
ABSTRACT
The gut microbiome has emerged as a contributing factor in non-communicable disease, rendering it a target of health-promoting interventions. Yet current understanding of the host-microbiome dynamic is insufficient to predict the variation in intervention outcomes across individuals. We explore the mechanisms that underpin the gut bacterial ecosystem and highlight how a more complete understanding of this ecology will enable improved intervention outcomes. This ecology varies within the gut over space and time. Interventions disrupt these processes, with cascading consequences throughout the ecosystem. In vivo studies cannot isolate and probe these processes at the required spatiotemporal resolutions, and in vitro studies lack the representative complexity required. However, we highlight that, together, both approaches can inform in silico models that integrate cellular-level dynamics, can extrapolate to explain bacterial community outcomes, permit experimentation and observation over ecological processes at high spatiotemporal resolution, and can serve as predictive platforms on which to prototype interventions. Thus, it is a concerted integration of these techniques that will enable rational targeted manipulations of the gut ecosystem.
Introduction
A growing body of evidence implicates the gut ‘microbiome’, the complex ecosystem comprising the human gut and the microorganisms inhabiting it, as a contributing factor in the etiology of non-communicable diseases,1–Citation8 thus positioning it as a potential therapeutic target. For instance, diet readily modulates the gut microbiome and could thus be used to intervene in microbiome–host interactions. However, whilst broad modulators of microbiome composition and metabolism are known, inter-individual variations complicate the design and targeting of beneficial interventions.Citation9,Citation10 People are unique organisms harboring individualized microbiomes, and their diverging intervention outcomes stem from variation in gut ecosystem constituents and processes. However, comprehensive understanding of the gut ecosystem is presently lacking and difficult to obtain. In vivo study lacks the necessary spatiotemporal sampling and observation capacity, and in vitro models cannot recapitulate the host’s complexity. Yet, they offer complementary perspectives, informing both mechanistic understanding of the microbiome’s influence on host health and cell-specific models. We propose that, by encapsulating this information, in silico models that enable the experimentation, insight, and predictive capacity needed to rationally design and target interventions are now possible. Whilst we focus primarily on bacterial taxa, as the most abundant and most studied portion of the microbiome, we note that archaea,Citation7 eukaryotes,Citation8,Citation11,Citation12 and virusesCitation6 (e.g. phages) that co-inhabit the gut are gaining attention and are being found to also influence host health status.
The microbiome’s impact on health outcomes is an emergent property manifesting from the collective activity of trillions of individual microbial cells vying for survival within their local gut environments (). Interventions alter cell local environments and drive changes in cell behavior. These behavioral changes cascade through the community, reconfiguring strain niches and fitness, intercellular interactions, community metabolic output, and ultimately the functional responses of the host. The host integrates these signals, and its responses feedback on local microbial environments and alter selective pressures. Thus, host and microbial processes are intertwined and co-responsive. Given such interactive complexity, individualized and diverging responses to intervention are to be expected. Importantly, reasoning about the microbiome at the level of the strain (and thus cell) and it’s co-possession of numerous genes and metabolic pathways is essential: genes (and metabolic pathways) are only expressed when housed within viable organisms, and cell viability spans several nutritional requirements that involve crosstalk (coordination) between multiple metabolic pathways. Environmental perturbations need only limit a cell’s access to one nutrient to impact all its metabolic activities. As such, analyzing the microbiome through genes alone, independently of one another and the strains co-possessing them, will be of limited insight.
Figure 1. Health outcomes emerge from individual microbes and their survival strategies. Nutrient availability, primarily dietary, but also host secretions, drive microbes to regulate their metabolic capabilities to survive. Individual microbes adapting to their nutritional environment reconfigures the gut microbiome metabolic network and community-level metabolic output. The resultant changes can impact on host health
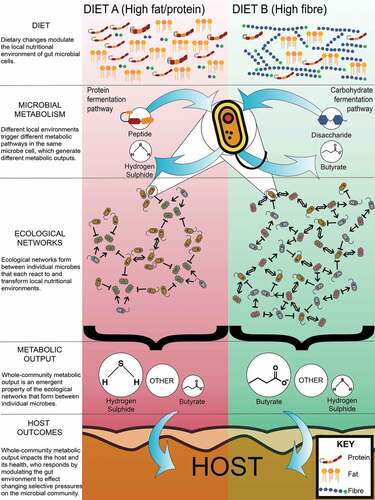
Two compositionally and functionally distinct microbiomes will adapt differently to a changing post-intervention environment and each host will respond differently to that adaptative process. Hence, to maximize effectiveness, interventions must be tailored to the individual. This ultimately entails (1) capturing the range of behaviors (‘dynamics’) each microbial strain in a community can exhibit across various environmental (including nutritional) contexts; (2) integrating these dynamics to extrapolate the resultant community-level outputs that can impact on host health; and (3) understanding host–microbiome interactions. Here, we consider how this can be achieved through an integration of modalities for studying the host-microbiome system.
In vivo studies associate host health outcomes with strains and molecular products, outcomes that originate from individual microbe-level behaviors. These factors form targets for control through intervention. In vitro culturing studies can reveal how specific strains respond to, and in turn modify, their environments. Understanding how and why interventions alter community-level emergent properties is not possible without detailed characterization of individual strain response dynamics. In silico modeling techniques deliver the integrative perspective of how cell-level behaviors scale up to the community-level phenomena that drive health outcomes. Models encode cell-level behaviors and replicate environment conditions, and then reveal spatiotemporal community outcomes.
In vivo studies integrate whole gut ecosystem processes to highlight intervention targets
Non-communicable diseases are host organism-level properties manifesting, in part, from how microbiome and host interact to shape one another.Citation13 In vivo study outcomes reflect an implicit integration of these factors. Of particular relevance is microbiome community composition. Host disease status has been associated with microbiome structural qualities including variation in strain relative abundancesCitation14–19 and/or microbial community diversity.Citation1–5,Citation19,Citation20 Effects have occasionally been ascribed to individual strains: Christensenella minuta was found enriched in lean twins relative to their obese siblings, and inoculation of this strain into “obese” microbiomes transferred into mice reduced subsequent adiposity gains in recipient animals.Citation21 However, isolating effects to specific microbial strains is difficult because strains overlap considerably in function. Yet it is pertinent to account for the effects of all strains, as an intervention may have no effect in a community if suppression of one strain elevates another of similar functional capacity.
Microbial community composition and metabolic output result from the growth substrates microbes have access to, and these primarily originate from host diet. Microbiomes shaped under high-fat and/or sugar diets have been associated with the etiology of diseases including diabetes,Citation22 obesity,Citation22 and hypertension.Citation23,Citation24 Conversely, dietary fiber consumption is associated with host health benefits,Citation25–27 in part due to microbial fermentation of fiber into short chain fatty acids (SCFA).Citation28 The direct dietary administration of SCFAs has conferred protection against induced colitis,Citation29 food allergies,Citation30 asthma,Citation31 and diabetesCitation32 in mice. However, associating diet–microbiome interactions with health outcomes is complicated. Firstly, microbial metabolic outputs vary with context. Substituting fiber with protein can increase microbial generation of pro-inflammatory metabolites, including hydrogen sulfide,Citation33,Citation34 ammonia,Citation35 and phenolic compounds.Citation36,Citation37 Yet, protein fermentation can also lead to the generation of anti-inflammatory compounds such as butyrateCitation38 and the polyamine agmatine.Citation39 Secondly, diet is compositional: a high fat diet necessitates low protein and/or carbohydrate, so which nutrient confers a given effect? Systematic variation is required for parsing of effects. One such study showed interactive effects of macronutrients on microbiome community composition which in turn corresponded with host immunometabolism and body composition status.Citation40 Dietary components are readily labeled as deleterious or beneficial to health, but in actuality effects are non-linear and wider context matters, yet this is difficult to account for.
Beyond diet, host-derived molecules including bile acids and mucus glycoproteins (mucins) secreted into the gut also impact the microbiome.Citation41,Citation42 Mucins shape mucosal microbiome communities as both a growth substrateCitation43 and anchoring matrixCitation44 for select commensals. For instance, Akkermansia municiphila and Bacteroides thetaiotaomicron can adhere to and hydrolyze mucin glycans and thus competitively colonize the gut mucosa.Citation44–47 The result is that luminal and mucosal communities are compositionallyCitation48 and metabolically distinct,Citation49 and as such these communities have different impacts on host health. The microbiome also metabolizes the primary bile acids cholic and chenodeoxycholic acids into numerous secondary bile acids that can actively regulate bacterial populationsCitation50,Citation51 at phylum-Citation50 and strain-levels.Citation52 Together, host diets and endogenous secretions interact in shaping the microbiome, and rational intervention design should account for both, lest they present as confounding factors.
