
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The challenging conditions encountered during long sea voyages increase the risk of health-threatening physiological and psychological stress for sailors compared with land-based workers. However, how the intestinal microbiota responds to a long sea voyage and whether there is a feasible approach for protecting gut health during sea voyage are still unexplored. Here, we designed a 30-d longitudinal study including a placebo group (n = 42) and a probiotic group (n = 40) and used shotgun metagenomic sequencing to explore the impacts of sea voyage on the intestinal microbiome of sailors. By comparing the intestinal microbiome of subjects in the placebo group at baseline (d 0) and at the end of the sea voyage (d 30), we observed an alteration in the intestinal microbiome during the long sea voyage based on the microbial structure; the results revealed an increase in the species Streptococcus gordonii and Klebsiella pneumoniae as well as a decrease in some functional features. However, the change in the microbial structure of sailors in the probiotic group between d 0 and d 30 was limited, which indicated a maintenance effect of probiotics on intestinal microbiome homeostasis. At the metagenomic strain level, a generally positive correlation was observed between probiotics and the strains belonging to Bifidobacterium longum and Bifidobacterium animalis, whereas a common negative correlation was observed between probiotics and Clostridium leptum; this result revealed the potential mechanism of maintaining intestinal microbiome homeostasis by probiotics. The present study provided a feasible approach for protecting gut health during a long sea voyage.
Introduction
Seafaring has a long history and greatly promotes the circulation of goods, cultural communication and territorial defense.Citation1 However, the challenging conditions encountered during long sea voyages increase the risk of disease and death for sailors compared with land-based workers. The ocean environment includes high humidity, high salinity, intense UV radiation, stormy waves, monotonous surroundings, altered circadian biorhythms, sleep deprivation, and an insufficient supply of fresh fruits and vegetables, causing significant health-threatening physiological and psychological stress.Citation2–Citation4 In addition to historically well-known septicemia caused by vitamin deficiency, chronic diseases of the immune system and digestive system have become the greatest risk to the health of sailorsCitation5 and have also become a potential threat to the development of seafaring.
The human intestinal microbiome is fundamental to the host immune system and is essential for maintaining human health.Citation6 In recent decades, emerging evidence has highlighted the importance of the diversity of intestinal microbial species and functional genes in various chronic metabolic diseases. Although much attention has been paid to the health management of sailors during long sea voyages,Citation5,Citation7 the research in this area remains limited. For example, the isolation of a land-based environment and the insufficient supply of fresh fruits and vegetables during a long sea voyage were all able to influence the intestinal microbiome of sailors. However, how the intestinal microbiota responds to a long sea voyage and how specific intestinal species or functional genes change are still unexplored.
Probiotics are defined as live microorganisms that confer health benefits to the host when present in adequate amountsCitation8 and commonly include bacterial strains of the genera Lactobacillus and Bifidobacterium.Citation9,Citation10 Currently, most probiotic-related studies have simply described fluctuations in intestinal microbiota profiles concluding that limited structural changes in the intestinal microbiota are a universal phenomenon that are accompanied by increased beneficial microbes and decreased pathogens after probiotic consumption.Citation11,Citation12 Some clinical studies have applied probiotics to treat a number of diseases, including liver disease,Citation13 cardiovascular diseases,Citation14 kidney diseaseCitation15 and irritable bowel syndrome.Citation16 Although not all of these studies were positive, we confirmed that probiotics exhibited common effects on intestinal microbiota regulation. Based on the consensus and the question we asked above, we wanted to know whether probiotics can maintain intestinal microbiome homeostasis in sailors during a long sea voyage. If so, how does this maintenance work?
Therefore, we designed a 30-d longitudinal study including two Chinese sailor cohorts (a placebo group, n= 42, and a probiotic group, n= 40) and used multi-probiotics, including Lactobacillus casei Zhang, Lactobacillus plantarumP-8, Lactobacillus rhamnosus M9, Bifidobacterium lactis V9 and Bifidobacterium lactis M8, which exhibited excellent probiotic characteristics in our previous studies,Citation17,Citation18 to explore the risk of a long sea voyage on the intestinal microbiome of sailors and reveal the potential mechanism of maintaining intestinal microbiome homeostasis by probiotics. The results of the present study will provide a feasible approach for protecting gut health during a long sea voyage and provide new insights into a personalized selection of probiotics based on population-level analyses.
Results
Comparative analysis of health-related physiological and psychological indexes between the placebo and probiotic groups
Because of the special ocean environment, most sailors suffer physiological and psychological stress during long sea voyages. To evaluate the potential effects of probiotics on improving a sailor’s fitness, we designed a questionnaire including scores for bowel movement (hardness of feces, volume of feces, constipation, diarrhea, bloody stools and defecation frequency), pain (stomachache, headache, pectoralgia, backache and muscle soreness) and stress/anxiety (flustered, dizziness, amnesia, annoyance, low activity, mistrustful, poor appetite, self-accusation, loneliness, palpitation and insomnia) at the end of the trip (Table S1). We set each subject’s score for the questionnaire content to 10 as baseline. Therefore, a decreased score represented the unfitness of the subject in the corresponding questionnaire content during the voyage (Figure S1(a–c)). By comparing the scores between the two cohorts, significant differences in the decreased score of stress and anxiety were observed (Figure S1(c)) in the placebo group, which indicated the potential role of probiotics in preventing anxiety during a long sea voyage.
