
ABSTRACT
Objective
The gut microbiome plays a key role in the development of acute graft-versus-host disease (GVHD) following allogeneic hematopoietic stem cell transplantation. Here we investigate the individual contribution of the pre- and post-transplant gut microbiome to acute GVHD using a well-studied mouse model.
Design
Wild-type mice were cohoused with IL-17RA–/ – mice, susceptible to hyperacute GVHD, either pre- or post-transplant alone or continuously (i.e., pre- and post-transplant). Fecal samples were collected from both WT and IL-17RA–/ – mice pre- and post-cohousing and post-transplant and the microbiome analyzed using metagenomic sequencing.
Results
Priming wild-type mice via cohousing pre-transplant only is insufficient to accelerate GVHD, however, accelerated disease is observed in WT mice cohoused post-transplant only. When mice are cohoused continuously, the effect of priming and exacerbation is additive, resulting in a greater acceleration of disease in WT mice beyond that seen with cohousing post-transplant only. Metagenomic analysis of the microbiome revealed pre-transplant cohousing is associated with the transfer of specific species within two as-yet-uncultured genera of the bacterial family Muribaculaceae; CAG-485 and CAG-873. Post-transplant, we observed GVHD-associated blooms of Enterobacteriaceae members Escherichia coli and Enterobacter hormaechei subsp. steigerwaltii, and hyperacute GVHD gut microbiome distinct from that associated with delayed-onset disease (>10 days post-transplant).
Conclusion
These results clarify the importance of the peri-transplant microbiome in the susceptibility to acute GVHD post-transplant and demonstrate the species-specific nature of this association.
Introduction
Graft-versus-host disease (GVHD) is a serious complication of hematopoietic stem cell transplantation occurring in 30–50% of cases.Citation1 The condition is characterized by T cell-mediated tissue damage to target organs, which include the skin, liver and gastrointestinal (GI) tract. In fact, in T-cell replete-unrelated stem cell transplants (SCT), the GI tract is involved in virtually all fatal cases of acute GVHD, with an overall survival at 2 years after the onset of stage 3–4 gut GVHD of 25%.Citation2 The gut microbiome was initially implicated as a key contributor to the development of intestinal GVHD following experiments demonstrating reduced disease severity in germ-free and antibiotic-treated mice.Citation3,Citation4 Conditioning regimens administered prior to transplant to ablate the immune system are necessary for successful engraftment, however, tissue injury resulting from these treatments permits translocation of microbes and microbial products (e.g., lipopolysaccharide) from the gastrointestinal lumen into circulation, triggering the release of inflammatory cytokines TNF, IL-1, and IL-6.Citation5–Citation8 This systemic circulation of danger/pathogen-associated molecular patterns and inflammatory cytokines results in the activation of host antigen-presenting cells, subsequent priming, differentiation and expansion of donor T-cells, and an overwhelming immune response resulting in destruction of target tissue.Citation9 Antibiotic treatment aimed at circumventing this process forms part of some conditioning regimens, however, concerns regarding antimicrobial resistance means the practice is not universal.Citation10 Disruption of the commensal microbiome, a system critical to human and animal health, may also impose further stress on the transplant recipient potentially impacting recovery.Citation11 Interestingly, we recently demonstrated a critical role for intestinal microbiota in influencing MHC class II presentation by intestinal epithelial cells leading to the initiation of GVHD.Citation12 As such, there exists a need to increase our understanding of the precise role of the gut microbiome in regulating the development of intestinal GVHD with a view to designing better targeted prophylactic or treatment strategies.
Investigation into the role of the gut microbiome in GVHD is complicated by observed microbial community fluctuations occurring both prior to and concomitant with the development of GVHD. At the time of disease, bacterial diversity within the gut decreases in both mice and humans in concert with large compositional shifts.Citation13 Members of the bacterial families Enterobacteriaceae, Lactobacillaceae and Enterococcaceae have all been shown to increase in abundance with disease development, often dominating the community as a whole.Citation13–Citation16 Decreased diversity in patients at the time of transplant, but prior to GVHD initiation, is associated with increased disease mortality,Citation17 in particular, decreased abundance of Blautia species has been linked to GVHD in humans.Citation18 Acute GVHD development is associated with increased abundance of Firmicutes in patients pre-transplant,Citation19,Citation20 as is an overall decreased level of diversity,Citation19 suggesting that the composition of the gut microbiota prior to transplant may also play a role. Understanding whether there is a defining period during which the gut microbiome is critical in the development of GVHD is vital to fully elucidating the nature of the relationship.
We recently observed an association between fecal microbiota and the development of hyper-acute GVHD in a mouse model of GVHD.Citation21 Following the description of a distinct microbial community within IL-17RA–/ – mice, a mouse genotype in which hyper-acute GVHD develops consistently post-transplant (survival post-transplant ≤10 days), it was discovered that accelerated disease could be induced in WT mice via cohousing with IL-17RA–/ – mice.Citation21 Analysis of the fecal microbiome before and after cohousing revealed transfer of community members primarily from IL-17RA–/ – to WT mice. However, as mice were cohoused throughout the experiment, it was unclear whether the pre-transplant cohousing was sufficient to induce accelerated disease or whether cohousing post-transplant also contributed to this observation. In order to ascertain whether there was an essential cohousing period, and by extension, an essential transfer of microorganisms, we undertook additional experiments using mice cohoused either pre- or post-transplant alone. These data demonstrate that sustained exposure to the disease-associated microbiota from IL-17 receptor-deficient mice is necessary for maximum induction of hyperacute GVHD in WT mice. Cohousing post-transplant only results in a rate of post-transplant disease progression intermediate between non-cohoused WT controls and continuously cohoused WT mice. Cohousing pre-transplant only is insufficient to significantly accelerate disease. The detrimental effect of pre-transplant priming is therefore only realized with post-transplant exposure to IL-17RA–/ – mice.
Results
Cohousing with a dysbiotic microbiome plays both a priming and exacerbating role in the development of acute GVHD in WT mice
In order to determine whether there was a critical period during which exposure to microbiota from IL-17 receptor-deficient mice was able to induce accelerated GVHD in WT mice, we compared the outcome of transplants involving mice cohoused continuously (I), pre-transplant only (II) or post-transplant only (III) (). Note that we previously published survival curves and a 16S rRNA-based analysis of Experiment I,Citation21 and in the present study used the DNA from the published experiment for metagenomic analysis. We hypothesized that pre-transplant cohousing primes the WT gut community for pathogen displacement and disease, and that post-transplant cohousing exacerbates disease progression by exposure to high pathogen load. Cohousing mice pre-transplant only (II) resulted in a reduced overall survival time, however, it was not significantly different to separately housed WT mice (median survival 33 versus 45 days, respectively; P = .3605) (). Cohousing mice post-transplant only (III) was more detrimental, with all WT mice succumbing to disease and a significantly accelerated rate of disease overall in comparison to separately housed WT mice (median survival 28 versus 45 days; P = .0015) (). However, the disease exacerbation in WT mice associated with post-transplant cohousing (III) was significantly delayed in comparison to that experienced by WT mice that were cohoused continuously (median survival 28 versus 9 days; P = .0057) (). While continuously cohoused WT mice (I) began to succumb to disease 4-days post-transplant, WT mice cohoused post-transplant but not pre-transplant survived for a minimum of 23 days. We suggest that this lag in disease initiation can be attributed to the absence of pre-transplant microbiome-dependent priming of the WT mice. Therefore, while the priming effect of cohousing pre-transplant is insufficient to accelerate disease in isolation, when combined with post-transplant exacerbation, primed mice are significantly more susceptible to accelerated GVHD than WT mice cohoused post-transplant only.
