
ABSTRACT
Escherichia coli Nissle (EcN), a probiotic bacterium, has been employed in treating inflammatory bowel disease, but the nature of its therapeutic effect is not fully understood. Intestinal inflammation alters the environment, exposing the microbial population to new stresses and eliciting transcriptional responses. We administered EcN to germ-free mice and then compared its transcriptional response between DSS-treated and untreated conditions using RNA-seq analysis to identify 187 differentially expressed genes (119 upregulated, 68 downregulated) and verifying a subset with qRT-PCR. The upregulated genes included many involved in flagella biosynthesis and motility, as well as several members of the formate hydrogenlyase complex. Despite prior evidence that these pathways are both transcriptionally regulated by nitric oxide, in vitro tests did not establish that nitric oxide exposure alone elicited the transcriptional response. The results provide new information on the transcriptional response of EcN to inflammation and establish a basis for further investigation of its anti-inflammatory activity.
Introduction
Inflammatory bowel disease (IBD), which includes Crohn’s disease and ulcerative colitis, results in inflammation in the human intestine. Although the exact mechanism that initiates IBD is still unclear, the general consensus is that it is caused by a combination of different factors including aberrant immune activation, the gut microbiota, environmental triggers, and genetic traits.Citation1,Citation2 In particular, dysbiosis of the gut microbiota has been suggested to play a role in the development of the disease.Citation3 Metagenomic analyses of intestinal microbes have shown that patients with IBD have significantly different microbial compositions compared to their non-IBD counterparts.Citation4,Citation5 Whether dysbiosis is a cause rather than an effect of IBD is unknown, but significant efforts have been made toward developing methods for altering the composition of the gut microbiota as a potential therapeutic option. Probiotics have become a promising treatment option for IBD patients. For example, the bacterium Escherichia coli Nissle has been one of the most widely studied probiotics and has been shown to have multiple potential clinical roles for the treatment of gastrointestinal diseases including IBD.Citation6,Citation7 Furthermore, administration of Escherichia coli Nissle to a DSS-mouse model of IBD helps reduce the severity of colitis.Citation8 Despite these promising findings, little is known about what confers the therapeutic properties of E. coli Nissle. One key category of missing information is the characterization and analysis of how this probiotic transcriptionally responds to its environment within the gastrointestinal tract. Metatranscriptomic analyses of the gut microbiota have shown that colitis changes the transcriptional response of the residing microbes.Citation9,Citation10 However, these studies have generally focussed on the community responses to colitis and not how individual species, like E. coli Nissle, would respond. Analyzing the transcriptional response to inflammation in E. coli Nissle would help us understand how the probiotic survives in these conditions, and lay the groundwork for an understanding of the mechanisms involved in creating its observed health benefits. Additionally, transcriptional markers of inflammation within this probiotic could be used to design whole-cell biosensors to detect IBD.Citation11,Citation12
In this work, we have investigated the transcriptional response of E. coli Nissle to inflammatory intestinal conditions by RNA-seq analysis, using a DSS-mouse model of colitis. Through differential expression analysis of RNA recovered from stool samples, we discovered 187 differentially expressed genes within the probiotic in response to colitis. Interestingly, genes involved in flagella assembly and formate oxidation were upregulated in response to colitis, suggesting a role for motility and anaerobic formate metabolism in the response to the inflammation. We further validated the transcriptional markers of inflammation by qRT-PCR and independently confirmed 12 of 17 of the differentially expressed genes in the RNA-seq dataset, including flagella assembly and formate hydrogenlyase genes. Finally, we tested the possibility that the transcriptional responses of E. coli Nissle were caused by an increase in a known biomarker for inflammation, nitric oxide. Despite evidence that nitric oxide does regulate flagella assembly and formate hydrogenlyase transcription, in-vitro treatment of E. coli Nissle with a nitric oxide donor did not replicate the transcriptional changes encountered within the inflamed gut environment, leading to the possibility that other factors must be responsible for its transcriptional response to colitis.