Targeted intervention design requires that we account for the effects of luminal and mucosal-associated strains on host health, and the various interacting dietary and host processes that shape the microbiome. Community and gut environmental contexts dictate which molecular products are produced, and what their contributions to health status are. In vivo study caries the advantage of integrating all such relevant factors in revealing host outcomes. Yet isolating specific causative factors is admittedly challenging as health status is rarely attributable to singular molecules and strains.
Intervention targets are multiple, with broad host-microbiome molecular exchanges impacting health
In vivo research has revealed the complexity of the host–microbiome signal exchange that modulates health outcomes, which interventions should seek to manipulate. For instance, whilst the SCFA butyrate, as a primary colonocyte energy source, is protective against colorectal cancer,Citation53,Citation54 it also delays wound repair during overt inflammation or damage to the intestinal mucosa.Citation55 Hydrogen sulfide, derived from bacterial metabolism,Citation56 can accelerate the ulcer healing process observed in colitis,Citation57 but also upregulate colorectal cancer cell division when compared to regular colonocytes.Citation58 Single metabolites are not ubiquitously deleterious or beneficial. Again, the broader context matters, and targeted interventions must account for this.
Microbe-associated molecular patterns (MAMPs) are cell structural components that influence host inflammatory status. Classic examples include peptidoglycan, lipopolysaccharide (LPS), and flagellin. These molecules are structurally complex and can vary considerably between strains.Citation59–61 Variants can modulate both innateCitation62 and adaptiveCitation63 arms of the immune system to both pro-inflammatory and tolerogenic effects.Citation64–66 For instance, LPS has been linked to the onset of obesity, diabetes, and cardiovascular disease.Citation67–69 Yet, Escherichia coli-derived LPS decreased autoimmune diabetes incidence in animal models relative to Bacteroides-derived LPS.Citation70 Thus, immune tone and disease status are a consequence of the balance of pro-and anti-inflammatory molecular signals.Citation71 Interventions should seek to target not only single MAMPs but shape the holistic profile of molecules generated, and this requires understanding of the ecology through which community composition arises.
The host responds to microbiome signals to maintain homeostasis, but tipping points exist beyond which microbiome–host interactions devolve into perpetuating inflammation that compromises gut barrier function.Citation72 A high-fat diet can induce this state by promoting overgrowth of gut bacteria with consequential over-production of pro-inflammatory cytokines by the host.Citation73 Localized inflammation increases gut permeability, a condition known as leaky gut,Citation74 followed by translocation of cytokines and MAMPs to portal circulation. Characterizing the boundaries at which tipping points transit between self-stabilizing beneficial and self-stabilizing deleterious host–microbiome interactions is critical, given interventions that seek to shift the ecosystem from one state into another.
Lastly, host–microbiome interactions vary both temporally and spatially within the gut. Being in closer proximity to host tissue, mucosa-associated strains interact more directly with the host, potentially exerting different and stronger pressures relative to those of luminal strains.Citation49 Accounting for this mucosal gut microbiome is relevant; however, mucosa-associated strains are underrepresented in fecal samples, and this could distort the relationships inferred between observed microbial communities and their consequences on host health. Further, both microbial populationsCitation75,Citation76 and environmental conditionsCitation77,Citation78 are distributed heterogeneously along the colon, and undergo functional and compositional diurnal oscillationsCitation79 in part reflecting host feeding patterns.Citation42,Citation80 Detailed spatiotemporal characterization of such patterns is highly invasive and thus infeasible in vivo, but such heterogeneity is highly relevant to host healthfor example, the localized inflammatory regions characterizing Crohn’s disease.
In summary, in vivo studies reveal system tipping points whose breech negatively impacts the host. The profile of molecular interactions between host and microbiome underlie these phenomena but are spatiotemporally variable. Further, no single molecule is uniformly beneficial or deleterious: context is key. Controlling these interactions is non-trivial as microbiome molecular output stems from community-wide ecological interactions, and these are difficult to study in vivo.
Complex community ecology underlies microbiome molecular output
Fermentative microbes have evolved a spectrum of survival strategies that rely on shorter/incomplete metabolic pathwaysCitation81 that output metabolites such as SCFAs,Citation81–83 that other microbes can further metabolize. A whole community engaging in such strategies yields complex inter-microbial dependencies. The profile of metabolites produced is difficult to forecast as it is sensitive to interactions among the microbial cells present, along with host diet and endogenous secretions. For instance, hydrogen sulfide can result from metabolism of endogenous and dietary cysteine and bile acids, thus its production depends on dietary proteinCitation84 and fat consumption,Citation33 and the presence of sulfur-reducing organisms including Clostridium and Enterobacter.Citation33,Citation34
The capacity to generate most metabolites is widely distributed among phylogenetically diverse strains, and such redundancy further complicates metabolite output forecasting as has been observed for butyrate.Citation85,Citation86 Over 25% of strains, spanning several phyla, in a given individual’s microbiome can generate butyrateCitation87 and four major butyrate production pathways with distinct substrates exist.Citation88 Further illustrating the consequences of such redundancy, microbiomes sampled from three individuals varied in the profile of SCFAs produced when cultured using identical growth media.Citation89 Thus, even in highly controlled culture conditions, a community’s possession of multiple pathways for generating a metabolite, and numerous strains supporting each pathway, render output prediction difficult.
Importantly, a strain’s presence and capacity for generating a given metabolite does not necessitate its actual engagement in the activity. Nutrient limitation will impact a cell’s capacity to grow. The nutritional environment is, in turn, modulated by host diet, which substrates metabolically active strains are presently consuming, and which intermediate metabolites they produce. Many microorganisms possess diverse strategies for satisfying their nutritional requirements. For example, Bacteroides thetaiotaomicron can degrade a wide range of complex dietary carbohydratesCitation90 and host-derived glycansCitation91 rendering it adaptive to varying nutritional contexts.Citation92 Thus, accurate prediction of community composition and metabolic output necessitates accounting for cell nutritional (and thus growth) statuses, their nutritional context and how they modify it.
Any intervention that modulates a strain’s growth dynamics, in terms of prevalence and output, has the potential for cascading effects through the microbial community. Nutrient availability and competition dynamics shift, as will the profile of metabolites contributed back into the environment (). These manifest as localized effects but can permeate more broadly through time and space. To rationally design interventions with predictable outcomes requires these ecological processes be characterized and their integrative consequences understood. This is extremely difficult to achieve in the real world, but possible in silico with support from in vitro studies that comprehensively map out strain-level growth dynamics.
Comprehensive mapping of strain growth dynamics through in vitro study
The growth dynamics of individual strains, and their interactions with one another and specific host processes, can be characterized in detail outside of the living host. Environments reflecting locales in the gut can be imposed, controlling for nutritional context, water content, pH and selective inclusion of particular host factors. Strain growth rates, substrate utilization, and metabolic output can be quantified. In vitro technologies afford a comprehensive mapping of how given strains respond to changing environments. Thus, mucin and high pH were identified as fundamental requirements for A. muciniphila colonization, explaining its prevalence in the distal colon.Citation93,Citation94 Capturing strain growth requirements is essential for intervention design, as they determine whether or not a particular strain can grow given a particular nutrient context, with downstream implications for community composition and metabolic output.
How strains adapt their behaviors under changing environments likewise influences intervention outcomes. Microbial substrate preferences can impact competition dynamics and have been uncovered. For instance, B. thetaiotaomicron prioritizes mannose over other monosaccharides,Citation95 and plant-derived polysaccharides over mucin carbohydrates.Citation96 Similarly, lactate-utilizing bacteria prefer glucose over lactate when both are available.Citation97 These substrate preferences are often strain-specific,Citation98 reflecting differences in metabolic pathways. Strain metabolic output also differs with environmental context. Certain bacteria can generate formate, a common intermediate metabolite, but under specific pH conditions the same microorganisms can further metabolize it and generate gaseous hydrogen instead.Citation99 In situations where formate-producing bacteria are paired with hydrogen-dependent microbes such as Blautia hydrogenotrophica, formate is also further metabolized to acetate and energy harvest is maximized.Citation100 Even in pure culture, the relationship between the factors of substrate availability, cell capability/attributes and the outcome of net metabolite production is complex since they result from multiple causal (and interfering) pathways that vary over time.