Figure 1. Continuous cohousing accelerates GVHD in WT mice post-transplant.
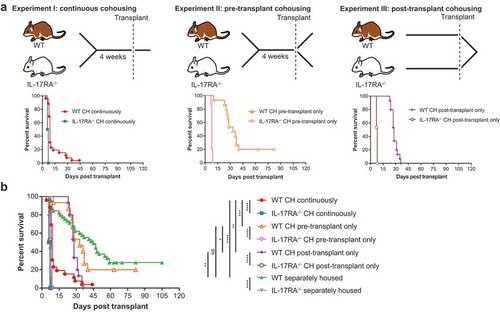
To obtain species-level resolution of transferred microbiota in addition to their functional potential we undertook metagenomic sequencing of fecal samples from one experimental replicate of each cohousing scenario, including the continuously cohoused mice analyzed previously by 16S rRNA amplicon sequencing.Citation21 Samples from the same mice were sequenced pre-transplant and at the time of sacrifice post-transplant, as well as pre-cohousing, where applicable. Separately housed (control) mice from each sequenced replicate were also analyzed pre- and post-transplant. To assess the bacterial community composition, we first undertook genome recovery, yielding a dereplicated set of 95 metagenome-assembled genomes (MAGs) representing 15 bacterial families (Table S1). Read mapping to these MAGs and a set of publicly available genomes confirmed the development of disease was associated with a substantial shift in the bacterial community (Figures S1 & S2 & Table S2). The composition of the disease-associated microbiome of WT and IL-17RA–/ – mice was distinguishable regardless of cohousing treatment (Figure S1B). Where cohousing occurred pre- or post-transplant only, ordination analysis suggested a more extensive difference in the disease-associated gut community between WT and IL-17RA–/ – mice as compared to continuously cohoused mice (Figure S1B). Continuous cohousing was also associated with a significant decrease in community diversity in WT mice post-transplant, whereas there was no significant decrease when cohousing occurred pre- or post-transplant only (Figure S3). These data suggest that WT mice cohoused pre- or post-transplant experience a more moderate community shift associated with disease onset than continuously cohoused WT mice, in agreement with the delayed disease phenotype observed in these mice ().
Members of the dominant bacterial family Muribaculaceae are transferred during cohousing and have the genetic potential to contribute to the GVHD phenotype
To investigate the putative priming effect of pre-transplant cohousing we compared both the functional potential and composition of the microbiome of cohoused WT mice to both their pre-cohousing composition and to separately housed WT mice at the same time point. At the functional level, there was no obvious distinction between pre-transplant cohorts, i.e. WT pre- and post-cohousing, nor between WT mice post-cohousing and separately housed control mice (Figure S4A-C). Only one annotation was identified as consistently differential between WT mice pre- and post-cohousing in both Experiment I and II, while also being significantly different from separately housed WT mice; tetracycline resistance monooxygenase (K18221) (Tables S3–5). The limited change in the functional potential of the microbiome during cohousing suggests that the priming effect is subtle and not detectable at the level of whole community comparison. Alternatively, the functional effect lies outside of the annotated portion of the metagenomes, or is occurring at the transcriptional/translational level.
At the compositional level, five species from the family Muribaculaceae (formerly uncultured lineage S24-7Citation22) increased during cohousing in WT mice from both Experiment I and II ( & Table S6). These species were also significantly more abundant in cohoused WT mice than in separately housed WT mice (Table S7). The only non-Muribaculaceae species to be enriched during cohousing in both experiments was Prevotella sp002933775; however, the average post-cohousing abundance of this species was substantially lower than that of the Muribaculaceae species (0.3% vs 1.7%; Table S6). Only two species were depleted consistently across both Experiment I and II: one from the family Muribaculaceae and one from the family Lachnospiraceae (Table S6). Notably, the pre-cohousing abundance of the depleted Lachnospiraceae species varied considerably between mice, ranging from 0.003% to 3.1%, and also did not differ in abundance between WT mice post-cohousing and separately housed WT mice (Table S7). In contrast, the depleted Muribaculaceae species was found to be significantly depleted in cohoused WT mice in comparison to separately housed WT mice (Table S7). We also undertook a broader analysis of all pre-transplant mice from all three experiments and identified a distinct microbiome composition between those typically susceptible to early-onset GVHD post-transplant (IL-17RA–/ – and pre-transplant cohoused WT mice) and those typically exhibiting delayed disease (separately housed WT mice) ( & Table S8). Comparison of MAGs associated with each disease type did not reveal any significant functional distinctions across the Pfam, KEGG and CAZy databases (Tables S9–11), further supporting the priming effect as occurring below the level of these functional categories or being potentially driven by variation in expression. Multivariate analysis of all pre-transplant WT mice also revealed a distinction between non-cohoused and cohoused WT mice (). Muribaculaceae species were consistently identified as critical to both this division and that distinguishing early- and delayed-onset disease ( and e & Table S8). These findings are in agreement with the results of our previous 16S rRNA gene sequencing analysis of pre- and post-transplant cohoused WT mice that identified members of the family Muribaculaceae as the dominant taxa with altered relative abundance via cohousing.Citation21
Figure 2. Species from the family Muribaculaceae are transferred during cohousing and are associated with disease susceptibility. (a) Heatmap displaying genomes identified as consistently significantly enriched or depleted in WT mice during pre-transplant cohousing across Experiments I and II (n = 4–5 per group per experiment). Complete analysis output and relative abundance values are contained in Table S6. (b) PCA displaying all pre-transplant mice across all three experiments, I, II and III, based on read mapping to a genome database composed of recovered MAGs plus genomes from NCBI (see methods). Dotted line indicates division between mice susceptible to early-onset (IL-17RA−/− and cohoused WT) and delayed-onset (separately housed WT) disease. Week 0 mice are pre-transplant mice from Experiment III and have undergone no cohousing pre-transplant. Week 4 mice have either been cohoused for 4 weeks or maintained separately for the same time period (control mice) and originate from Experiment I and II. (c) Heatmap displaying genomes with significantly different abundance between early-onset and delayed-onset GVHD susceptible mice, using division indicated in (b). Complete analysis output and relative abundance values are contained in Table S8. (d) Multivariate sparse partial least squares discriminant analysis (sPLS-DA) based on read mapping to genome database composed of recovered MAGs plus genomes from NCBI of all pre-transplant WT mice from Experiments I, II and III. (e) Genome-based signature contributing to separation along component 1 of (D). Color indicates group in which median genome abundance is highest. Bar length corresponds to loading weight. CH: cohoused, SH: separately housed.