Alteration of the intestinal microbiome in sailors during a long sea voyage
To address our initial questions, how intestinal microbiota respond to a long sea voyage and how specific intestinal species and functional genes change, we compared the intestinal microbiome of subjects in the placebo group at baseline (d 0) and at the end of the sea voyage (d 30) by shotgun metagenomic sequencing (Table S3). A principal coordinate analysis (PCoA) was performed based on the Aitchison and Bray–Curtis distances of the metagenomic sequencing profiles at the species level (,)). The intestinal microbiota from the subjects in the placebo group on d 0 and d 30 were distinct with respect to organismal structure. To quantify the difference, we calculated the P values (Wilcoxon rank-sum tests) of PC1 based on Aitchison distances (P =.014) and Bray–Curtis (P =.058), which indicated that the impacts of a long sea voyage on the intestinal microbiota of sailors were significant. Meanwhile, it was observed that the microbial Bray–Curtis and Aitchison distances of sailors in the placebo group between d 0 and d 30 were significantly larger than those of sailors in the probiotic group ()). The results were confirmed by microbial alpha diversity analysis, in which we observed a sharp decrease in microbial alpha diversity between the subjects at baseline and at the end of the sea voyage (), Table S4).
Figure 1. The impacts of a long sea voyage on the intestinal microbiota of sailors. (a,b) The metagenomic species-based Aitchison and Bray–Curtis distances principal coordinate analysis (PCoA) of sailors in the placebo and probiotic groups between the baseline (d 0) and the end of the voyage (d 30). The points in different colors represent the intestinal microbial structure of the subjects in each group. PC1 was extracted and compared using a boxplot, and the P value represents the significance between the two groups (Wilcoxon rank-sum tests). (c) Comparison of the intestinal microbial Aitchison and Bray–Curtis distances between the subjects at baseline and at the end of the voyage in both the placebo and probiotic groups (Wilcoxon rank-sum tests). “Pla” represents the placebo group, and “Pro” represents the probiotic group. Both the Aitchison and Bray–Curtis distances in the placebo group were significantly larger than those of sailors in the probiotic group, which indicated a significant change in intestinal microbiota during the sea voyage of subjects in the placebo group. (d) The Shannon index and Simpson index represented the microbial alpha diversity of sailors both in the placebo and probiotic groups at baseline and at the end of the sea voyage.
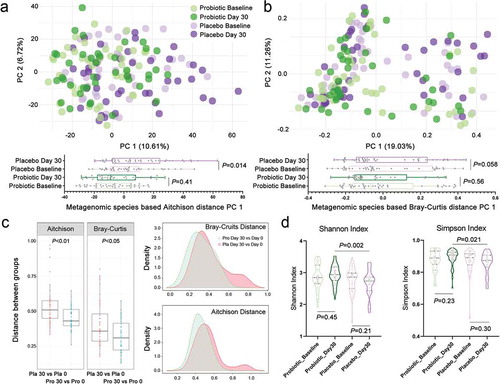
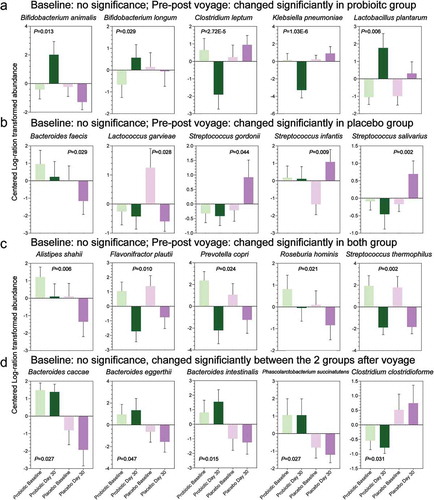
At the taxonomic level ( and S2, Tables S5 and S6), we focused on the intestinal species that had no significant difference between the placebo and the probiotic groups at baseline but changed significantly at the end of the voyage (d 30) in the placebo group. To correct the potential bias introduced by the relative microbial abundance, we performed a centered log-ratio (CLR) transformation of the relative abundance matrix (Table S6). By comparing the species abundance at baseline, we observed that the CLR abundances of the species Bacteroides faecis and Roseburia hominis decreased significantly in sailors of the placebo group at the end of the sea voyage, whereas the CLR abundances of the species Streptococcus infantis, Streptococcus gordonii and Streptococcus salivarius were enriched (,); Wilcoxon rank-sum tests). Meanwhile, we compared the difference in species abundance between the placebo and probiotic groups at the end of the voyage and found that the species Bacteroides caccae, Bacteroides eggerthii, Bacteroides intestinalis and Phascolarctobacterium succinatutens decreased sharply in the placebo group (); Wilcoxon rank-sum tests).