Intricate metabolic interactions take place between gut microbes. Diverse ecological relationships, such as cross-feeding (mutualism), amensalism, and competition coexist in the gut to shape the microbiome and its metabolic output. Cross-feeding has been extensively studied owing to its scope for expanding a strain’s growth niche. For instance, in co-culture, Ruminococcus bromii produces formate which B. hydrogenotrophica consumes, and B. hydrogenotrophica reciprocates with panthothenate which R. bromii requires for optimal growth but cannot synthesize.Citation100 This relationship also involves hydrogen utilization by B. hydrogenotrophica as described above. Concomitantly, R. bromii and B. hydrogenotrophica compete for the vitamin thiamine in this context, exemplifying how complex interactions in the gut can be. Microbes form a complex web of interactions and variations in microbial membership or activity can trigger a cascade of effects across the network. The units of biological activity in a community are not necessarily cells or strains – syntrophic dependencies mean multi-organism networks are more appropriate for modeling some outputs.
In vitro technological advancements are also facilitating detailed investigation of isolated host–microbiome interaction pathways. These methods cannot fully recapitulate the host but can elucidate the ‘behavioral building blocks’ from which whole-scale host–microbiome interactions emerge. Cell culture models employ specific cell lines to replicate the intestinal epithelium,Citation101 capturing processes including mucin excretion, cell migration, and signaling. This has enabled study of bacterial adherence to epithelial cells and the former’s response to cytokine generation.Citation102 Microfluidic technologies combined with cellular culturing have enabled the study of how peristalsis and intraluminal flow impact the gut microbiome.Citation103,Citation104 Multi-stage continuous fermentation models reveal spatiotemporal dynamics, e.g. that the impact of a dietary intervention was not equally distributed along the gut.Citation105 For instance, the Simulator of the Human Intestinal Microbial Ecosystem (SHINE), a modular multi-stage continuous fermentation model, was extended to study mucosa-adherent microbiome dynamics (M-SHINE), specifically the recovery of Lactobacillus strains following an antibiotic pulse.Citation106 Microbial-host interactions fundamentally shape microbial behaviors and in vitro approaches that trace and objectively measure them are important to rational intervention development. Advances in techniques to culture cells and measure physiology mean it is possible to effectively describe the range of cell states in response to variation in physio-chemical context (at least in much more detail than previously).Citation107
These in vitro approaches represent ‘reductionist’ science: isolating and manipulating pathways and microbes to probe their dynamics. They offer insights that cannot be accurately generated through other means, but they also face limitations of scale. Complete characterization of strain dynamics necessitates wide, systematic exploration of environmental contexts and strain responses. It is impractical, even with advances in modern robotic fermentation systems, to conduct such broad experimentation for each of the thousands of strains occupying the gut. When compounded with the range of unique consortia that can be assembled and investigated, the experimental burden is staggering. For instance, to map the growth requirements around the eight B vitamins would entail 255 unique inclusion/exclusion combinations, which if applied to 400 strains (approximate richness of the human microbiomeCitation108) entails 102,000 cultures. A second key challenge is one of complexity; comprehensive characterization of strain dynamics is vital to understanding the principles of community organization and function, but there remains a sizable leap in scaling from these strain-level dynamics to predicting community outcomes. Reassembling and integrating these pathways to study how they deliver emergent behaviors and system-wide consequences is a ‘constructionist’ task. In silico techniques, fusing biological data with modeling, can accomplish this.
In silico models: from individual microbes to community outcomes
In silico modeling relies on computer algorithms that recapitulate microbial and host dynamics. Such models integrate data from different modes of study and at varying biological scales, spanning metabolites, cells, communities, and the entire gut ecosystem with the aim of reproducing outcomes observed in real-world systems. In so doing, they seek to explain these emergent outcomes as manifesting from individual molecule- and cell-level dynamics. Validated models can be tremendously insightful: being computer code, they present no limits for intervention or spatiotemporal observation scope. Putative targets for intervention can be rapidly and systematically explored for their effects. Models can be used in a predictive capacity, exploring possible interventions to achieve a desired outcome.
Models can aid understanding of a system’s operation, elucidating the fundamental mechanistic principles underpinning the system through simplification to the most essential components and their interactions. Simplifying can mean modeling a subset of the full system, and can entail amalgamating distinct components (cells, molecules, pathways) with similar function or that constitute a module of given function (e.g. multi-organism networks) into a single-component types.Citation109
Agent-based modeling (ABM) is one such ‘abstractive’ technology. ABMs can capture spatially explicit, heterogeneous environments occupied by discrete, dynamic agents whose interactions give rise to complex emergent outcomes.Citation109 Modeled agents encode behavioral responses to environmental stimuli and interactions with one another which are dependent on an agent’s state: the result of its past experiences. With microbiome phylogenetic diversity exceeding functional diversity,Citation110 findings reported in terms of phylogenetic taxonomies have become increasingly complicated to interpret.Citation111 Interpretation through functional units and their interactions may prove more tractable and informative. ABM was used to abstract the gut microbiome into six trophic guilds that represented distinct nutritional strategies,Citation40 thus distilling hundreds of stains into far fewer and functionally distinct terms. This model demonstrated that microbial strategies for acquiring nitrogen from either dietary or endogenous sources could explain the variation in community composition observed across a broad range of mouse diets. The model revealed ecosystem dynamics, explaining changes in community composition in terms of which nutrients were limiting the growth of each microbial cell; those cells’ nutritional environments as resulting from diet, host digestion, and endogenous substrate provision; and how effectively cells were competing for nutrients. Such mechanistic insight would have proven difficult to obtain through the lenses of phylogenetic and gene cataloging. This study also exemplifies the high-throughput of in silico modeling: 250 ‘mice’ administered varying diets were readily simulated within hours, whereas the corresponding in vivo work was a huge undertaking spanning many months.Citation40 By offering unparalleled tracing of spatiotemporal dynamics, ABMs have shown how modifications in host epithelial secretions shift the microbial communityCitation112 and how isolated and aggregated feedback mechanisms (toxin-antitoxin, substrate sharing and antibiotic resistance dynamics) impacted gut microbiome resilience to common stressors (e.g. antibiotic therapy).Citation113 These insights would have proven complicated to extract from real-world observations. Thus, ABMs can trace microbial dynamics to reveal the governing ecological principles at the single-cell level and what their community-level consequences are.
Not all modeling approaches strive for simplification, and models emphasizing capture of real-world detail seek to quantitatively explain real-world phenomena in those same terms. Genome Scale Models (GSMs) are one such technology. They are reconstructions of a cell’s genome-encoded metabolic pathways and networks,Citation114 and simulations of communities are emerging.Citation115 A common GSM application is to estimate a configuration of ‘fluxes’ (throughputs) through metabolic pathways that maximize organism growth rate(s) in a given metabolic environment: ‘flux balance analysis’.Citation116,Citation117 GSMs offer ease of systematic exploration of the nutritional dimension, allowing the sampling of hundreds of media formulations in a matter of hours. The technology can substitute for arduous in vitro culturing work to determine a strain’s nutritional growth requirements. For instance, an A. muciniphila GSM predicted the commensal could utilize a number of monosaccharides derived from mucin, findings that were subsequently validated in vitro.Citation118 Similarly, GSMs have been used to identify the minimal media needed for Faecalibacterium prausniitziiCitation119 and Bacteroides caccae,Citation120 resulting in their successful culturing. Beyond determining strain niches, growth rates and metabolic output yields are potentially determinable through GSMs, but these are sensitive to bounds on nutritional uptake and biomass generation rates that are presently unknown for most strains. If these upper rate bounds can be determined through targeted in vitro cultures, and are otherwise invariant to nutritional context, then growth and metabolic output rates for very broad combinations of media could also be rapidly estimated in silico and thus save considerable in vitro effort.
GSMs can now be simulated in consortia and have provided insight into the ecological principles that underpin emergent community outcomes. This technology enables tracing of strain-level metabolic activities, and how their survival strategies vary with changing community contexts. For instance, using GSMs, a behavior was observed wherein one microbial guild’s reduction of growth rate (70% of its community-specific maximum growth rate) when paired with another guild allowed for an increased total community biomass, thus improving community-level fitness.Citation121 Similarly, over 800 GSM microbial communities were assembled and assessed in terms of competition and cooperation dynamics and showed that whilst competition was the predominant interaction across the community, modules of up to four cooperating strains persisted across communities.Citation122 The metabolites that were cooperatively exchanged were also identifiable.Citation122 Such understanding is key for rational intervention design as it explains the underlying ecology that is difficult to otherwise interrogate through non-in silico means. Lastly, GSM consortia’s capture of community ecology has enabled successful prediction of intervention outcomes. This was accomplished for two groups of human patients differing in insulin resistance profiles;Citation123 GSM consortia reflecting patient microbiomes correctly predicted changes in stool amino acid and SCFA levels under a dietary intervention that was subsequently administered to the humans. Thus, there is a potential role for GSM technology to guide intervention design and choice. Although useful, GSM technology caries some caveats, namely omitting genome regulatory networks and equating the genotype and phenotype. Yet, GSM technology is rapidly advancing,Citation114,Citation124 and is well poised to revolutionize our understanding of the gut ecology.