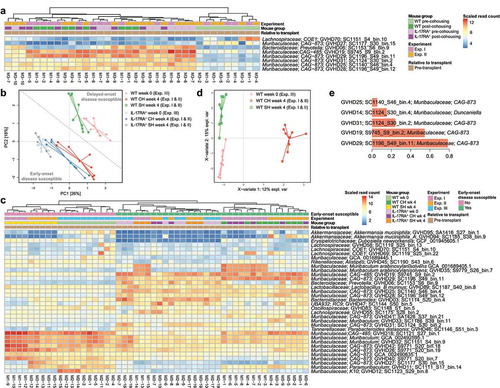
Of the enriched Muribaculaceae species, four are members of the genus CAG-873 and one is a member of the genus CAG-485, both genera currently lacking cultured representatives (Figure S5). The Muribaculaceae species depleted during pre-transplant cohousing also belonged to genus CAG-873 (GVHD27), suggesting priming effects must be at least species-specific. Two species (CAG-485 sp. GVHD19 and CAG-873 sp. GVHD29) displayed a relative abundance pattern of particular interest; both increased consistently in WT mice during cohousing experiments and were also observed in WT mice following disease onset (Figure S6). To assess whether these species displayed similar functional profiles we compared their representative genomes to others from the family Muribaculaceae using functional annotation of predicted proteins, including all species enriched in WT mice during cohousing (Figure S7A). The enriched genomes did not appear distinct amongst the family, indicating their broad metabolic profile was typical of Muribaculaceae. Based on their carbohydrate active enzyme complement, both GVHD19 and GVHD29 are part of the α-amylase trophic guild,Citation23 a guild defined by a reduced set of glycoside hydrolases (Figure S7B). No Muribaculaceae species enriched in WT mice during cohousing were members of the host glycan guild, characterized by increased abundance of enzymes associated with the degradation of mucin,Citation23 suggesting that they are not priming the gut for disease by promoting barrier dysfunction and subsequent pathogen translocation.Citation24
We subsequently undertook a comparison of orthologous proteins amongst Muribaculaceae genomes, specifically seeking those present in species enriched in WT mice during pre-transplant cohousing but rare in other species and with a predicted function contributing to capacity for disease priming such as adhesion or protein interaction domains, peptidases or surface antigens (Table S12). Multiple C10 peptidases were identified in the enriched genomes that displayed limited sequence conservation within the family Muribaculaceae (Table S12). The C10 protease family includes streptococcal pyrogenic exotoxin B (SpeB) from Streptococcus pyogenes capable of degrading multiple components of the host immune system.Citation25 Within GVHD19, several C10 peptidases are encoded directly adjacent to a putative serine protease inhibitor (serpin) that may act as a regulator of peptidase expression.Citation26 GVHD19 also encodes two-class C25 peptidases, of which gingipain produced by Porphyromonas gingivalis is the reference enzyme, and both GVHD19 and GVHD29 encode M6 peptidases, metalloendopeptidases shown to be important in degrading immune system elements.Citation25 Other proteins of interest present in the enriched genomes include homologs of potential adhesins such as the leucine-rich repeat encoding internalin J, most closely resembling that of Porphyromonas spp. (Figure S8A), which may play a role in biofilm formation,Citation27 and the cleaved adhesin domain carrying hemagglutinin, resembling that of Prevotella spp. (Figure S8B-D) which confers capacity to adhere to host cells.Citation28 Eukaryotic-like domains such as leucine-rich repeats, tetratricopeptide repeats and fibronectin type III are present in multiple additional proteins representing further candidates with potential for host interaction.Citation29
Continuous cohousing with a dysbiotic microbiome promotes a disease-associated bloom of Enterobacteriaceae
Due to the large shift in gut microbiome composition associated with the onset of GVHD (Figure S1A), we initially examined the disease-associated microbiome within each group of WT mice in comparison to their pre-transplant composition at the bacterial family level. The family Enterobacteriaceae was significantly enriched in cohoused WT mice at the time of sacrifice regardless of when cohousing occurred (Table S13), with continuous cohousing associated with a substantially greater enrichment of this family ( & Figure S2). WT mice undergoing continuous cohousing also displayed a significant enrichment in the family Acutalibacteraceae as well as a significant depletion of Muribaculaceae upon onset of GVHD (Table S13). No significant change in either family was observed in WT mice displaying GVHD from the alternative cohousing scenarios, although a similar trend was visible (Table S13, ). Separately housed WT mice that developed GVHD also displayed a significant increase in Enterobacteriaceae with disease (Table S14) and there was no significant difference in the abundance of Enterobacteriaceae between cohoused and separately housed WT mice with GVHD in any experiment (Table S15). Enterobacteriaceae blooms therefore appear characteristic of disease regardless of the cohousing scenario.
Figure 3. Increased abundance of Enterobacteriaceae is associated with disease. (a) Relative abundance of bacterial families Enterobacteriaceae, Acutalibacteraceae and Muribaculaceae across each experiment, before and after transplant. (b) Multivariate sparse partial least squares discriminant analysis (sPLS-DA) based on read mapping to genome database composed of recovered MAGs plus genomes from NCBI (see methods) of post-transplant WT mice. (c) Genome-based signature contributing to component 1 of (B). Genomes with frequency >0.75 across validation replicates displayed. Color indicates group in which median genome abundance is highest. Bar length corresponds to loading weight. CH: cohoused, SH: separately housed.
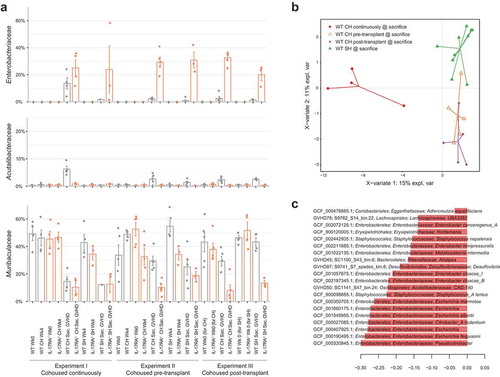
At the species level, Escherichia coli comprised the majority of the significant increase in abundance of Enterobacteriaceae in cohoused WT mice post-transplant in all three cohousing scenarios (Table S16). Other members of the family were present at low abundance, with the exception of Enterobacter hormaechei subsp. steigerwaltii, which increased significantly in all experiments and markedly more so with continuous cohousing (maximum 14% relative abundance). The relative abundance of Bacteroides vulgatus, Lactobacillus murinus and a member of the genus CAG-180 from the family Acutalibacteraceae also increased significantly with disease progression in cohoused WT mice in all three cohousing scenarios (Table S16). Separately housed WT mice also experienced significant increases in each of these species with the exception of CAG-180, which only increased significantly in one experimental replicate (Table S17). Despite these similar microbiome transitions amongst WT mice from different cohousing scenarios, multivariate comparison of the disease-associated microbiome of all WT mice clearly indicated separation of continuously and partially (pre- or post-transplant) cohoused mice based on higher relative abundance of Enterobacteriaceae species in the former group ( and c). Continuous cohousing may therefore promote acute disease through increased expansion of Enterobacteriaceae species beyond that experienced by mice under the alternative cohousing scenarios.