Materials and methods
Bacteria strain and growth conditions
The bacteria strain E. coli Nissle 1917 (EcN) was used for all experiments. A chloramphenicol resistant strain was generated by genomically integrating the chloramphenicol-resistant marker within an intergenic region of EcN (dps::glnH) using Recombineering technique and protocols,Citation13 and is available upon request. Bacteria were grown in standard LB media (1% tryptone, 0.5% yeast extract, 1% sodium chloride) with chloramphenicol (10 µg/mL) at 37°C with shaking. EcN was grown overnight in LB media to OD600 of 1 (8E+08 cells) and resuspended in PBS for oral gavage. Spermine NONOate (Sigma) was used as a nitric oxide donor and diluted into water at the desired concentration prior to use.
Germ-free mice protocol
Germ-free C57BL/6 J mice were maintained under standard germ-free conditions by the Division of Comparative Medicine (DCM) at the University of Toronto. Germ-free mice were monitored for microbial contamination by aerobic and anaerobic bacterial culturing and qPCR for microbial DNA. Male and female germ-free mice were then taken outside the isolators, administered with ~1E+09 EcN by oral gavage, then maintained under standard specific-pathogen-free conditions using out-of-the-isolator husbandry, as previously described.Citation14 Briefly, the mice were housed in pre-sterilized polyester filter-top Allentown cages with sterile (irradiated) rodent chow and autoclaved water. Cages were housed in racks separated from other cages within the specific-pathogen-free facility. Mice were handled minimally and weighed in autoclaved weigh boxes. Originally, there were nine mice total included in the experiment, but one control mouse (control mouse 4) reacted negatively to the oral gavage. Control mouse 4 lost significant weight and was found to have higher amounts of non-EcN taxa relative to other samples (Figure S1(a,b)); results from this mouse were therefore excluded from the final analysis. Mice were placed on chloramphenicol water (10 µg/ml) for 5 days and switched to autoclaved drinking water prior to dextran sulfate sodium (Fisher, DSS) induced colitis. Mice were given 2% DSS in autoclaved drinking water for 5 days to induce colitis and reverted to normal drinking water afterward. Mice were sacrificed 2 days following the end of DSS treatment, which coincided with the peak inflammatory response while also allowing enough time to reduce DSS contamination in the stool samples––a known inhibitor of downstream RNA applications.Citation15
RNA, genomic DNA extraction, and RNA-seq
Total RNA from stool samples recovered from the distal colon section of each mice was extracted using the RNeasy® PowerMicrobiome® kit (Qiagen). Briefly, stool samples were resuspended in the lysis buffer into capped tubes with 0.1 mm glass beads and vortexed for 10 min. The soluble extracts from the homogenized stool samples of both DSS-treated and untreated mice were loaded into RNA purification columns, washed and treated on-column with DNAse before eluting in RNase free water and stored at −80°C without any further purification or precipitation procedures. For genomic DNA extraction, the same procedure was used to extract the nucleic acids but without DNaseI treatment. Purified RNA samples were submitted for library preparation (including bacterial ribosomal RNA depletion) and Next-Generation Sequencing at The Center for Applied Genomics (TCAG, Toronto, Ontario, http://www.tcag.ca/) using their Illumina HiSeq2500 platform (125 bp paired-end reads). Raw sequencing reads are available in the NCBI SRA archive, accession number PRJNA695566. Quality control of raw reads was performed using FastQC v0.11.9 to assess read quality of samples. Sequence alignment of paired-end reads was performed using the STAR pipeline v2.7.1a using assembly GCF_003546975.1 of Escherichia coli Nissle 1917 as a reference. R package DESeq2 v1.24.0 was used to perform differential gene expression analysis between the DSS-treated samples (DSS mouse 1, 2, 3, 4, 5) and the control samples (control mouse 1, 2, 3). The default settings as described by Love et al. were used,Citation16 which include the normalization of raw counts followed by gene-wise dispersion estimates and fitting to a negative binomial generalized linear model. False discovery rate of differential expression testing was controlled at a 5% cutoff using the Benjamini–Hochberg procedure.Citation17 Taxonomic classification of reads was assigned by processing paired end FASTQ reads of each mouse stool sample using the MetaPro pipeline under the default settings.Citation18 Low-quality sequences were filtered, followed by host and vector removal through BWA alignment, then rRNA removal was completed using Infernal. From our initial analyses, we identified control mouse 4 as being a clear outlier (Figure S1(c,d)), which, with the previously mentioned problems, was therefore removed from subsequent analyses. After differential expression analysis, gene sequences of differentially expressed genes were annotated to the most closely aligned gene name of the E. coli K-12 strain using the National Center for Biotechnology Information (NCBI) alignment tool Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Pathway and biological function analyses of differentially expressed genes were completed using DAVID bioinformatics online tools (https://david.ncifcrf.gov)Citation19.