Figure 2. The metagenomic species was not significantly different in the centered log-ratio (CLR)-transformed abundance at baseline between the placebo and probiotic groups but significantly changed at the end of the voyage (d 30) (Wilcoxon rank-sum tests). (a) The species had no significant difference at baseline between the placebo and probiotic groups but changed significantly only in the probiotic group during the sea voyage. (b) The species changed significantly only in the placebo group during the sea voyage. (c) The species changed significantly both in the placebo and probiotic groups during the sea voyage. (d) The species had no significant difference at baseline between the placebo and probiotic groups but changed significantly between the two groups at the end of the voyage.
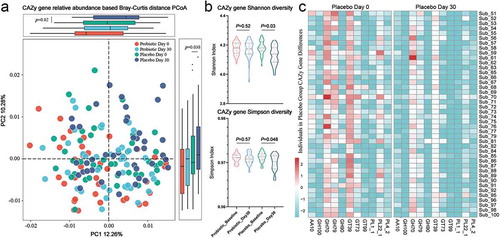
To investigate the observed differences in the functional profiles of the intestinal microbiota during the long sea voyage, high-quality reads from all samples were assembled and annotated for protein-coding genes. Based on the results, a collective, non-redundant intestinal microbiota gene catalog was created. Next, for each sample, the reads were mapped to the collective gene catalog to reconstruct sample-specific gene profiles, and metabolic pathways were also generated with the Kyoto Encyclopedia of Genes and Genomes Orthology database (Tables S7 and S8). PCoA was performed based on the Bray–Curtis distances of the intestinal microbial functional gene profiles (Figure S3), and the specific changes in the microbial metabolic pathway were represented by a decreased toluene degradation ability and ubiquinol and glycogen synthesis ability (). Additionally, a significant shift from the intestinal microbial carbohydrate-active enzyme (CAZy) gene profile (Table S9) based on the Bray–Curtis distances ()) and a sharp decline in the alpha diversity of microbial CAZy genes ()) were observed at the end of the voyage in the placebo group, which were represented by a decrease in the relative abundance of the gene families glycoside hydrolases (GH), glycosyltransferases (GT) and polysaccharide lyases (PL) ()). These results indicated that the long sea voyage not only disordered the balance of the intestinal microbiota of sailors but also reduced the diversity of functional features of intestinal microbiota.
Table 1. The significantly different pathways in the placebo group between d 0 and d 30.
Figure 3. The alteration of the intestinal microbial carbohydrate-active enzyme (CAZy) genes during the long sea voyage. (a) The Bray–Curtis distance-based intestinal microbial CAZy gene profile of sailors during the long sea voyage. The points in different colors represent the intestinal microbial structure of the subjects in each group. A comparison analysis of PC1 and PC2 was visualized with a boxplot (Wilcoxon rank-sum tests). (b) The alpha diversity, including Shannon and Simpson indexes, of the intestinal microbial CAZy genes of sailors both in the placebo and probiotic groups at baseline and at the end of the sea voyage. (c) The heatmap revealed a significant decrease in the relative abundance of CAZy gene families, including glycoside hydrolases (GH), glycosyltransferases (GT) and polysaccharide lyases (PL), in the placebo group during the sea voyage.
Probiotics maintained intestinal microbiome homeostasis during the long sea voyage
Since we observed that the long sea voyage had a significant impact on the intestinal microbiome, we further addressed our second question, whether probiotics can maintain intestinal microbiome homeostasis in sailors during a long trip. The structures of the intestinal microbiota of subjects in the probiotic group were compared (Aitchison and Bray–Curtis distances based on metagenomic species level, ,)), but we did not observe any significant difference, and the P values (Wilcoxon rank-sum tests) of PC1 based on Aitchison and Bray–Curtis distances were all greater than 0.05. Additionally, the microbial Aitchison and Bray–Curtis of sailors in the probiotic group between d 0 and d 30 were significantly less than those of sailors in the placebo group ()). To confirm our observation, we further compared the microbial structure of sailors between the placebo group and the probiotic group on d 0 and d 30. Interestingly, no significant difference was found at baseline between the two groups, but the compositions of the intestinal microbiota of the two groups were highly distinct at the end of the sea voyage, which confirmed the positive impacts of the probiotics consumed. Even though the change in microbial structure was limited, we could also observe some specific changes at the microbial species level. The CLR abundances of the species Bifidobacterium longum, Bifidobacterium animalis and Lactobacillus plantarum increased significantly in sailors at the end of the sea voyage, whereas those of the species Clostridium leptum, Klebsiella pneumoniae and Prevotella copri decreased (); Wilcoxon rank-sum tests). Similarly, the change in the intestinal microbial metabolic pathway was limited in the probiotic group during the sea voyage (), and no significant change was observed in the diversity of microbial CAZy genes (,)). Taken together, these results suggest that the probiotics maintained intestinal microbiome homeostasis during the long sea voyage.
Table 2. The significantly different pathways in the probiotic group between d 0 and d 30.