Disease-associated Enterobacteriaceae have no conspicuous GVHD-specific virulence factors
To determine whether the association observed between E. coli and E. hormaechei subsp. steigerwaltii and GVHD was driven by a specific set of virulence factors we undertook comparative analysis between all MAGs representing these species (non-dereplicated) and 21 high quality reference genomes not known to be associated with GVHD. A total of 28 E. coli and two E. hormaechei subsp. steigerwaltii MAGs (>90%-estimated completeness) were recovered across all experiments (Figure S9A-B). We additionally recovered four E. coli MAGs from publicly available datasets from patients undergoing hematopoietic stem cell transplantation; one adult that developed acute GVHD and three pediatric patients that did not.Citation30,Citation31 A further E. hormaechei subsp. steigerwaltii MAG was also obtained from a pediatric patient that developed acute GVHD.Citation31 Ordination analysis suggested that the virulence profile was driven more by phylogeny than by disease association, as MAGs from the current dataset clustered with their phylogenetic counterparts (Figure S9C). Inspection of individual virulence factors in the GVHD-associated E. coli MAGs also support differences being driven by phylogeny rather than disease association, e.g. iron acquisition capability and type VI secretion systems differed between the phylotypes (Figures S9A-B & S10, Table S18). Virulence factors that were shared across the majority of GVHD MAGs did not appear GVHD-specific; e.g., FdeC, capable of mediating adhesion to mammalian cells and found at higher prevalence in pathogenic subtypes,Citation32 was also found in non-GVHD reference genomes and was absent from the clinical GVHD E. coli MAG (Figure S10). Unbinned contigs homologous to a public set of E. coli genomes were also analyzed to identify potential plasmid elements not initially clustered with the MAGs; however, no further virulence factors were identified (Table S18). The lack of a clear GVHD-associated virulence profile supports E. coli and E. hormaechei subsp. steigerwaltii as having an opportunistic role in disease progression.
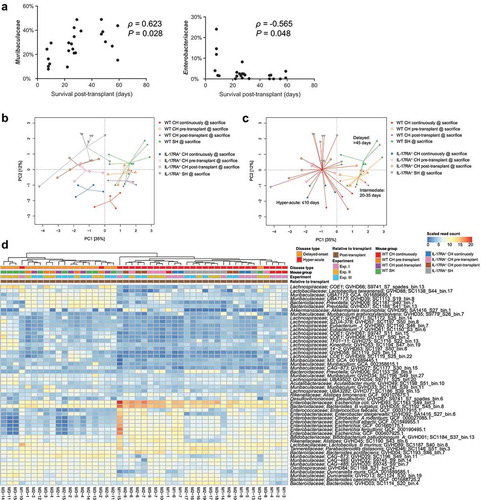
Survival post-transplant is associated with microbiome composition
Across all three cohousing scenarios we encountered a range of survival times post-transplant amongst WT mice; some succumbed to GVHD rapidly, similar to IL-17RA deficient mice, while others did not develop disease for over a month (). Using this distribution of length of survival post-transplant, we identified a negative correlation between the abundance of Enterobacteriaceae and survival of WT mice post-transplant, and positive correlation of the abundance of Muribaculaceae with survival (, Table S19). These trends were also observable at the genome level; however, there was clear variation in abundance between mice with similar survival times (Table S20). We subsequently defined three disease types for further comparison based on observable clustering of survival times post-transplant: hyper-acute (survival ≤10 days), intermediate (survival 20–35 days) and delayed (survival 45+ days) (). Ordination analysis suggested a distinction between the microbiome composition with hyper-acute disease (both WT and IL-17RA–/–) and those with intermediate or delayed disease onset ( and c). Hyper-acute GVHD was associated with a significantly higher abundance of E. coli and B. vulgatus (, Table S21). Intermediate or delayed disease was associated with higher abundance of members of the families Muribaculaceae and Lachnospiraceae, consistent with a greater retention of commensal organisms.
Figure 4. Length of survival post-transplant is associated with altered microbiome composition. (a) Correlation between relative abundance of Muribaculaceae and Enterobacteriaceae in WT mice at the time of sacrifice due to GVHD with length of survival post-transplant. Benjamini-Hochberg adjusted P values shown. (b) PCA displaying all mice sacrificed due to the development of GVHD across the three experiments (n = 4–5 per group per experiment) based on read mapping to genome database composed of recovered MAGs plus genomes from NCBI (see methods). (c) As per (B) with colored lines connecting mice within each survival timeframe. (d) Heatmap displaying genomes identified as significantly different between hyper-acute and delayed-onset GVHD at the time of sacrifice. Genomes with relative abundance >1% in at least one mouse displayed. Full list contained in Table S21. CH: cohoused, SH: separately housed.
Discussion
Here we investigate the effect of the pre- and post-transplant gut microbiome on the development of hyper-acute GVHD. Whilst we had previously observed accelerated disease development in WT mice following cohousing with IL-17RA–/ – mice,Citation21 it was unknown whether the phenotype was induced by exposure to the microbiome from IL-17RA–/ – mice before or after transplant. Pre-transplant priming of the gut microbiome via cohousing was insufficient to significantly accelerate disease in isolation, while cohousing post-transplant resulted in a significantly higher morbidity of WT mice versus non-cohoused controls. However, disease onset following cohousing post-transplant and not pre-transplant was delayed in comparison to WT mice cohoused continuously, indicating pre-transplant priming is also necessary for generating the hyper-acute phenotype. These novel data support both a priming and exacerbating role for the gut microbiome in disease development. We observed transfer of members of the bacterial family Muribaculaceae during cohousing pre-transplant from IL-17RA–/ – mice to WT mice and a bloom of members of the family Enterobacteriaceae post-transplant that associated with disease. Continuous cohousing was associated with an exaggeration of the expansion of Enterobacteriaceae, including increased abundance of both E. coli and E. hormaechei subsp. steigerwaltii.