Recently, supervised machine learning has shown promise in predicting physiological outcomes (e.g., post-prandial glucose responseCitation125) of dietary manipulation, essentially treating microbiome–host interactions and dynamics as a ‘black box’. These approaches demonstrate that intervention outcomes can be predictable and therefore targeted at individuals. Yet they have limitations. These models are opaque and do not necessarily learn the true mechanisms underpinning the biology. Consequently, they may prove inaccurate in predicting outcomes for cases outside the training data’s range of intervention. Lastly, it is debatable how much penetration machine learning efforts will have if they are not accompanied by a well-understood mechanistic foundation.
In silico approaches offer continuous observation, at high spatiotemporal resolution, of the nutritional environment, cellular interactions, internal cell state and competition dynamics, and facilitate simulation of multifactorial interventions. They cannot (yet) simulate ‘health outcomes’, as these are emergent from the holistic host at a breadth current models do not capture. But they can model the profile of health-impacting community composition and metabolic output and the ecological dynamics responsible for them. GSMs are close to predicting these profiles for previously unobserved contexts, which would prove transformative for targeted intervention discovery.Citation123 This could be accomplished through systematic exploration of intervention space (e.g. the space of possible dietary interventions). For example, a computer model of an individual’s gut microbiome (profiling of in vivo communities) could be constructed and used to predict the outcomes for an intervention based on the fiber inulin, accounting for: which strains can catabolize it (based on in vitro experimentation) and are they competitive to do so given the community; which metabolites will be generated from inulin based on this profile; how these metabolites will impact other strains and how this will ultimately impact the community’s composition and metabolic output. Such study is easily repeated with variation to investigate how outcomes relate, potentially non-linearly, with fiber dosage. To conclude, evidence-based in silico methods can offer unparalleled experimental resolution and traceability at the spatial-temporal and biological levels.
Practical considerations for in silico modeling
ABM encompasses considerable flexibility in how the target biology is represented within the model, and this translates to models that require substantial custom computer code to implement. Which cells, pathways, molecules, and environmental features a given model captures is typically entirely bespoke to the given research context. Such flexibility complicates the generation of ‘one size fits all’ generalized simulation frameworks – lacking a standardization of what should be simulated and how, such frameworks would need to be vastly complicated in catering for all possibilities, and thus they instead typically represent very abstract and general tools that can require considerable custom code to adopt into a specific simulation.Citation126 Our lab’s ARTIMMUSCitation127 and MotiliSimCitation128 agent-based models are built atop the MASON agent-based simulation framework,Citation129 and yet encompass several thousand lines of custom Java code each. The GutSim simulation is entirely custom code, written in Python.Citation40 Whilst existing ABM models can certainly be adapted to different research questions, engaging with the ABM paradigm can necessitate a considerable aptitude for writing computer code.
A key consideration for ABM technology, stemming from the flexibility it affords in which biological features are represented and how, is ensuring that ABM models correctly capture the biology and that they are well-calibrated against existing real-world data. By necessity of simulating a system that is incompletely understood, ABMs often encompass parameters that control agent behaviors for which appropriate values are not exactly known. Calibration aims to infer these values from existing data by exploring a range of putative parameter values with an aim of reproducing known results. Agent-based model calibration has been reviewed extensively elsewhere,Citation130 and we summarize the salient points here. Calibration requires data describing target system behaviors, and there is no minimum threshold for this – more is better but with a diminishing return on additional data. Ideally, multiple real-world experiments will be used in calibration,Citation131 as these perturb the real-world system (and thus the simulation in attempting to reproduce real-world results) in different ways, targeting various cells, pathways, genes, molecules, etc. This encourages the full range of each agent’s possible behaviors to be exercised and assessed. Thus, ideally, data or knowledge describing the behaviors, rates, probabilities, timings, etc., of each individual model component (e.g. strain) can be provided, but failing this, higher-level data (e.g. at community composition-level) describing how the biological system behaves both ‘normally’ and under perturbation can facilitate calibration efforts.
ABM models are not necessarily computationally expensive to run (). The scale of the simulation, in terms of number of agents (e.g. cells), size, complexity and detail of the physical simulated space can often be adjusted, and these are the chief determinants of how much computational power and time is needed for the simulations to execute. In our lab, we have often prototyped models on personal computers before deploying them on a high-performance computing (HPC) facility to execute many more replicates and experimental variations at a larger-scale than a personal computer could accommodate. Access to HPC facilities is rarely prohibitive these days, with most research institutes either providing them directly or facilitating access to shared utilities. Thus, the biggest resource needed in engaging with ABM technology is time and personnel in building the models. Relative to this, experimentation with the models is relatively swift and the software tools are either cheap or free to use. Model building is rarely done in a timeframe less than months, with calibration and exploration of which biological features to include, and how, taking the most time – writing code is easy, demonstrating and arguing that the code correctly captures the target biology is what takes time.Citation137 This is an important (and easily overlooked) consideration, as without it results are misleading rather than insightful. Planning experiments to verify model predictions (whatever they may be; this is problem-specific) is a worthwhile activity to budget for and if successful can hugely elevate the work’s impact.
Table 1. List of recommended computational resources for gut microbiome in silico modeling
Growing support around GSM has contributed a rich, accessible, and growing ecosystem of tools for the further development and application of this technology. Free online tools such as AutoKEGGRegCitation134 and ModelSEEDCitation135 allow the generation of draft GSMs based on annotated genomes. Curated GSMs are commonly made freely available by researchers specialized in the matter, and the tools to simulate them are likewise freely accessible (). Curation can be the most time-consuming step of the GSM assembly process. GSMs are complicated, and manual critical inspection and verification against known organism behavior by a specialist cannot be avoided. Most readily available gut microbiome GSMs have been rigorously curated at a qualitative level of which metabolic reactions a given strain is capable of. However, we are not aware of any GSMs for which the upper bounds on reaction rates have been comprehensively calibrated. This is an important omission, as these reaction rate bounds ultimately constrain maximum growth rates, and thus community composition and metabolic output outcomes. Not knowing these upper bounds limits the extent to which GSMs can be used as faithful surrogates for real communities in predicting outcomes to putative interventions (i.e. where these outcomes are not already known and thus cannot be calibrated against). There is enormous upside in establishing these reaction rate limits, as the search for interventions that deliver targeted outcomes could then be automated and run in a massively parallel fashion on a HPC using GSM technology as faithful surrogates for the real community. Importantly, for a given GSM/strain, these rates could be inferred through a combination of in vitro culturing where specific growth rates, substrate uptake, and metabolite production rates can be established across a variety of media formulations, and through automated calibration methodologies that find parameter values that best recapitulate all these in vitro-observed strain behaviors simultaneously.Citation131
GSM technology can be relatively straightforward to engage with. GSMs are built upon individual strain genomes, independently of one another, forming modules that can then be simulated individually or combined into communities in a ‘plug and play’ fashion. The COBRACitation132 and COBRApyCitation133 toolboxes cater for individual GSM simulations, and frameworks such as MICOMCitation115 and µbialSimCitation138 support GSM simulations of communities. Coding proficiency in Python or MATLAB is required. Simulations of individual GSMs or simple consortia can be conducted on a personal computer, but HPC access is advisable for large communities or very broad experimental designs, such as extensive systematic exploration of nutritional variations or community memberships.
Conclusion
The gut microbiome impacts host health and has emerged as a therapeutic target. The therapeutic value of currently available interventions, e.g. prebiotics, probiotics, or diet, is contingent on how they integrate within a host’s existing gut ecosystem.Citation9 Yet this differs between hosts, and as such outcomes are divergent. Better design and targeting of interventions requires improved understanding of the ecological processes underlying microbiome composition and function. In vivo approaches have revealed the breadth and nature of interactions between host and microbiome; these are multiple and have non-linear, context-dependent effects. These interactions are a culmination of intertwined processes that shape both host and microbiome. However, in vivo approaches struggle to isolate and probe individual ecological processes at fine spatiotemporal resolution. Through in vivo approaches we understand which microbial outputs impact host health, but not why or how such outputs emerge as key factors across differing contexts and individuals. In vitro approaches permit isolation and study of particular microbes and pathways in broad environmental contexts. They reveal the cellular behaviors upon which an ecology is built. Yet the number of microbes, unique consortia and unique environmental contexts that exist represent a combinatorial explosion that is insurmountable to study purely in vitro. Further, in vitro technologies cannot capture the breadth of complexity within the host, and thus cannot integrate all relevant pathways, or reveal critical ecological parameters such as cellular nutritional state and competition dynamics, without disturbing or destroying the system. We have argued here that in silico approaches can span this divide. Genome-scale models can broadly supplement in vitro culturing efforts, though tuning and corroboration through in vitro means are necessary. In silico models enable observation and experimentation that reveal how cell-level behaviors underpin ecological processes to generate community-level outputs of relevance to the host (). In silico techniques are undergoing rapid advancement and their full potential is as yet untapped. Efforts to functionally annotate genomes are accelerating, giving rise to increasing numbers of bacteria GSMs.Citation120 Yet, their adoption is sparse relative to in vitro and in vivo experimental research.