The potential effective mechanism underlying the interaction between probiotics and intestinal microbes
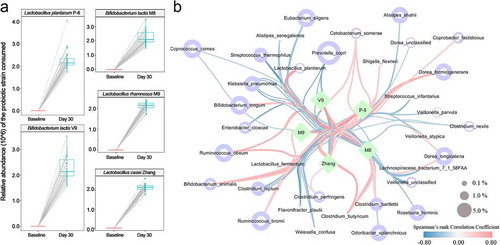
After confirming that the probiotics were able to maintain intestinal microbiome homeostasis during the long sea voyage, we were eager to explore the third question: how do the probiotics work? To address this question, we had to elucidate the correlation and the interaction between the probiotics consumed and the host symbiotic intestinal microbes. Effective persistence in the host gut is crucial for the health-promoting characteristics of consumed robiotics. Accordingly, by mapping to the whole genomes of the consumed probiotics, we calculated the relative abundance of the five strains (), Table S10), and the five probiotics exhibited excellent colonization ability. Then, we further explored the correlations between the consumed probiotics and the other intestinal microbes by constructing a bundle network based on the determined Spearman’s rank correlation coefficients (), Table S11). As shown in ), a generally positive correlation was observed between the strains Lactobacillus casei Zhang, Lactobacillus plantarumP-8, Lactobacillus rhamnosus M9, Bifidobacterium lactis V9 and Bifidobacterium lactis M8 and the species Bifidobacterium longum, Bifidobacterium animalis, Bacteroides eggerthii, Clostridium butyricum, Lactobacillus fermentum and Lactobacillus plantarum, whereas a common negative correlation was observed between the five probiotics and the species Clostridium leptum, Alistipes shahii, Klebsiella pneumoniae, Streptococcus salivarius and Prevotella copri.
Figure 4. The proliferation of the probiotics consumed and their correlation with the intestinal microbes of the host. (a) The proliferation of the probiotics consumed represented the relative abundance (10^6) of the mapped reads, namely, Lactobacillus casei Zhang, Lactobacillus plantarumP-8, Lactobacillus rhamnosus M9, Bifidobacterium lactis V9 and Bifidobacterium lactis M8. (b) The correlation network between the probiotics consumed and the specific metagenomic species. The edge width and color (red, positive; blue, negative) are proportional to the correlation strength by Spearman’s rank correlation coefficient (bundle style). The node size is proportional to the mean abundance in the respective population.
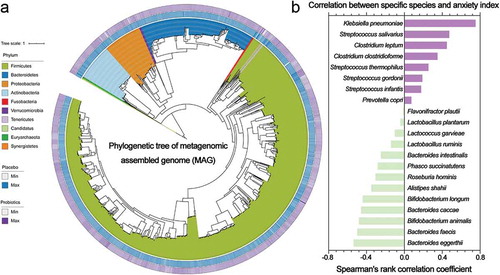
Finally, since we observed the potential role of probiotics in stress and anxiety improvement during a long sea voyage as well as the correlation between the consumed probiotic and specific intestinal microbes, we further explored the relationship between the probiotic-derived changes in intestinal bacterial populations and the anxiety index of the questionnaire results. Using a co-abundance algorithm, we assembled all the shotgun metagenomic sequencing data into 1498 metagenome-assembled genomes (MAGs) (Table S12), assigned specific taxonomic levels and constructed a phylogenetic tree ()). Spearman’s rank correlation coefficient was calculated to quantify the correlation. We observed a significant positive correlation (R > 0.4) between the metagenomic species Klebsiella pneumoniae, Streptococcus salivarius and Clostridium leptum and the anxiety index, whereas the metagenomic species Bifidobacterium longum, Bifidobacterium animalis, Bacteroides eggerthii, Bacteroides caccae and Bacteroides faecis were significantly negatively (R > 0.4) correlated with the anxiety index. In summary, these results indicated that probiotic consumption maintained intestinal microbiome homeostasis and introduced some positive changes in intestinal species, which also prevented sailor anxiety during a long sea voyage.
Figure 5. (a) Phylogenetic tree of the metagenomic assembled genome (MAG) from metagenomic samples. The heatmap of the tree shows the log2-fold change in the relative abundance of the MAGs during the long sea voyage (outer, probiotic group; inner, placebo group). (b) Spearman’s rank correlation coefficient between the probiotic-correlated species and the anxiety index. An R-value greater than 0.4 or less than −0.4 indicated that the correlation was significant.
Discussion
In our present research, we found that the intestinal microbiome of sailors in the placebo group was seriously disordered during a long sea voyage, as indicated by the changes in some important intestinal species and the significant decrease in carbohydrate-active enzyme genes. The rich diversity of host intestinal microbes as well as their CAZy-represented functional genes is the foundation for maintaining intestinal microbiome homeostasis and host fitness.Citation19 It is well known that most of the fiber, polysaccharide and polyphenol in our daily diet cannot be degraded and utilized by our own cells. Carbohydrate-active enzymes encoded by the human gut microbiome catalyze the breakdown of glycoconjugates, oligosaccharides and polysaccharides to fermentable monosaccharides. Therefore, the vast number of CAZy genes in intestinal microbes, which outnumbers that of humans by 100 times, helps us degrade these complex carbohydrates into short chain fatty acids (SCFAs) directly or by a cross-feeding mechanism,Citation20 which improves our gut health fitness. In contrast, a low diversity of intestinal microbes with CAZy-represented functional genes is closely related to the proliferation of pathogenic and conditional pathogens, which leads to various chronic diseases.