As a dominant family in fecal microbiomes of laboratory mice,Citation23 it was unsurprising to see transition occurring amongst members of Muribaculaceae during cohousing. This family is regularly noted to vary in abundance in perturbation studies,Citation33,Citation34 however, it is currently unclear whether the group plays a role in disease development. Functional studies have been impeded by a lack of cultured Muribaculaceae representatives with the first isolates only recently being reported.Citation22,Citation35 Our use of metagenomic sequencing in the current study enabled us to identify, with species-level resolution, which members of the family were changing in abundance during cohousing. The improved resolution of metagenomic sequencing in comparison with amplicon sequencing means that species assignment will be directly comparable with future studies, including with new isolates as they become available. We used comparative genomics to compare the species of interest with available genomes from the family to compile a list of candidate genes with the potential to play a role in priming the gut to drive acute GVHD. A number of peptidases in this list suggest one possible priming mechanism is active degradation of components of the immune system rendering mice more susceptible to blooms of opportunistic pathogens post-transplant, a mechanism with potential to also operate in the human gut. While members of Muribaculaceae are found in the human gut, their prevalence is low (~7%).Citation35 However, the functions identified here as GVHD-associated are not exclusive to Muribaculaceae and therefore may be playing a similar role in other gut species. We identified peptidases from the families C10, C25 and M6, each of which has been demonstrated to have immunomodulatory activity and are identified in a variety of bacterial species.Citation25 We also observed a potential regulatory arrangement between peptidase and inhibitor similar to that described in Bacteroides fragilis where the gene pair is co-transcribed and responsive to environmental stimuli, particularly oxygen.Citation26 Increased oxygen availability associated with epithelial damage induced by transplant conditioning may therefore provide stimulus for peptidase expression while also promoting expansion of facultative anaerobes such as E. coli.Citation36
The bloom of E. coli we observed associated with the development of acute GVHD is consistent with previous observations both in mice and humans.Citation14,Citation30 Expansion of E. hormaechei subsp. steigerwaltii has not been described previously, although unclassified Enterobacter spp. have been associated with bloodstream infection in patients post-transplant in connection with acute GVHD.Citation37,Citation38 Using genome level analysis of the associated populations we confirmed potential pathogenicity of the identified E. coli supporting an active role in disease progression. While we observed a correlation between the abundance of Enterobacteriaceae and length of survival post-transplant, one limitation of these data is that they are only taken at the end-point of disease. There is a possibility that the species responsible for exacerbating acute GVHD bloomed early post-transplant and then diminished. In contrast, GVHD itself may directly modulate the microbiome and potentially hide preceding species of interest. For example, Paneth cells decrease in number and are damaged during GVHD, potentially contributing to altered microbiota via decreased production of the antimicrobial peptide α-defensin.Citation13,Citation14 However, sampling more frequently post-transplant is difficult due to the severity of GVHD and the consequence of this on the overall health of the mice.
In the broader disease context, these data support both the pre- and post-transplant gut microbiome as critical factors in the susceptibility to acute GVHD. Multiple factors affect the gut microbiome during stem cell transplant complicating the design and application of approaches for management of the bacterial community. Prior to transplant, antibiotics may be administered prophylactically; however, despite clear beneficial effects in randomized studies,Citation39,Citation40 some negative effects have been also been suggested in retrospective cohort analysis,Citation41 perhaps consistent with the fact that the microbiome varies significantly between individuals.Citation42 Conditioning regimens also affect pre-transplant microbiota, however, to what degree is difficult to establish due to prior and/or concomitant antibiotic administration.Citation43–Citation45 Antibiotic treatment is often initiated post-transplant in response to infectious complications and may negatively impact overall prognosis depending on the antibiotics used.Citation46,Citation47 As an alternative to microbiome depletion, prophylactic post-transplant manipulation of the microbiota via fecal microbiota transplantation (FMT) is currently under investigation as a means of restoring commensal organisms, thereby improving bacterial diversity. Whilst this research is only emerging and still largely experimental, early clinical data showing feasibility, safety and some efficacy of FMT to treat refractory acute GVHD in the GI tract is encouraging.Citation48–Citation50 Restoration and/or maintenance of a diverse microbiome may increase resistance to blooms of opportunistic pathogens such as E. coli.Citation51 Our results demonstrate association of specific species with disease outcome, where other members of the same genus appear unassociated, indicating a necessary resolution for manipulation that is unlikely to be achievable using antibiotics. Therefore, promoting bacterial diversity using methods such as FMT may be a more successful strategy for reducing acute GVHD development following stem cell transplantation.
Methods
Mice and allogeneic stem cell transplantation
Wild-type C57BL/6 mice (referred to as WT herein) were purchased from the Animal Resource Center (Perth, Western Australia). IL-17RA–/ – mice on a C57BL/6 background (Amgen, Washington, USA) were bred in-house. Animal procedures were undertaken using protocols approved by the QIMR Berghofer Animal Ethics committee. Mice were transplanted and monitored as described previously.Citation21,Citation52 Briefly, recipient mice received total body irradiation of 1000 cGy. Recombinant human Granulocyte Colony Stimulating Factor (G-CSF; Amgen Inc., Thousand Oaks, CA, USA) was administered to donor mice subcutaneously at 10 µg/dose/animal for 6 days. Mice were transplanted with either 25 × 106 T-cell replete or 20 × 106 T-cell depleted (TCD) G-CSF mobilized splenocytes. All transplanted mice were housed in sterilized microisolator cages and received acidified autoclaved water. GVHD was assessed using an established scoring systemCitation53 and mice with clinical scores ≥6 were sacrificed in accordance with institutional guidelines. The data from the continuous cohousing experiments (Experiment I in ) has been published previously.Citation21
Metagenomic data processing, assembly and MAG recovery
Mouse reads were removed by mapping against the Mus musculus genome (GRCm38.p5) using BWA v0.7.12Citation54 requiring a minimum alignment length of 30 bases and maximum of 15 clipped bases for reads to be considered of mouse origin. Non-mouse reads from each sample were assembled independently using metaSPAdes v3.12.0.Citation55 Coverage based binning was performed by read mapping of a subset of six samples to each resulting assembly using BamM v1.7.3 (https://github.com/ecogenomics/BamM) with bins recovered using MetaBAT v2.12.1Citation56 with a minimum contig length of 2000 bases. Contamination and completeness of resulting bins was assessed using CheckM v1.0.12.Citation57 Bins with completeness >75% and contamination <7% were dereplicated using dRep v2.2.3Citation58 at 95% minimum secondary average nucleotide identity (-sa 0.95). The taxonomic affiliation of recovered MAGs was determined using GTDB-Tk v0.3.0 with the Genome Taxonomy Database (GTDB) Release 04-RS89.Citation59
Metagenomic community profiling
Reads for each sample were mapped to a dereplicated set (dRep, 95%) of 16,958 bacterial and archaeal genomes from GTDB Release 03-RS86 (>80% complete, <7% contamination) using BamM with a minimum seed length of 25. Genomes with >1x coverage of >1% of the genome and overall coverage of 0.01X, as determined using Mosdepth v0.2.3,Citation60 were retained and combined with the dereplicated set of recovered MAGs for assessment of community composition. The final genome set of 237 comprised 95 MAGs recovered from this study plus 142 genomes from NCBI (NCBI accessions are contained in Table S23). Read counts for the final genome set were determined for each sample via mapping using BamM with minimum seed length of 25 bases and subsequent filtering for minimum mapping percentage identity of 95%. Per genome read counts were scaled to account for genome size whilst maintaining the raw unmapped read percentage for each sample as a reflection of unrepresented diversity. Relative abundance was calculated using scaled read counts as a fraction of total non-host reads per sample. Alpha-diversity was calculated using QIIME v1.8.0Citation61 with counts normalized using the size factor method implemented within the R package DESeq2 v1.20.0.Citation62 MAGs were annotated using Prokka v1.12.Citation63
Functional annotation
Functional annotation of raw reads and predicted proteins from MAGs was undertaken via alignment with HMMER v3.1b2Citation64 to the hidden Markov model databases dbCAN CAZy v6,Citation65 Pfam r32Citation66 and TIGRFAM v15Citation67 (MAGs only) with maximum e-value cutoff of 1e-10. KEGG orthology was determined via BLAST v2.8.1Citation68 alignment to UniProt UniRef100 database downloaded July 2017Citation69 with maximum e-value of 1e-10 and subsequent extraction of associated KO terms.