Figure 2. In vivo, in vitro and in silico methods each contribute complementary insights that are collectively necessary to understand gut ecology and manipulate it to achieve specific outcomes
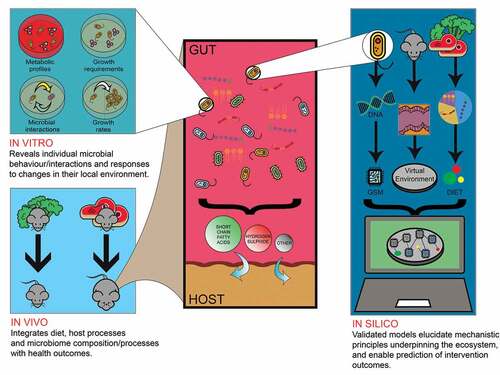
There are aspects of the gut microbiome that influence health outcomes and are difficult to interrogate experimentally, but which could be amendable to study through computational modeling technologies. The non-bacterial (archaea, fungal, viral) gut microbiome is gaining recognition for its impact on host health,Citation7,Citation8 though such investigations are as yet scarce. Since in silico investigations rely on in vitro- and in vivo-derived data, exploration of the non-bacterial gut microbiome through computational modeling approaches will take some time to become established at scale. However, there is progress: GSMs of gut methanogenic archaea species have been added to AGORACitation120 and GSMs of (non-gut) protozoa can be found as part of the BiGG Models knowledgebase.Citation139 Similarly, GSMs of fungal gut commensals and opportunistic pathogens have emerged in recent years.Citation140,Citation141 GSM has limited capacity to represent phages, however, as these viruses are not metabolically self-sufficient organisms. ABMs could represent the effects of phage-infection, and other behavior-modifying phenomena such as horizontal gene transfer (HGT). For instance, an ABM that explores bacteria-phage interactions at the community level and their effect on antibiotic resistance has already been developed,Citation142 but this did not concern the gut microbiome specifically. Similarly, modeling efforts have been made toward the study of bacteria–bacteria HGT at the community level.Citation143–144 Furthermore, ABM has scope for the study of spatiotemporal dynamics of mucosa-associated microbiome, offering some advantages over in vitro continuous fermentation models. For example, M-SHINE relied on static mucin-covered microcosms to replicate a mucin layer,Citation106 whereas GutSim readily simulated real-world continuous mucin secretion patterns.Citation40 We are unaware of any agent-based models that explicitly distinguish mucosal from luminal microbial communities, though it is not conceptually difficult to do so by, e.g., simulating an additional spatial dimension in GutSim. Detailed spatiotemporal analysis, beyond what is possible in vivo, would thus be possible.
Scientific investigations that integrate in vitro, in vivo and in silico perspectives are key to enable the knowledge that would move the field of personalized interventions forward. However, such efforts are exceedingly rare as research groups seldom possess deep capacity in all three. This is necessarily a broadly inter-disciplinary venture encompassing clinical and animal studies, anaerobic microbial culturing and bioengineering, omics, mathematics, and computer science. Dialog between these disciplines is not always easily established but should be encouraged, as should collaboration between research groups that specialize in these techniques. Education is already shifting to i) give biologists and health-care practitioners exposure to data and modeling, and ii) give a greater focus on clinical and biological application in engineering programs. The forthcoming convergence between disciplines together with our proposed integration of in vivo, in vitro, and in silico technologies will reveal the mechanistic underpinnings required for the design of rational interventions targeting the diet-gut microbiome-host system.
List of abbreviations
SCFAs: Short-chain fatty acids
MAMPs: Microbe-associated molecular patterns
LPS: Lipopolysaccharide
SHINE: Simulator of the human intestinal microbial ecosystem
M-SHINE: Mucin-simulator of the human intestinal microbial ecosystem
ABM: Agent-based modeling/model
GSM: Genome-scale modeling/model
ARTIMMUS: artificial murine multiple-sclerosis simulation
MASON: Multiagent simulator of neighborhoods
COBRA: Constraint-based reconstruction analysis
KEGG: Kyoto encyclopedia of genes and genomes
AGORA: Assembly of gut organisms through reconstruction and analysis
HGT: Horizontal gene transfer
Authors’ contributions
MNR conceived of the study. JPMO reviewed the literature. JPMO and MNR wrote the manuscript with input from all authors.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105(24):1907–19. doi:10.1093/jnci/djt300.
- Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637. doi:10.1136/gut.2010.223263.
- Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, Schoos AMM, Kunøe A, Fink NR, Chawes BL, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018;9(1):141. doi:10.1038/s41467-017-02573-2.
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455(7216):1109. doi:10.1038/nature07336.
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480. doi:10.1038/nature07540.
- Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, Silver PA, Gerber GK. Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe. 2019 e5;25(6):803–814. doi:10.1016/j.chom.2019.05.001.
- Jhangi S, Gandhi R, Glanz B, Cook S, Nejad P, Ward D, Li N, Gerber G, Bry L, Weiner H. Increased Archaea species and changes with therapy in gut microbiome of multiple sclerosis subjects (S24.001). Neurology. 2014;82(10):S24.001.
- Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574(7777):264–267. doi:10.1038/s41586-019-1608-2.
- Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5(2):220–230. doi:10.1038/ismej.2010.118.
- Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee Y, De Vadder F, Arora T, Hallen A, Martens E, Björck I, Bäckhed F, et al. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of. Prevotella Cell Metabolism. 2015;22(6):971–982. doi:10.1016/j.cmet.2015.10.001.
- Jain U, Ver Heul AM, Xiong S, Gregory MH, Demers EG, Kern JT, Lai C-W, Muegge BD, Barisas DAG, Leal-Ekman JS, et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science. 2021;371(6534):1154–1159. doi:10.1126/science.abd0919.
- Limon JJ, Tang J, Li D, Wolf AJ, Michelsen KS, Funari V, Gargus M, Nguyen C, Sharma P, Maymi VI, et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25(3):377–388. doi:10.1016/j.chom.2019.01.007.
- Read MN, Holmes AJ. Towards an integrative understanding of diet–host–gut microbiome interactions. Front Immunol. 2017;8:538.
- Lambeth SM, Carson T, Lowe J, Ramaraj T, Leff JW, Luo L, Bell CJ, Shah VO. Composition, diversity and abundance of gut microbiome in prediabetes and type 2 diabetes. Journal of Diabetes and Obesity. 2015;2(3):1.
- Lewis JD, Chen E, Baldassano R, Otley A, Griffiths A, Lee D, Bittinger K, Bailey A, Friedman E, Hoffmann C, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe. 2015;18(4):489–500. doi:10.1016/j.chom.2015.09.008.
- Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, Zeng L, Chen J, Fan S, Du X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry. 2016;21(6):786–796. doi:10.1038/mp.2016.44.
- Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, Hardt PD. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18(1):190–195. doi:10.1038/oby.2009.167.
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi:10.1038/4441022a.
- Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):1–11. doi:10.1038/s41598-017-13601-y.
- Sepehri S, Kotlowski R, Bernstein CN, Krause DO. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(6):675–683. doi:10.1002/ibd.20101.
- Goodrich JK, Waters J, Poole A, Sutter J, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell J, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi:10.1016/j.cell.2014.09.053.
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481.
- Durgan DJ, Ganesh BP, Cope JL, Ajami NJ, Phillips SC, Petrosino JF, Hollister EB, Bryan Jr RM. Role of the gut microbiome in obstructive sleep apnea–induced hypertension. Hypertension. 2016;67(2):469–474.
- Zoll J, Read MN, Heywood SE, Estevez E, Marshall JP, Kammoun HL, Allen TL, Holmes AJ, Febbraio MA, Henstridge DC. Fecal microbiota transplantation from high caloric-fed donors alters glucose metabolism in recipient mice, independently of adiposity or exercise status. American Journal of Physiology-Endocrinology and Metabolism. 2020.