We supposed that the human intestinal microbiome was determined mainly by the factors of host genotype, environmental microbes and daily diet.Citation21 The environment and diet, but not the host genotype, changed during the long sea voyage. It has been widely reported that the microbes in the soil are extremely abundant and that the environmental microbes in mainland areas come mainly from the soil,Citation22 but the diversity of microbes at the surface of the sea is great.Citation23 Accordingly, sailors are unable to interact with diverse microbes during sea voyages as they would on the mainland, which may greatly impact the intestinal microbiota of sailors. At the same time, diverse fresh vegetables and fruits are limited during long sea voyages, which may be a second factor explaining the reduction in the diversity of the intestinal microbes.Citation24 Additionally, some studies also reported that gut microbial communities are bidirectionally plastic and resilient across a long stay in the space situational environment or with multiple dietary shifts,Citation25,Citation26 but these studies lasted at least one half year, and they also highlighted significant changes in intestinal microbiota within months.
In the probiotic group, by supplying a mixed probiotic product containing Lactobacillus casei Zhang, Lactobacillus plantarumP-8, Lactobacillus rhamnosus M9, Bifidobacterium lactis V9 and Bifidobacterium lactis M8, we observed that the CLR abundances of the species Bifidobacterium longum, Bifidobacterium animalis and some species belonging to Bacteroides increased significantly in sailors at the end of the sea voyage; however, the CLR abundances of the species Clostridium leptum, Klebsiella pneumoniae and Prevotella copri decreased (); Wilcoxon rank-sum tests). The species Bifidobacterium and Bacteroides encode various CAZy genes that are able to degrade complex carbohydrates into SCFAs.Citation27 The species Bifidobacterium longum carries key genes encoding glycoside hydrolases and carbohydrate transport systems involved in the metabolism of 4-galactosyl-kojibiose and lactulosucrose, and acetic acid is the main metabolic end product followed by lactic and formic acids.Citation28 SCFAs are the main energy source of cells. They can regulate epithelial barrier function and reduce the translocation of bacterial endotoxins across itCitation29 and promote the secretion of mucin, defensin and antibacterial peptides. Accordingly, the consumed probiotic promoted the growth of beneficial microbes and inhibited the proliferation of pathogenic and conditional pathogens, which were essential in maintaining host health by stimulating natural immunity and contributing to the balance of the microbiota.Citation30
The emergence of a microbiota-gut-brain axis to describe the complex networks and relationship between the gastrointestinal microbiota and host reflects the major influence this environment may have in brain health and disorders of the central nervous system (CNS).Citation31 Similar to our finding of intestinal microbial disorder in the placebo group at the end of the voyage, recent evidence also suggested that perturbations of the gut microbial community may play a key role in neuropsychiatric disorders, such as depression and anxiety. However, the potential intestinal species correlated with depression and anxiety reported in different studies were not consistent. For instance, we highlighted the beneficial species Bifidobacterium longum and Bifidobacterium animalis in our research. However, a recent intestinal microbiome study of a generalized anxiety disorder cohort focused on the low prevalence of five genera: Faecalibacterium, Eubacterium rectale, Lachnospira, Butyricicoccus and Sutterella.Citation32 Although we highlighted the different microbes in the intestine, the disorder in intestinal microbiome homeostasis (especially the microbial metabolites) was consistent for individuals who suffered from anxiety. The author also suggested that these genera could be relevant to mental health due to their documented production of short-chain fatty acid compounds.Citation32 Accordingly, the reduced SCFA production in the anxiety population could result in intestinal barrier dysfunction,Citation33 which could compromise proper immune responses and ultimately contribute to brain dysfunction. Meanwhile, we found that probiotic consumption prevented the potential anxiety of sailors during the long sea voyage. Currently, there is considerable evidence suggesting that the intake of probiotics may contribute to decreased anxiety and depression-like behaviors in both human and mouse models. For instance, a probiotic mix of Lactobacillus helveticus and Bifidobacterium longum given to healthy human volunteers for 1 month alleviated psychological distress in comparison to a control group, consistent with the positive results seen in rats.Citation34 This result indicated that there is significant potential for probiotic treatment to maintain intestinal microbiome homeostasis and to prevent anxiety and depression in sailors during long sea voyages.
By performing a longitudinal study containing two cohorts, we draw two main conclusions from the present research. The first is that the long sea voyage not only disordered the balance of the intestinal microbiota of sailors but also reduced the diversity of functional features of intestinal microbiota and increased the stress and anxiety of sailors. The second conclusion was that the probiotics maintained intestinal microbiome homeostasis and further prevented anxiety during the long sea voyage. The results of the present study will provide a feasible approach for protecting gut health during long sea voyages and provide new insights into a personalized selection of probiotics based on population-level analyses.