Muribaculaceae MAG analysis
A Muribaculaceae genome tree was constructed using recovered MAGs plus publicly available genomes based on alignment of 120 single-copy marker genes.Citation59 A bootstrapped maximum-likelihood tree was inferred using IQ-TREE v1.6.9Citation70 (100 replicates, non-parametric) using model LG+C10 + F + G with posterior mean site frequency approximationCitation71 based on alignment positions containing a residue within ≥50% of sequences (38,690 positions). Trophic guilds were assigned using the method described in ref.Citation23 Orthologous proteins were identified using Proteinortho v5.16bCitation72 with an e-value cutoff 1e-6. Gene trees were constructed using homologues identified within the recovered Muribaculaceae MAGs and the GTDB Release 03-RS86 by GeneTreeTK v0.0.14 (https://github.com/dparks1134/GeneTreeTk) with default settings. Bootstrapped maximum-likelihood trees (100 replicates, non-parametric) were constructed using IQ-TREE v1.6.9Citation70 with ModelFinderCitation73 used for model selection. Alignment sites with a minimum similarity of 30% were used for phylogenetic inference. ARBCitation74 was used for alignment filtering and tree curation.
Escherichia coli MAG analysis
Virulence factor prediction in binned and unbinned E. coli contigs was undertaken via BLAST search of recovered MAGs and public genomes against the VFDB core databaseCitation75 (accessed February 2019) with maximum e-value cutoff 1e-10, minimum alignment identity of 30% and minimum alignment fraction of 70%. Virulence factor classes were obtained from reference genomes downloaded from VFDB VFanalyzer. Unbinned contigs from all mice at sacrifice were clustered using CD-HITCitation76 using the cd-hit-est command with 99% identity (-c 0.99) and 90% coverage of the shorter sequence (-aS 0.9) thresholds. Representative contigs homologous to E. coli were identified via BLAST search of the NCBI nt database (downloaded January 2019) with maximum e-value cutoff 1e-10 followed by filtering for sequence description containing ‘Escherichia coli’, minimum alignment identity of 97% and minimum alignment fraction of 50% of the query contig. Additional E. coli MAGs were recovered using the same methodology described above from SRA experiments SRR3340629, SRR5050584, SRR5050585, and SRR5050587. Bootstrapped maximum-likelihood tree was inferred using IQ-TREE as described above.
Statistical analysis and graphical presentation
Survival curves were plotted using Kaplan-Meier estimates and compared by log-rank analysis. The Wilcoxon rank-sum test was used for the statistical analysis of alpha-diversity values with Benjamini-Hochberg adjustment for multiple comparisons. P < .05 was considered statistically significant. Dot plots are presented as mean ± standard error of the mean. Principal component analysis was conducted using the R package vegan v2.5–1Citation77 on data normalized using log cumulative-sum-scaling (log-CSS) implemented within metagenomeSeq v1.22.0.Citation78 Differential abundance of bacterial taxa and functional annotations (raw reads) between sample groups was assessed using the Wald test within DESeq2 v1.20.0Citation62 based on total annotated read counts or read counts per genome scaled to account for genome size with the Benjamini-Hochberg adjustment for multiple comparisons. P < .001 was considered statistically significant. sPLS-DA analysis was conducted using the R package mixOmics v6.3.2Citation79 using centered log-ratio transformed relative abundance values (pseudocount 1e-07) with 50xM-fold cross-validation (fivefold pre-transplant WT, threefold post-transplant WT). Comparison of functional annotations within MAGs associated with early-onset and delayed-onset GVHD was undertaken using EnrichM v0.5.0 (https://github.com/geronimp/enrichM) with Fisher’s exact test employed to compare the number of MAGs encoding/not-encoding each functional category (i.e., each individual CAZy, KO, or Pfam classification) associated with each disease type (with Benjamini-Hochberg adjustment for multiple comparisons). Spearman’s rho calculated using ‘corr.test’ function within R package psych v1.8.12Citation80 with the Benjamini-Hochberg adjustment for multiple comparisons using centered log-ratio transformed relative abundance values. Bacterial families filtered for those with minimum relative abundance ≥0.05%. P value of <0.05 was considered statistically significant. Heatmaps were produced using the R package pheatmap v1.0.10,Citation81 box plots produced using the R package ggplot2 v2.2.1Citation82 with extension GGally v1.4.0Citation83 and dot plots of family abundance generated within GraphPad Prism v8.0.1 (GraphPad Software).
Data availability
Raw sequencing data and recovered MAGs are available via NCBI BioProject PRJNA544874. Sample accessions are provided in Table S22.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download PDF (5 MB)Supplemental Material
Download MS Excel (3.7 MB)Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Zeiser R, Blazar BR. Acute graft-versus-host disease — biologic process, prevention, and therapy. N Engl J Med. 2017;377:2167–2179.
- Castilla-Llorente C, Martin PJ, McDonald GB, Storer BE, Appelbaum FR, Deeg HJ, Mielcarek M, Shulman H, Storb R, Nash RA. Prognostic factors and outcomes of severe gastrointestinal GVHD after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2014;49:966. doi:10.1038/bmt.2014.69.
- Jones JM, Wilson R, Bealmear PM. Mortality and gross pathology of secondary disease in germfree mouse radiation chimeras. Radiat Res. 1971;45(3):577–588. doi:10.2307/3573066.
- Bekkum D, Roodenburg J, Heidt PJ, Waaij D. Mitigation of secondary disease of allogeneic mouse radiation chimeras by modification of the intestinal microflora. JNCI: J National Cancer Inst. 1974;52(2):401–404. doi:10.1093/jnci/52.2.401.
- Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JLM. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90(8):3204–3213. doi:10.1182/blood.V90.8.3204.
- Kennedy GA, Varelias A, Vuckovic S, Le Texier L, Gartlan KH, Zhang P, Thomas G, Anderson L, Boyle G, Cloonan N, et al. Addition of interleukin-6 inhibition with tocilizumab to standard graft-versus-host disease prophylaxis after allogeneic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 2014;15(13):1451–1459. doi:10.1016/S1470-2045(14)71017-4.
- Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, Rossignol DP, Ferrara JL. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest. 2001;107(12):1581–1589. doi:10.1172/JCI12156.
- Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, Ferrara JL. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8(6):575–581. doi:10.1038/nm0602-575.
- Koyama M, Hill GR. Alloantigen presentation and graft-versus-host disease: fuel for the fire. Blood. 2016;127(24):2963–2970. doi:10.1182/blood-2016-02-697250.
- Whangbo J, Ritz J, Bhatt A. Antibiotic-mediated modification of the intestinal microbiome in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2016;52:183–190. doi:10.1038/bmt.2016.206.
- Khoruts A, Hippen KL, Lemire AM, Holtan SG, Knights D, Young JAH. Toward revision of antimicrobial therapies in hematopoietic stem cell transplantation: target the pathogens, but protect the indigenous microbiota. Transl Res. 2017;179:116–125. doi:10.1016/j.trsl.2016.07.013.
- Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hülsdünker J, Varelias A, Vetizou M, Kuns RD, Robb RJ, Zhang P, et al. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity. 2019;51(5):885–898
- Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, Liu C, West ML, Singer NV, Equinda MJ, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med. 2012;209(5):903–911. doi:10.1084/jem.20112408.