- Menni C, Jackson MA, Pallister T, Steves CJ, Spector TD, Valdes AM. Gut microbiome diversity and high-fibre intake are related to lower long-term weight gain. Int J Obes. 2017;41(7):1099–1105.
- Dash S, Clarke G, Berk M, Jacka FN. The gut microbiome and diet in psychiatry: focus on depression. Curr Opin Psychiatry. 2015;28(1):1–6.
- Zeng H, Lazarova DL, Bordonaro M. Mechanisms linking dietary fiber, gut microbiota and colon cancer prevention. World J Gastrointest Oncol. 2014;6:41.
- Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, De Los Reyes-gavilán CG, Salazar N. Intestinal short chain fatty acids and their link with diet and human health. Front Microbiol. 2016;7:185.
- Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, Maruya M, McKenzie CI, Hijikata A, Wong C, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6(p):6734.
- Tan J, McKenzie C, Vuillermin P, Goverse G, Vinuesa C, Mebius R, Macia L, Mackay C. Dietary fiber and bacterial SCFA enhance oral tolerance and protect against food allergy through diverse cellular pathways. Cell Rep. 2016;15(12):2809–2824. doi:10.1016/j.celrep.2016.05.047.
- Thorburn AN, McKenzie CI, Shen S, Stanley D, Macia L, Mason LJ, Roberts LK, Wong CHY, Shim R, Robert R, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun. 2015;6(1):1–13. doi:10.1038/ncomms8320.
- Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, McKenzie C, Kranich J, Oliveira AC, Rossello FJ, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. 2017;18(5):552–562. doi:10.1038/ni.3713.
- Blachier F, Beaumont M, Kim E. Cysteine-derived hydrogen sulfide and gut health: a matter of endogenous or bacterial origin. Curr Opin Clin Nutr Metab Care. 2019;22(1):68–75. doi:10.1097/MCO.0000000000000526.
- Hale VL, Jeraldo P, Mundy M, Yao J, Keeney G, Scott N, Cheek EH, Davidson J, Greene M, Martinez C, et al. Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods. 2018;149:59–68. doi:10.1016/j.ymeth.2018.04.024.
- Blachier F, Mariotti F, Huneau JF, Tomé D. Effects of amino acid-derived luminal metabolites on the colonic epithelium and physiopathological consequences. Amino Acids. 2007;33(4):547–562. doi:10.1007/s00726-006-0477-9.
- Pedersen G, Brynskov J, Saermark T. Phenol toxicity and conjugation in human colonic epithelial cells. Scand J Gastroenterol. 2002;37(1):74–79. doi:10.1080/003655202753387392.
- McCall IC, Betanzos A, Weber DA, Nava P, Miller GW, Parkos CA. Effects of phenol on barrier function of a human intestinal epithelial cell line correlate with altered tight junction protein localization. Toxicol Appl Pharmacol. 2009;241(1):61–70. doi:10.1016/j.taap.2009.08.002.
- Beyer-Sehlmeyer G, Glei M, Hartmann E, Hughes R, Persin C, Böhm V, Schubert R, Jahreis G, Pool-Zobel BL. Butyrate is only one of several growth inhibitors produced during gut flora-mediated fermentation of dietary fibre sources. British Journal of Nutrition. 2003;90(6):1057–1070. doi:10.1079/BJN20031003.
- Auguet M, Viossat I, Marin J-G, Chabrier P-E. Selective inhibition of inducible nitric oxide synthase by agmatine. The Japanese Journal of Pharmacology. 1995;69(3):285–287. doi:10.1254/jjp.69.285.
- Holmes AJ, Chew YV, Colakoglu F, Cliff JB, Klaassens E, Read MN, Solon-Biet SM, McMahon AC, Cogger VC, Ruohonen K, et al. Diet-microbiome interactions in health are controlled by intestinal nitrogen source constraints. Cell Metab. 2017;25(1):140–151. doi:10.1016/j.cmet.2016.10.021.
- Marcobal A, Southwick AM, Earle KA, Sonnenburg JL. A refined palate: bacterial consumption of host glycans in the gut. Glycobiology. 2013;23(9):1038–1046. doi:10.1093/glycob/cwt040.
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi:10.1038/nature12820.
- Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012;3(4):289–306. doi:10.4161/gmic.19897.
- Corfield AP. Mucins: a biologically relevant glycan barrier in mucosal protection. Biochimica Biophysica Acta (BBA)-General Subjects. 2015;1850:236–252.
- Juge N. Microbial adhesins to gastrointestinal mucus. Trends Microbiol. 2012;20(1):30–39. doi:10.1016/j.tim.2011.10.001.
- Sonnenburg JL, Angenent LT, Gordon JI. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat Immunol. 2004;5(6):569. doi:10.1038/ni1079.
- Van Passel MW, Kant R, Zoetendal EG, Plugge CM, Derrien M, Malfatti SA, Chain PSG, Woyke T, Palva A, de Vos WM, et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PloS One. 2011;6(3):e16876. doi:10.1371/journal.pone.0016876.
- Carroll IM, Ringel-Kulka T, Keku TO, Chang Y-H, Packey CD, Sartor RB, Ringel Y. Molecular analysis of the luminal-and mucosal-associated intestinal microbiota in diarrhea-predominant irritable bowel syndrome. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2011;301(5):G799–G807. doi:10.1152/ajpgi.00154.2011.
- Jones RB, Zhu X, Moan E, Murff HJ, Ness RM, Seidner DL, Sun S, Yu C, Dai Q, Fodor AA, et al. Inter-niche and inter-individual variation in gut microbial community assessment using stool, rectal swab, and mucosal samples. Sci Rep. 2018;8(1):1–12. doi:10.1038/s41598-018-22408-4.
- Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773–1781. doi:10.1053/j.gastro.2011.07.046.
- Wahlström A, Sayin S, Marschall H-U, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24(1):41–50. doi:10.1016/j.cmet.2016.05.005.
- Ibrahim SA, Bezkorovainy A. Survival of Bifidobacteria in the presence of bile salt. J Sci Food Agric. 1993;62(4):351–354. doi:10.1002/jsfa.2740620407.
- O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13(12):691. doi:10.1038/nrgastro.2016.165.
- Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288–3294.
- Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, Stappenbeck TS. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. 2016;165(7):1708–1720. doi:10.1016/j.cell.2016.05.018.
- Barton LL, Ritz NL, Fauque GD, Lin HC. Sulfur cycling and the intestinal microbiome. Dig Dis Sci. 2017;62(9):2241–2257. doi:10.1007/s10620-017-4689-5.
- Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009 e1;137(2):569–578. doi:10.1053/j.gastro.2009.04.012.
- Szabo C, Hellmich MR. Endogenously produced hydrogen sulfide supports tumor cell growth and proliferation. Cell Cycle. 2013;12(18):2915. doi:10.4161/cc.26064.
- Terashima H, Kawamoto A, Morimoto YV, Imada K, Minamino T. Structural differences in the bacterial flagellar motor among bacterial species. Biophysics and Physicobiology. 2017;14:191–198. doi:10.2142/biophysico.14.0_191.
- Turner RD, Vollmer W, Foster SJ. Different walls for rods and balls: the diversity of peptidoglycan. Mol Microbiol. 2014;91(5):862–874. doi:10.1111/mmi.12513.
- Wang X, Quinn PJ. Lipopolysaccharide: biosynthetic pathway and structure modification. Prog Lipid Res. 2010;49(2):97–107. doi:10.1016/j.plipres.2009.06.002.
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi:10.1038/nri1391.
- Tough DF, Sun S, Sprent J. T cell stimulation in vivo by lipopolysaccharide (LPS). J Exp Med. 1997;185(12):2089–2094. doi:10.1084/jem.185.12.2089.
- Bryn T, Yaqub S, Mahic M, Henjum K, Aandahl EM, Tasken K. LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+ T cells through a COX-2–PGE2-dependent mechanism. Int Immunol. 2008;20(2):235–245. doi:10.1093/intimm/dxm134.
- Hajam IA, Dar PA, Shahnawaz I, Jaume JC, Lee JH. Bacterial flagellin—a potent immunomodulatory agent. Exp Mol Med. 2017 e373-e373;49(9):e373–e373. doi:10.1038/emm.2017.172.
- Whang H, Mayer H, Schmidt MG, Neter E. Strain-related differences in immunosuppressive effects of Enterobacteriaceae and their lipopolysaccharides on production in rabbits of antibody to enterobacterial common antigen. Infect Immun. 1976;13(4):1074–1079. doi:10.1128/iai.13.4.1074-1079.1976.
- Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31:817–844.