Materials and methods
Experimental design and subject recruitment
We designed a 30-d longitudinal experiment that included 2 Chinese sailor cohorts in the same warship: a placebo cohort (n= 42) and a probiotic-consuming cohort (n= 40). During the trial, the sailors in the two groups were performing a 30-d cruising task in the Yellow Sea. After being informed of the experimental guidelines and details, the volunteers agreed to participate in the subsequent experiment. All the subjects in the probiotic group were asked to consume a 2 g package containing mixed probiotics including 9.70 Log 10 CFU of Lactobacillus casei Zhang, 9.70 Log 10 CFU of Lactobacillus plantarumP-8, 9.70 Log 10 CFU of Lactobacillus rhamnosus M9, 9.88 Log 10 CFU of Bifidobacterium lactis V9 and 9.88 Log 10 CFU of Bifidobacterium lactis M8 once daily for 30 d. Meanwhile, the subjects in the placebo group were asked to consume a placebo of the same weight and the same taste once daily for 30 d. The probiotic and the placebo formulations (lyophilized cells) were provided by the Key Laboratory of Dairy Biotechnology and Engineering, Ministry of Education, Inner Mongolia Agricultural University. Fecal samples were collected at baseline (d 0) and at the end of the long sea voyage (d 30). An individual health-related questionnaire that included physiological and psychological stress was obtained from every subject at the end of the sea voyage (Table S2). We set each subject’s score for the questionnaire content to a baseline of 10. Therefore, a decreased score represented the unfitness of the subject in the corresponding questionnaire during the voyage. Meanwhile, we recorded the weekly food supply on the cruise, and the menu from Sunday to Saturday was not the same but was repeated every week. The food supply on the cruise was in buffet form. Therefore, the subjects in the placebo and probiotic groups had the same choice of food in every meal. The subjects could eat according to their willingness, but they were suggested to eat at least two kinds of staple food, five kinds of entrées and two to three kinds of fruit every day for their nutritional needs. The study was reviewed and approved by the Ethics Committee of the Inner Mongolia Agricultural University (IMAU-EC-2018017, Hohhot, China), and informed consent was obtained from all volunteers before they enrolled in the study. The participants provided written informed consent to participate in the study. Sampling and all described subsequent steps were conducted in accordance with the approved guidelines. Fecal samples were collected from each subject in the morning before the first meal. After the weight of the fecal samples was determined, a sample protector (CW0592M, CWBIO, China) was added at a ratio of one-part fecal sample to five-part sample protectors, after which the samples were stored at −20°C until further processing.
Shotgun metagenomic sequencing and quality control
All the samples were subjected to shotgun metagenomic sequencing by using an Illumina HiSeq 2500 instrument. Libraries were prepared with a fragment length of approximately 300 bp. Paired-end reads were generated using 100 bp in the forward and reverse directions. The reads were trimmed using Sickle and were subsequently aligned to the human genome to remove the host DNA fragments. An average of 7.51 gigabases (Gb) of high-quality paired-end reads was obtained for each sample, totaling 1232.42 Gb of high-quality data that were free of human DNA and adaptor contaminants (Table S3).
Non-redundant gene catalog construction and calculation of gene abundance
The shotgun reads were assembled into contigs and scaffolds using IDBA-UD,Citation35 and then the contigs were used to predict the functional genes with MetaGeneMark.Citation36 Finally, a non-redundant gene catalog was constructed using CD-HIT.Citation37
The abundances of genes were determined by aligning the reads to the gene catalog using Bowtie 2.Citation38 Subsequently, for any sample N, we calculated the abundance as follows:
Step 1: Calculation of the copy number of each gene:
Step 2: Calculation of the relative abundance of gene i
ai: the relative abundance of gene i
bi: the copy number of gene i from sample N
Li: the length of gene i
xi: the number of mapped reads
Metagenomic assembled genome (MAG)
The microbial taxonomic profile was constructed by the software MetaPhlAn2.Citation39 Moreover, because the whole genomes of the 5 probiotics in the present study were sequenced, we could easily annotate and calculate the relative abundances of these probiotics in samples on d 30 by bowtie2. For metagenomic species analysis, the software MetaBATCitation40 was applied, and the co-abundance principle and canopy clustering algorithm were performed to generate MAGs by binning shotgun reads. After reassembling, the MAGs were assigned to a given genome when more than 80% of the subgene matched the same genome using BLASTn at a threshold of 95% identity over 90% of the gene length. If greater than 80% of the genes from an MAG had the same taxonomic level of assignment, then MGS was identified as the same microbe.
The centered log-ratio (CLR) of microbial relative abundance and the microbial Aitchison distance
It was debatable by using the microbial relative abundance in microbiome research. The total microbial load or absolute copy gene number of the whole microbes should be the efficient methods. Here, we performed a widely used centered log ratio of microbial relative abundance to verify our analysis results.Citation41 The “zcomposition” package in the R program was used to resolve the value of 0, and the “composition” package in R was applied to realize the relative abundance profile CLR transformation. The Aitchison distance could be directly calculated from the Euclidean distance of the CLR-transformed data.