- Eriguchi Y, Takashima S, Oka H, Shimoji S, Nakamura K, Uryu H, Shimoda S, Iwasaki H, Shimono N, Ayabe T, et al. Graft-versus-host disease disrupts intestinal microbial ecology by inhibiting Paneth cell production of α-defensins. Blood. 2012;120(1):223–231. doi:10.1182/blood-2011-12-401166.
- Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, Steinhoff U, Tchaptchet S, Thiel E, Freudenberg MA, et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut. 2010;59(8):1079–1087. doi:10.1136/gut.2009.197434.
- Holler E, Butzhammer P, Schmid K, Hundsrucker C, Koestler J, Peter K, Zhu W, Sporrer D, Hehlgans T, Kreutz M, et al. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. Biol Blood Marrow Transplant. 2014;20(5):640–645. doi:10.1016/j.bbmt.2014.01.030.
- Taur Y, Jenq RR, Perales M-A, Littmann ER, Morjaria S, Ling L, No D, Gobourne A, Viale A, PB D, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;124(7):1174–1182. doi:10.1182/blood-2014-02-554725.
- Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, Littmann ER, Ling L, Gobourne AC, Miller LC, et al. Intestinal Blautia is associated with reduced death from graft-versus-host disease. Biol Blood Marrow Transplant. 2015;21(8):1373–1383. doi:10.1016/j.bbmt.2015.04.016.
- Biagi E, Zama D, Nastasi C, Consolandi C, Fiori J, Rampelli S, Turroni S, Centanni M, Severgnini M, Peano C, et al. Gut microbiota trajectory in pediatric patients undergoing hematopoietic SCT. Bone Marrow Transplant. 2015;50(7):992–998. doi:10.1038/bmt.2015.16.
- Doki N, Suyama M, Sasajima S, Ota J, Igarashi A, Mimura I, Morita H, Fujioka Y, Sugiyama D, Nishikawa H, et al. Clinical impact of pre-transplant gut microbial diversity on outcomes of allogeneic hematopoietic stem cell transplantation. Ann Hematol. 2017;96(9):1517–1523. doi:10.1007/s00277-017-3069-8.
- Varelias A, Ormerod KL, Bunting MD, Koyama M, Gartlan KH, Kuns RD, Lachner N, Locke KR, Lim CY, Henden AS, et al. Acute graft-versus-host disease is regulated by an IL-17–sensitive microbiome. Blood. 2017;129(15):2172–2185. doi:10.1182/blood-2016-08-732628.
- Lagkouvardos I, Pukall R, Abt B, Foesel BU, Meier-Kolthoff JP, Kumar N, Bresciani A, Martínez I, Just S, Ziegler C, et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol. 2016;1:16131. doi:10.1038/nmicrobiol.2016.131.
- Ormerod KL, Wood DLA, Lachner N, Gellatly SL, Daly JN, Parsons JD, Dal’Molin CGO, Palfreyman RW, Nielsen LK, Cooper MA, et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome. 2016;4(1):1–17. doi:10.1186/s40168-016-0181-2.
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016;167(5):1339–1353.e1321. doi:10.1016/j.cell.2016.10.043.
- Potempa J, Pike RN. Corruption of innate immunity by bacterial proteases. J Innate Immun. 2009;1(2):70–87. doi:10.1159/000181144.
- Thornton RF, Murphy EC, Kagawa TF, O’Toole PW, Cooney JC. The effect of environmental conditions on expression of Bacteroides fragilis and Bacteroides thetaiotaomicron C10 protease genes. BMC Microbiol. 2012;12(1):190. doi:10.1186/1471-2180-12-190.
- Capestany CA, Kuboniwa M, Jung I-Y, Park Y, Tribble GD, Lamont RJ. Role of the Porphyromonas gingivalis InlJ protein in homotypic and heterotypic biofilm development. Infect Immun. 2006;74(5):3002–3005. doi:10.1128/IAI.74.5.3002-3005.2006.
- Sakai E, Naito M, Sato K, Hotokezaka H, Kadowaki T, Kamaguchi A, Yamamoto K, Okamoto K, Nakayama K. Construction of recombinant hemagglutinin derived from the gingipain-encoding gene of Porphyromonas gingivalis, identification of its target protein on erythrocytes, and inhibition of hemagglutination by an interdomain regional peptide. J Bacteriol. 2007;189(11):3977–3986. doi:10.1128/JB.01691-06.
- Frank AC. Molecular host mimicry and manipulation in bacterial symbionts. FEMS Microbiol Lett. 2019;366:4. doi:10.1093/femsle/fnz038.
- Kaysen A, Heintz-Buschart A, Muller EEL, Narayanasamy S, Wampach L, Laczny CC, Graf N, Simon A, Franke K, Bittenbring J, et al. Integrated meta-omic analyses of the gastrointestinal tract microbiome in patients undergoing allogeneic hematopoietic stem cell transplantation. Transl Res. 2017;186:79–94.e71. doi:10.1016/j.trsl.2017.06.008.
- Simms-Waldrip TR, Sunkersett G, Coughlin LA, Savani MR, Arana C, Kim J, Kim M, Zhan X, Greenberg DE, Xie Y, et al. Antibiotic-induced depletion of anti-inflammatory Clostridia is associated with the development of graft-versus-host disease in pediatric stem cell transplantation patients. Biol Blood Marrow Transplant. 2017;23(5):820–829. doi:10.1016/j.bbmt.2017.02.004.
- Nesta B, Spraggon G, Alteri C, Gomes Moriel D, Rosini R, Veggi D, Smith S, Bertoldi I, Pastorello I, Ferlenghi I, et al. FdeC, a novel broadly conserved Escherichia coli adhesin eliciting protection against urinary tract infections. mBio. 2012;3(2):e00010–00012. doi:10.1128/mBio.00010-12.
- Schroeder BO, Birchenough GMH, Ståhlman M, Arike L, Johansson MEV, Hansson GC, Bäckhed F. Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe. 2018;23(1):27–40.e27. doi:10.1016/j.chom.2017.11.004.
- Borton MA, Sabag-Daigle A, Wu J, Solden LM, O’Banion BS, Daly RA, Wolfe RA, Gonzalez JF, Wysocki VH, Ahmer BMM, et al. Chemical and pathogen-induced inflammation disrupt the murine intestinal microbiome. Microbiome. 2017;5(1):47. doi:10.1186/s40168-017-0264-8.
- Lagkouvardos I, Lesker TR, Hitch TCA, Gálvez EJC, Smit N, Neuhaus K, Wang J, Baines JF, Abt B, Stecher B, et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome. 2019;7(1):28. doi:10.1186/s40168-019-0637-2.
- Rigottier-Gois L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. Isme J. 2013;7:1256. doi:10.1038/ismej.2013.80.
- Kung AL, Robinson C, George D, Karamehmet E, Garvin JH, Sosna J, Pinkney K, Foca MD, Bhatia M, Satwani P, et al. Acute gastrointestinal graft-vs-host disease is associated with increased enteric bacterial bloodstream infection density in pediatric allogeneic hematopoietic cell transplant recipients. Clin Infect Dis. 2015;61(3):350–357. doi:10.1093/cid/civ285.