- Hersoug LG, Møller P, Loft S. Gut microbiota‐derived lipopolysaccharide uptake and trafficking to adipose tissue: implications for inflammation and obesity. Obesity Reviews. 2016;17(4):297–312. doi:10.1111/obr.12370.
- Lerouge I, Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant–microbe interactions. FEMS Microbiol Rev. 2002;26(1):17–47. doi:10.1111/j.1574-6976.2002.tb00597.x.
- Vatanen T, Kostic A, d’Hennezel E, Siljander H, Franzosa E, Yassour M, Kolde R, Vlamakis H, Arthur T, Hämäläinen A-M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165(4):842–853. doi:10.1016/j.cell.2016.04.007.
- Steimle A, Autenrieth IB, Frick J-S. Structure and function: lipid A modifications in commensals and pathogens. International Journal of Medical Microbiology. 2016;306(5):290–301. doi:10.1016/j.ijmm.2016.03.001.
- Ha CW. Mechanistic links between gut microbial community dynamics, microbial functions and metabolic health. World Journal of Gastroenterology: WJG. 2014;20(44):16498–16517. doi:10.3748/wjg.v20.i44.16498.
- Mokhtari Z, Gibson DL, Hekmatdoost A. Nonalcoholic fatty liver disease, the gut microbiome, and diet. Advances in Nutrition. 2017;8(2):240–252. doi:10.3945/an.116.013151.
- Hollander D. Intestinal permeability, leaky gut, and intestinal disorders. Curr Gastroenterol Rep. 1999;1(5):410–416. doi:10.1007/s11894-999-0023-5.
- Sheth RU, Li M, Jiang W, Sims PA, Leong KW, Wang HH. Spatial metagenomic characterization of microbial biogeography in the gut. Nat Biotechnol. 2019;37(8):877–883. doi:10.1038/s41587-019-0183-2.
- Sarma-Rupavtarm RB, Ge Z, Schauer DB, Fox JG, Polz MF. Spatial distribution and stability of the eight microbial species of the altered schaedler flora in the mouse gastrointestinal tract. Appl Environ Microbiol. 2004;70(5):2791–2800. doi:10.1128/AEM.70.5.2791-2800.2004.
- Evans D, Pye G, Bramley R, Clark AG, Dyson TJ, Hardcastle JD. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut. 1988;29(8):1035–1041. doi:10.1136/gut.29.8.1035.
- Smith EA, Macfarlane GT. Studies on amine production in the human colon: enumeration of amine forming bacteria and physiological effects of carbohydrate and pH. Anaerobe. 1996;2(5):285–297. doi:10.1006/anae.1996.0037.
- Thaiss CA, Levy M, Korem T, Dohnalová L, Shapiro H, Jaitin DA, David E, Winter DR, Gury-BenAri M, Tatirovsky E, et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell. 2016;167(6):1495–1510. e12. doi:10.1016/j.cell.2016.11.003.
- Zarrinpar A, Chaix A, Yooseph S, Panda S. Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014;20(6):1006–1017. doi:10.1016/j.cmet.2014.11.008.
- Kreft J-U, Griffin BM, González-Cabaleiro R. Evolutionary causes and consequences of metabolic division of labour: why anaerobes do and aerobes don’t. Curr Opin Biotechnol. 2020;62:80–87. doi:10.1016/j.copbio.2019.08.008.
- Harcombe WR, Riehl W, Dukovski I, Granger B, Betts A, Lang A, Bonilla G, Kar A, Leiby N, Mehta P, et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014;7(4):1104–1115. doi:10.1016/j.celrep.2014.03.070.
- Carlson RP, Beck AE, Phalak P, Fields MW, Gedeon T, Hanley L, Harcombe WR, Henson MA, Heys JJ. Competitive resource allocation to metabolic pathways contributes to overflow metabolisms and emergent properties in cross-feeding microbial consortia. Biochem Soc Trans. 2018;46(2):269–284. doi:10.1042/BST20170242.
- Beaumont M, Andriamihaja M, Lan A, Khodorova N, Audebert M, Blouin J-M, Grauso M, Lancha L, Benetti P-H, Benamouzig R, et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: the adaptive response. Free Radic Biol Med. 2016;93:155–164. doi:10.1016/j.freeradbiomed.2016.01.028.
- Boets E, Deroover L, Houben E, Vermeulen K, Gomand SV, Delcour JA, Verbeke K. Quantification of in vivo colonic short chain fatty acid production from inulin. Nutrients. 2015;7(11):8916–8929.
- Duncan SH, Belenguer A, Holtrop G, Johnstone AM, Flint HJ, Lobley GE Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl Environ Microbiol. 2007;73(4):1073–1078.
- Vital M, Karch A, Pieper DH. Colonic butyrate-producing communities in humans: an overview using omics data. Msystems. 2017;2(6.
- Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. MBio. 2014;5(2.
- Bourriaud C, Robins RJ, Martin L, Kozlowski F, Tenailleau E, Cherbut C, Michel C. Lactate is mainly fermented to butyrate by human intestinal microfloras but inter‐individual variation is evident. J Appl Microbiol. 2005;99(1):201–212. doi:10.1111/j.1365-2672.2005.02605.x.
- Reeves AR, Wang GR, Salyers AA. Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron . Journal of Bacteriology. 1997;179(3):643–649. doi:10.1128/jb.179.3.643-649.1997.
- Hooper LV. Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291(5505):881–884. doi:10.1126/science.291.5505.881.
- Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. Journal of Biological Chemistry. 2006;281:36269–36279.
- Van Herreweghen F, Van den Abbeele P, De Mulder T, De Weirdt R, Geirnaert A, Hernandez-Sanabria E, Vilchez-Vargas R, Jauregui R, Pieper DH, Belzer C, et al. In vitro colonisation of the distal colon by Akkermansia muciniphila is largely mucin and pH dependent. Benef Microbes. 2017;8(1):81–96. doi:10.3920/BM2016.0013.
- Van Herreweghen F, De Paepe K, Roume H, Kerckhof F-M, Van de Wiele T. Mucin degradation niche as a driver of microbiome composition and Akkermansia muciniphila abundance in a dynamic gut model is donor independent. FEMS Microbiol Ecol. 2018;94(12):fiy186. doi:10.1093/femsec/fiy186.
- Degnan B, Macfarlane G. Carbohydrate utilization patterns and substrate preferences in Bacteroides thetaiotaomicron. Anaerobe. 1995;1(1):25–33. doi:10.1016/S1075-9964(95)80392-0.
- Lynch JB, Sonnenburg JL. Prioritization of a plant polysaccharide over a mucus carbohydrate is enforced by a Bacteroides hybrid two-component system. Mol Microbiol. 2012;85(3):478–491. doi:10.1111/j.1365-2958.2012.08123.x.
- Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol. 2004;70(10):5810–5817. doi:10.1128/AEM.70.10.5810-5817.2004.
- LoCascio RG, Ninonuevo MR, Freeman SL, Sela DA, Grimm R, Lebrilla CB, Mills DA, German JB. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. J Agric Food Chem. 2007;55(22):8914–8919. doi:10.1021/jf0710480.
- Voolapalli RK. Hydrogen production in anaerobic reactors during shock loads—influence of formate production and H2 kinetics. Water Res. 2001;35(7):1831–1841. doi:10.1016/S0043-1354(00)00441-3.
- Laverde Gomez JA, Mukhopadhya I, Duncan SH, Louis P, Shaw S, Collie-Duguid E, Crost E, Juge N, Flint HJ. Formate cross‐feeding and cooperative metabolic interactions revealed by transcriptomics in co‐cultures of acetogenic and amylolytic human colonic bacteria. Environ Microbiol. 2019;21(1):259–271. doi:10.1111/1462-2920.14454.
- Fois CAM, Le TYL, Schindeler A, Naficy S, McClure DD, Read MN, Valtchev P, Khademhosseini A, Dehghani F. Models of the gut for analyzing the impact of food and drugs. Adv Healthcare Mater. 2019;8(21):1900968. doi:10.1002/adhm.201900968.
- Bahrami B, Child MW, Macfarlane S, Macfarlane GT. Adherence and cytokine induction in Caco-2 cells by bacterial populations from a three-stage continuous-culture model of the large intestine. Appl Environ Microbiol. 2011;77(9):2934–2942. doi:10.1128/AEM.02244-10.
- Sanderson IR. The physicochemical environment of the neonatal intestine. Am J Clin Nutr. 1999;69(5):1028s–1034s. doi:10.1093/ajcn/69.5.1028s.
- Kim HJ, Huh D, Hamilton G, Ingber DE. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip. 2012;12(12):2165–2174. doi:10.1039/c2lc40074j.