Functional annotation and metabolic pathway analysis
The annotated amino acid sequences were aligned against the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases using BLASTp (e-value≤1e-5 with a bit-score higher than 60). The annotated sequences were assigned to the KEGG orthologue group (KO) according to the highest score. CAZymes (carbohydrate-active enzymes)Citation42 were predicted from amino acid sequences by generating alignments with family-specific HMMs of CAZymes in the dbCAN database using the program Hmmscan in the HMMER 3.0 package.Citation43 Reporter Z-scores were calculated to reveal the differences in enriched metabolic pathways between the control and PCOS groups, as previously described. Accordingly, a reporter score of > 2.3 (90% confidence according to the normal distribution) was used as a detection threshold to significantly differentiate between pathways.Citation13
Statistical analysis
All statistical analyses were performed using R software. PCA analysis was performed in R using the ade4Citation44 package. The differential abundances of genera, genes and KOs were tested with the Wilcoxon rank-sum test and were considered significantly different at p < .05. For boxplot construction, the package ggpubr was used. The heatmap was constructed using the “pheatmap” package, and the Sankey diagram was built using the “riverplot” package. The CLR transformation was realized using the “composition” package. The networks were calculated by the Spearman rank correlation coefficient and visualized by networks in Cytoscape (Version 3.7.1).
Data and code availability
The sequence data reported in this paper have been deposited in the NCBI database (metagenomic sequencing data: PRJNA554501). All analyses can be found under https://github.com/zhjch321123/long-sea-voyage-program.git.
Disclosure of potential conflicts of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The study was reviewed and approved by the Ethics Committee of the Inner Mongolia Agricultural University (IMAU-EC-2018017, Hohhot, China), and informed consent was obtained from all volunteers before they enrolled in the study.
Supplemental Material
Download MS Excel (3.7 MB)Supplemental Material
Download PDF (793.6 KB)Acknowledgments
We sincerely thank all the volunteers for their participation.
Supplementary materials
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Rydstedt LW, Lundh M. An ocean of stress? The relationship between psychosocial workload and mental strain among engine officers in the Swedish merchant fleet. Int Marit Health. 2010;62:168–175.
- O’Halloran CL, Silver MW, Colford JM Jr. Acute stress symptoms among US ocean lifeguards. Wilderness Environ Med. 2015;26:442–443. doi:10.1016/j.wem.2015.02.010.
- Carel RS, Carmil D, Keinan G. Occupational stress and well-being: do seafarers harbor more health problems than people on the shore? Isr J Med Sci. 1990;26:619–624.
- Elo AL. Health and stress of seafarers. Scand J Work Environ Health. 1985;11:427–432. doi:10.5271/sjweh.2204.
- Xie S, Lin H, Meng Y, Zhu J, Zhang Y, Zhang L, Li G. Analysis and determinants of Chinese navy personnel health status: a cross-sectional study. Health Qual Life Outcomes. 2018;16:138. doi:10.1186/s12955-018-0961-4.
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. Strains, functions and dynamics in the expanded human microbiome project. Nature. 2017;550:61–66. doi:10.1038/nature23889.
- Zheng W, Zhang Z, Liu C, Qiao Y, Zhou D, Qu J, An H, Xiong M, Zhu Z, Zhao X, et al. Metagenomic sequencing reveals altered metabolic pathways in the oral microbiota of sailors during a long sea voyage. Sci Rep. 2015;5:9131. doi:10.1038/srep09131.
- Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506–514. doi:10.1038/nrgastro.2014.66.
- Derrien M, van Hylckama Vlieg JE. Fate, activity, and impact of ingested bacteria within the human gut microbiota. Trends Microbiol. 2015;23:354–366. doi:10.1016/j.tim.2015.03.002.
- Sanchez B, Delgado S, Blanco-Miguez A, Lourenco A, Gueimonde M, Margolles A. Probiotics, gut microbiota, and their influence on host health and disease. Mol Nutr Food Res. 2017;61:1600240. doi:10.1002/mnfr.v61.1.
- Shin JH, Nam MH, Lee H, Lee JS, Kim H, Chung MJ, Seo JG, et al. Amelioration of obesity-related characteristics by a probiotic formulation in a high-fat diet-induced obese rat model. Eur J Nutr. 2018;57:2081–2090. doi:10.1007/s00394-017-1481-4.
- Xu H, Huang W, Hou Q, Kwok LY, Laga W, Wang Y, Ma H, Sun Z, Zhang H. Oral administration of compound probiotics improved canine feed intake, weight gain, immunity and intestinal microbiota. Front Immunol. 2019;10:666. doi:10.3389/fimmu.2019.00666.
- Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. doi:10.1038/nature13568.
- Liu TX, Niu HT, Zhang SY. Intestinal Microbiota Metabolism and Atherosclerosis. Chin Med J (Engl). 2015;128:2805–2811. doi:10.4103/0366-6999.167362.
- Nallu A, Sharma S, Ramezani A, Muralidharan J, Raj D. Gut microbiome in chronic kidney disease: challenges and opportunities. Transl Res. 2017;179:24–37. doi:10.1016/j.trsl.2016.04.007.
- Wlodarska M, Kostic AD, Xavier RJ. An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe. 2015;17:577–591. doi:10.1016/j.chom.2015.04.008.
- Zhang J, Sun Z, Jiang S, Bai X, Ma C, Peng Q, Chen K, Chang H, Fang T, Zhang H, et al. Probiotic Bifidobacterium lactis V9 regulates the secretion of sex hormones in polycystic ovary syndrome patients through the gut-brain Axis. mSystems. 2019;4. doi:10.1128/mSystems.00017-19
- Zhang J, Wang L, Guo Z, Sun Z, Gesudu Q, Kwok L, Zhang H. 454 pyrosequencing reveals changes in the faecal microbiota of adults consuming Lactobacillus casei Zhang. FEMS Microbiol Ecol. 2014;88:612–622. doi:10.1111/fem.2014.88.issue-3.