- Sano H, Hilinski JA, Qayed M, Applegate K, Newton JG, Watkins B, Chiang KY, Horan J. Early blood stream infection following allogeneic hematopoietic stem cell transplantation is a risk factor for acute grade III-IV GVHD in children and adolescents. Pediatr Blood Cancer. 2018;65:2. doi:10.1002/pbc.26821.
- Beelen DW, Elmaagacli A, Müller K-D, Hirche H, Schaefer UW. Influence of intestinal bacterial decontamination using metronidazole and ciprofloxacin or ciprofloxacin alone on the development of acute graft-versus-host disease after marrow transplantation in patients with hematologic malignancies: final results and long-term follow-up of an open-label prospective randomized trial. Blood. 1999;93:3267–3275.
- Beelen DW, Haralambie E, Brandt H, Linzenmeier G, Muller KD, Quabeck K, Sayer HG, Graeven U, Mahmoud HK, Schaefer UW. Evidence that sustained growth suppression of intestinal anaerobic bacteria reduces the risk of acute graft-versus-host disease after sibling marrow transplantation. Blood. 1992;80(10):2668–2676. doi:10.1182/blood.V80.10.2668.2668.
- Routy B, Letendre C, Enot D, Chénard-Poirier M, Mehraj V, Séguin NC, Guenda K, Gagnon K, Woerther P-L, Ghez D, et al. The influence of gut-decontamination prophylactic antibiotics on acute graft-versus-host disease and survival following allogeneic hematopoietic stem cell transplantation. OncoImmunology. 2017;6(1):e1258506. doi:10.1080/2162402X.2016.1258506.
- Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJ, Huttenhower C. Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci U S A. 2015;112(22):E2930–2938. doi:10.1073/pnas.1423854112.
- Dun CAJ, de Bont ESJM, van Vliet MJ, Kamps WA, Tissing WJE, Harmsen HJM, Meessen NEL. Chemotherapy treatment in pediatric patients with acute myeloid leukemia receiving antimicrobial prophylaxis leads to a relative increase of colonization with potentially pathogenic bacteria in the gut. Clin Infect Dis. 2009;49(2):262–270. doi:10.1086/599346.
- Montassier E, Batard E, Massart S, Gastinne T, Carton T, Caillon J, Le Fresne S, Caroff N, Hardouin JB, Moreau P, et al. 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb Ecol. 2014;67(3):690–699. doi:10.1007/s00248-013-0355-4.
- Montassier E, Gastinne T, Vangay P, Al-Ghalith GA, Bruley Des Varannes S, Massart S, Moreau P, Potel G, de La Cochetière MF, Batard E, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther. 2015;42(5):515–528. doi:10.1111/apt.2015.42.issue-5.
- Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, Tsai JJ, Slingerland AE, Smith OM, Young LF, Gupta J, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med. 2016;8(339):339ra371–339ra371. doi:10.1126/scitranslmed.aaf2311.
- Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, Lee YJ, Dubin KA, Socci ND, Viale A, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55(7):905–914. doi:10.1093/cid/cis580.
- DeFilipp Z, Peled JU, Li S, Mahabamunuge J, Dagher Z, Slingerland AE, Del Rio C, Valles B, Kempner ME, Smith M, et al. Third-party fecal microbiota transplantation following allo-HCT reconstitutes microbiome diversity. Blood Adv. 2018;2(7):745–753. doi:10.1182/bloodadvances.2018017731.
- Kaito S, Toya T, Yoshifuji K, Kurosawa S, Inamoto K, Takeshita K, Suda W, Kakihana K, Honda K, Hattori M, et al. Fecal microbiota transplantation with frozen capsules for a patient with refractory acute gut graft-versus-host disease. Blood Adv. 2018;2(22):3097–3101. doi:10.1182/bloodadvances.2018024968.
- Taur Y, Coyte K, Schluter J, Robilotti E, Figueroa C, Gjonbalaj M, Littmann ER, Ling L, Miller L, Gyaltshen Y, et al. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci Transl Med. 2018;10(460):eaap9489. doi:10.1126/scitranslmed.aap9489.
- Stecher B. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol Spectr. 2015;3:3.
- Varelias A, Bunting MD, Ormerod KL, Koyama M, Olver SD, Straube J, Kuns RD, Robb RJ, Henden AS, Cooper L, et al. Recipient mucosal-associated invariant T cells control GVHD within the colon. J Clin Invest. 2018;128(5):1919–1936. doi:10.1172/JCI91646.
- Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr., Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The Roles of Minor H Antigens and Endotoxin. Blood. 1996;88:3230–3239.
- Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324.
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27(5):824–834.
- Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165. doi:10.7717/peerj.1165.
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi:10.1101/gr.186072.114.
- Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. Isme J. 2017;11(12):2864–2868. doi:10.1038/ismej.2017.126.
- Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996. doi:10.1038/nbt.4229.
- Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2017;34(5):867–868.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi:10.1038/nmeth.f.303.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8.
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153.
- Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41(12):e121. doi:10.1093/nar/gkt263.
- Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(WebServer issue):W445–W451. doi:10.1093/nar/gks479.
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(D1):D222–D230. doi:10.1093/nar/gkt1223.
- Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31(1):371–373. doi:10.1093/nar/gkg128.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi:10.1016/S0022-2836(05)80360-2.
- Suzek BE, Huang HZ, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23(10):1282–1288. doi:10.1093/bioinformatics/btm098.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–274. doi:10.1093/molbev/msu300.
- Wang H-C, Minh BQ, Susko E, Roger AJ. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst Biol. 2017;67(2):216–235. doi:10.1093/sysbio/syx068.
- Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12(1):1–9. doi:10.1186/1471-2105-12-124.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587. doi:10.1038/nmeth.4285.
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Kumar Y, Buchner A, Lai T, Steppi S, Jobb G, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32(4):1363–1371. doi:10.1093/nar/gkh293.
- Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40(D1):D641–D645. doi:10.1093/nar/gkr989.
- Li W, Godzik A. CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659.
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H 2015. vegan: community ecology package. R package version 2.3-1. http://CRAN.R-project.org/package=vegan.
- Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10(12):1200–1202. doi:10.1038/nmeth.2658.
- Rohart F, Gautier B, Singh A, Lê Cao K-A. mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol. 2017;13:e1005752.
- Revelle W 2019. psych: procedures for psychological, psychometric, and personality research. R package version 1. 8.12. https://CRAN.R-project.org/package=psych.
- Kolde R 2015. pheatmap: pretty heatmaps. R package version 1.0.7. http://CRAN.R-project.org/package=pheatmap.
- Wickham H, Chang W, Henry L, Pedersen TL, Takahashi K, Wilke C, Woo K 2019. ggplot2: create elegant data visualisations using the grammar of graphics. R package version 3.1.1. https://CRAN.R-project.org/package=ggplot2.
- Schloerke B, Crowley J, Cook D, Briatte F, Marbach M, Thoen E, Elberg A, Larmarange J 2018. GGally: extension to ‘ggplot2ʹ. R package version 1.4.0. https://CRAN.R-project.org/package=GGally.