- McBain A, Macfarlane G. Investigations of bifidobacterial ecology and oligosaccharide metabolism in a three-stage compound continuous culture system. Scand J Gastroenterol. 1997;32(sup222):32–40. doi:10.1080/00365521.1997.11720715.
- Van den Abbeele P, Roos S, Eeckhaut V, MacKenzie DA, Derde M, Verstraete W, Marzorati M, Possemiers S, Vanhoecke B, Van Immerseel F, et al. Incorporating a mucosal environment in a dynamic gut model results in a more representative colonization by lactobacilli. Microb Biotechnol. 2012;5(1):106–115. doi:10.1111/j.1751-7915.2011.00308.x.
- Lagier J-C, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nature Microbiology. 2016;1(12):16203. doi:10.1038/nmicrobiol.2016.203.
- Guarner F, Malagelada J-R. Gut flora in health and disease. The Lancet. 2003;361(9356):512–519. doi:10.1016/S0140-6736(03)12489-0.
- Cosgrove J, Butler J, Alden K, Read M, Kumar V, Cucurull‐Sanchez L, Timmis J, Coles M. Agent‐based modeling in systems pharmacology. CPT: Pharmacometrics & Systems Pharmacology. 2015;4(11):615–629.
- Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T., Bacterial community assembly based on functional genes rather than species. Proceedings of the National Academy of Sciences, 2011. 108: p. 14288–14293.
- The Human Microbiome Project, C., et al., Structure, function and diversity of the healthy human microbiome. Nature, 2012. 486: p. 207. 7402. doi:10.1038/nature11234
- Schluter J, Foster KR, Ellner SP. The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol. 2012;10(11):e1001424. doi:10.1371/journal.pbio.1001424.
- Shashkova T, Popenko A, Tyakht A, Peskov K, Kosinsky Y, Bogolubsky L, Raigorodskii A, Ischenko D, Alexeev D, Govorun V, et al. Agent based modeling of human gut microbiome interactions and perturbations. PLoS One. 2016;11(2):e0148386. doi:10.1371/journal.pone.0148386.
- Magnusdottir S, Thiele I. Modeling metabolism of the human gut microbiome. Curr Opin Biotechnol. 2017;51:90–96. doi:10.1016/j.copbio.2017.12.005.
- Diener C, Gibbons SM, Resendis-Antonio O. MICOM: metagenome-scale modeling to infer metabolic interactions in the gut microbiota. Msystems. 2020;5(1.
- Lee D-S, Burd H, Liu J, Almaas E, Wiest O, Barabasi A-L, Oltvai ZN, Kapatral V. Comparative Genome-Scale Metabolic Reconstruction and Flux Balance Analysis of Multiple Staphylococcus aureus Genomes Identify Novel Antimicrobial Drug Targets. J Bacteriol. 2009;191(12):4015–4024. doi:10.1128/JB.01743-08.
- Khannapho C, Zhao H, Bonde BK, Kierzek AM, Avignone-Rossa CA, Bushell ME. Selection of objective function in genome scale flux balance analysis for process feed development in antibiotic production. Metab Eng. 2008;10(5):227–233. doi:10.1016/j.ymben.2008.06.003.
- Ottman N, Davids M, Suarez-Diez M, Boeren S, Schaap PJ, Martins Dos Santos VAP, Smidt H, Belzer C, de Vos WM. Genome-Scale Model and Omics Analysis of Metabolic Capacities of Akkermansia muciniphila Reveal a Preferential Mucin-Degrading Lifestyle. Appl Environ Microbiol. 2017;83(18):e01014–17. doi:10.1128/AEM.01014-17.
- Heinken A, Khan MT, Paglia G, Rodionov DA, Harmsen HJ, Thiele I. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. Journal of Bacteriology. 2014;196(18):3289–3302.
- Magnusdottir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, Greenhalgh K, Jäger C, Baginska J, Wilmes P, et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2017;35(1):81–89. doi:10.1038/nbt.3703.
- Zomorrodi AR, Maranas CD, Rao CV. OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol. 2012;8(2):e1002363. doi:10.1371/journal.pcbi.1002363.
- Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proceedings of the National Academy of Sciences, 2015. 112: p. 6449–6454.
- Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, de Wouters T, Juste C, Rizkalla S, Chilloux J, et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015;22(2):320–331. doi:10.1016/j.cmet.2015.07.001.
- Oberhardt MA, Palsson BØ, Papin JA. Applications of genome‐scale metabolic reconstructions. Mol Syst Biol. 2009;5(1.
- Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M, et al. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163(5):1079–1094. doi:10.1016/j.cell.2015.11.001.
- Abar S, Theodoropoulos GK, Lemarinier P, O’Hare GMP. Agent based modelling and simulation tools: a review of the state-of-art software. Computer Science Review. 2017;24:13–33. doi:10.1016/j.cosrev.2017.03.001.
- Read M, Andrews PS, Timmis J, Williams RA, Greaves RB, Sheng H, Coles M, Kumar V. Determining disease intervention strategies using spatially resolved simulations. PloS One. 2013;8(11):e80506. doi:10.1371/journal.pone.0080506.
- Read MN, Bailey J, Timmis J, Chtanova T. Leukocyte motility models assessed through simulation and multi-objective optimization-based model selection. PLoS Comput Biol. 2016;12(9.
- Luke S, Cioffi-Revilla C, Panait L, Sullivan K, Balan G. Mason: a multiagent simulation environment. Simulation. 2005;81(7):517–527. doi:10.1177/0037549705058073.
- Read MN, Alden K, Timmis J, Andrews PS. Strategies for calibrating models of biology. Brief Bioinform. 2020;21(1):24–35.
- Read MN, Alden K, Rose LM, Timmis J. Automated multi-objective calibration of biological agent-based simulations. Journal of the Royal Society Interface. 2016;13(122):20160543. doi:10.1098/rsif.2016.0543.
- Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A, Haraldsdóttir HS, Wachowiak J, Keating SM, Vlasov V, et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat Protoc. 2019;14(3):639–702. doi:10.1038/s41596-018-0098-2.
- Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: constraints-based reconstruction and analysis for python. BMC Syst Biol. 2013;7(1):1–6. doi:10.1186/1752-0509-7-74.
- Karlsen E, Schulz C, Almaas E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinform. 2018;19(1):1–11. doi:10.1186/s12859-018-2472-z.
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28(9):977–982. doi:10.1038/nbt.1672.
- Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, Dehal P, Ware D, Perez F, Canon S, et al. KBase: the United States department of energy systems biology knowledgebase. Nat Biotechnol. 2018;36(7):566–569. doi:10.1038/nbt.4163.
- Alden K, Andrews PS, Polack FA, Veiga-Fernandes H, Coles MC, Timmis J. Using argument notation to engineer biological simulations with increased confidence. Journal of the Royal Society Interface. 2015;12(104):20141059. doi:10.1098/rsif.2014.1059.
- Popp D, Centler F. μbialSim: constraint-based dynamic simulation of complex microbiomes. Frontiers in Bioengineering and Biotechnology. 2020;8:574.
- King ZA, Lu J, Dräger A, Miller P, Federowicz S, Lerman JA, Ebrahim A, Palsson BO, Lewis NE. BiGG Models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016;44(D1(D1):D515–D522. doi:10.1093/nar/gkv1049.
- Viana R, Dias O, Lagoa D, Galocha M, Rocha I, Teixeira MC. Genome-scale metabolic model of the human pathogen Candida albicans: a promising platform for drug target prediction. Journal of Fungi. 2020;6(3):171. doi:10.3390/jof6030171.
- Sánchez BJ, Zhang C, Nilsson A, Lahtvee P-J, Kerkhoven EJ, Nielsen J. Improving the phenotype predictions of a yeast genome‐scale metabolic model by incorporating enzymatic constraints. Mol Syst Biol. 2017;13(8):935. doi:10.15252/msb.20167411.
- de Sousa JA, Alsaadi A, Haaber J, Ingmer H, Rocha EP. Modelling phage-bacteria interactions driving predation and horizontal gene transfer. In: BioRxiv. 2018. p. 291328.
- van Dijk B, Hogeweg P, Doekes HM, Takeuchi N. Slightly beneficial genes are retained by bacteria evolving DNA uptake despite selfish elements. Elife. 2020;9:e56801. doi:10.7554/eLife.56801.
- Prensky H, Gomez‐Simmonds A, Uhlemann A-C, Lopatkin AJ. Conjugation dynamics depend on both the plasmid acquisition cost and the fitness cost. Mol Syst Biol. 2021;17(3):e9913. doi:10.15252/msb.20209913.