- Kriss M, Hazleton KZ, Nusbacher NM, Martin CG, Lozupone CA. Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. Curr Opin Microbiol. 2018;44:34–40. doi:10.1016/j.mib.2018.07.003.
- So D, Whelan K, Rossi M, Morrison M, Holtmann G, Kelly JT, Shanahan ER, Staudacher HM, Campbell KL. Dietary fiber intervention on gut microbiota composition in healthy adults: a systematic review and meta-analysis. Am J Clin Nutr. 2018;107:965–983. doi:10.1093/ajcn/nqy041.
- Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. Cross-species comparisons of host genetic associations with the microbiome. Science. 2016;352:532–535. doi:10.1126/science.aad9379.
- Fierer N. Embracing the unknown: disentangling the complexities of the soil microbiome. Nat Rev Microbiol. 2017;15:579–590. doi:10.1038/nrmicro.2017.87.
- Moran MA. The global ocean microbiome. Science. 2015;350:aac8455. doi:10.1126/science.aac8455.
- Fang S, Evans RM. Microbiology: wealth management in the gut. Nature. 2013;500:538–539. doi:10.1038/500538a.
- Turroni S, Rampelli S, Biagi E, Consolandi C, Severgnini M, Peano C, Quercia S, Soverini M, Carbonero FG, Bianconi G, et al. Temporal dynamics of the gut microbiota in people sharing a confined environment, a 520-day ground-based space simulation, MARS500. Microbiome. 2017;5:39. doi:10.1186/s40168-017-0256-8.
- Liu H, Han M, Li SC, Tan G, Sun S, Hu Z, Yang P, Wang R, Liu Y, Chen F, et al. Resilience of human gut microbial communities for the long stay with multiple dietary shifts. Gut. 2019;68:2254–2255. doi:10.1136/gutjnl-2018-317298.
- Ndeh D, Gilbert HJ. Biochemistry of complex glycan depolymerisation by the human gut microbiota. FEMS Microbiol Rev. 2018;42:146–164. doi:10.1093/femsre/fuy002.
- Ruiz-Aceituno L, Esteban-Torres M, James K, Moreno FJ, van Sinderen D. Metabolism of biosynthetic oligosaccharides by human-derived Bifidobacterium breve UCC2003 and Bifidobacterium longum NCIMB 8809. Int J Food Microbiol. 2019;316:108476. doi:10.1016/j.ijfoodmicro.2019.108476.
- Turroni F, Milani C, Duranti S, Mahony J, van Sinderen D, Ventura M. Glycan utilization and cross-feeding activities by bifidobacteria. Trends Microbiol. 2018;26:339–350. doi:10.1016/j.tim.2017.10.001.
- Turroni F, Milani C, Duranti S, Mancabelli L, Mangifesta M, Viappiani A, Lugli GA, Ferrario C, Gioiosa L, Ferrarini A, et al. Deciphering bifidobacterial-mediated metabolic interactions and their impact on gut microbiota by a multi-omics approach. Isme J. 2016;10:1656–1668. doi:10.1038/ismej.2015.236.
- Rieder R, Wisniewski PJ, Alderman BL, Campbell SC. Microbes and mental health: A review. Brain Behav Immun. 2017;66:9–17. doi:10.1016/j.bbi.2017.01.016.
- Jiang HY, Zhang X, Yu ZH, Zhang Z, Deng M, Zhao JH, Ruan B. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130–136. doi:10.1016/j.jpsychires.2018.07.007.
- Morris G, Berk M, Carvalho A, Caso JR, Sanz Y, Walder K, Maes M. The role of the microbial metabolites including tryptophan catabolites and short chain fatty acids in the pathophysiology of immune-inflammatory and neuroimmune disease. Mol Neurobiol. 2017;54:4432–4451. doi:10.1007/s12035-016-0004-2.
- Messaoudi M, Lalonde R, Violle N, Javelot H, Desor D, Nejdi A, Bisson J-F, Rougeot C, Pichelin M, Cazaubiel M, et al. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br J Nutr. 2011;105:755–764. doi:10.1017/S0007114510004319.
- Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi:10.1093/bioinformatics/bts174.
- Zhu WH, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38:e132. doi:10.1093/nar/gkq275.
- Li WZ, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi:10.1093/bioinformatics/btl158.
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi:10.1186/gb-2009-10-3-r25.
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–903. doi:10.1038/nmeth.3589.
- Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165. doi:10.7717/peerj.1165.
- Martino C, Morton JT, Marotz CA, Thompson LR, Tripathi A, Knight R, Zengler K. A novel sparse compositional technique reveals microbial perturbations. mSystems. 2019;4. doi:10.1128/mSystems.00016-19.
- Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–51. doi:10.1093/nar/gks479.
- Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inf. 2009;23:205–211.
- Zapala MA, Schork NJ. Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc Natl Acad Sci U S A. 2006;103:19430–19435. doi:10.1073/pnas.0609333103.