Typhimurium-Induced Diarrhea. Infect Immun. 2003;71(1):1–12. doi:10.1128/iai.71.1.1-12.2003.
- Brien CL, Pavli P, Gordon DM, Allison GE. Detection of bacterial DNA in lymph nodes of Crohn’s disease patients using high throughput sequencing. Gut. 2014;63(10):1596. doi:10.1136/gutjnl-2013-305320.
- Berg RD. Bacterial translocation from the gastrointestinal tract. Adv Exp Med Biol. 1999;473:11–30.
- Michielan A, D’Incà R. Intestinal Permeability in Inflammatory Bowel Disease: pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflamm. 2015;2015:628157. doi:10.1155/2015/628157.
- Deitch EA. Gut-Origin sepsis; evolution of a concept. Surgeon. 2012;10(6):350–356. doi:10.1016/j.surge.2012.03.003.
- Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, D’Amato M, Bonfiglio F, McDonald D, Gonzalez A, et al. Dynamics of the human gut microbiome in Inflammatory Bowel Disease. Nat Microbiol. 2017;2:17004. doi:10.1038/nmicrobiol.2017.4.
- Günzel D, Yu ASL. Claudins and the Modulation of Tight Junction Permeability. Physiol Rev. 2013;93(2):525–569. doi:10.1152/physrev.00019.2012.
- van der Walle CF, Schmidt E. Chapter 9 - Modulation of the Intestinal Tight Junctions Using Bacterial Enterotoxins. In: Van Der Walle C, editor. Peptide and Protein Delivery. Boston (MA): Academic Press; 2011. p. 195–219.
- Ochieng JB, Boisen N, Lindsay B, Santiago A, Ouma C, Ombok M, Fields B, Stine OC, Nataro JP. Serratia marcescens is injurious to intestinal epithelial cells. Gut Microbes. 2014;5(6):729–736. doi:10.4161/19490976.2014.972223.
- Cheung CH, Moore EW, Prystowsky JB, Rege RV. TNFα stimulates mucus secretion in gallbladder epithelial cells. Gastroenterology. 1998;114:A1225.
- Mac CM. The movements of bones and joints; the mechanical structure of articulating cartilage. J Bone Joint Surg Br. 1951;33b(2:251–257.
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol Gastrointest Liver Physiol. 2004;286(3):G367–G376. doi:10.1152/ajpgi.00173.2003.
- Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361. doi:10.1038/nrc2628.
- Rutgeerts P, Van Assche G, Sandborn WJ, Wolf DC, Geboes K, Colombel J-F, Reinisch W, Kumar A, Lazar A, Camez A, et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: data from the EXTEND trial. Gastroenterology.2012;142(5):1102–1111.e2.doi:10.1053/j.gastro.2012.01.035.
- Modi SR, Collins JJ, Relman DA. Antibiotics and the gut microbiota. J Clin Invest. 2014;124(10):4212–4218. doi:10.1172/JCI72333.
- Rehal S, von der Weid PY. TNFDeltaARE Mice Display Abnormal Lymphatics and Develop Tertiary Lymphoid Organs in the Mesentery. Am J Pathol. 2017;187(4):798–807. doi:10.1016/j.ajpath.2016.12.007.
- Rehal S, Stephens M, Roizes S, Liao S, von der Weid P-Y. Acute small intestinal inflammation results in persistent lymphatic alterations. Am J Physiol Gastrointest Liver Physiol. 2017;314(3):G408–G417. doi:10.1152/ajpgi.00340.2017.