Quantitative PCR and RT-PCR
For RNA samples, 2 µg was reverse transcribed into cDNA using a reverse transcriptase kit (Applied Bioscience). For the standard curve, genomic DNA was extracted from E. coli Nissle and serially diluted 1/5 in water 8 times. Primers against each E. coli Nissle gene were pre-validated for specificity and amplification efficiency using the genomic DNA standard curve and are listed in Supplementary file 1. NEB Luna qPCR 2X master mix reagent was used for the qPCR reactions. We used 5 µL reactions of which 0.5 µL of an appropriately diluted cDNA or genomic DNA sample were used as template. qRT-PCR reactions were done in 384 well plates (BioRad); within each plate included the standard curve in technical duplicates and as many samples as could fit in technical duplicates for each target genes and two reference genes (16S rRNA and cysG). Real-time PCR was completed using BioRad Real-Time PCR machine and BioRad CFX Maestro analysis software. For the analysis, the Ct values of each sample were adjusted automatically to the threshold set by the software using the standard curve for each target primer pairs. The average of Ct values from two technical replicates were normalized to the geometric averages of the two reference genes: 16srRNA and cysG. The average Ct values from two technical replicates were normalized to the geometric averages of the two reference genes using the Pfaffl method [equation 3].Citation20 Mean gene expression across biological replicates is relative to the mean normalized Ct value of the control samples. Raw data and qPCR analysis are available in Supplementary file 1.
Results
Experimental overview of E. coli Nissle within a germ-free DSS mouse model of colitis
Mice held initially in germ-free conditions were administered a chloramphenicol resistant E. coli Nissle strain by oral gavage before being transferred to non-sterile facilities, as described in,Citation14 then separated into two groups: 1) control group (3 mice) and 2) DSS-treated group (five mice) (Figure S2(a)). We confirmed that the probiotic successfully colonized all eight mice and remained present at roughly 1E+09 to 1E+10 CFUs per gram of stool throughout the course of the experiment (Figure S2(b)). We also determined the relative bacterial content of genomic DNA in stool collected from post-DSS treated mice using primers specific toward the 16S rDNA of selected bacterial groups.Citation21 For the most part, the mice contained almost exclusively 16S rDNA of the Enterobacteriaceae group of bacteria that contains E. coli Nissle (Figure S2(c)). We also verified that our specific E. coli Nissle strain was present in these samples by using primers specific toward our modified chloramphenicol-resistant E. coli Nissle strain (data not shown). Together with the CFU counts, this confirms that E. coli Nissle is not only present in the mice but is also the most abundant bacteria within their stool content. Both the relative weight changes and lipocalin-2 (Lcn-2) stool levels were monitored over the course of the experiment as positive markers for inflammation.Citation22 Although the relative weight change of the DSS-treated mice (93%) was slightly greater than the control group (96%), those differences were not statistically significant (Figure S2(d)). However, Lcn-2 levels were much higher in the DSS-treated mice (65 058 pg/mg) compared to control mice (293 pg/mg) suggesting colitis had been induced in the treatment group (Figure S2(e)).
Transcriptomic profiling of E. coli Nissle reveals that flagella genes and formate hydrogenlyase genes are upregulated in response to colitis
RNAs were extracted from the recovered colon stool samples from both treatment groups, depleted of bacterial ribosomal RNA, converted into a cDNA library, and sequenced by RNA-seq. The paired-end reads were trimmed and processed before aligning them against the complete genome sequence of E. coli Nissle (see materials and methods for details on the RNA-seq analysis). On average, there were 26 million paired end reads per sample with an average of 24% of the reads uniquely mapped with the E. coli Nissle genome (Table S1). Despite only being administered with a single bacterial strain, there were a number of reads that did not align with E. coli Nissle. Taxonomic classification of the sequenced reads against a database of known bacterial references revealed that only a small yet consistent portion of the putative mRNA reads (8.1% on average) belonged to bacterial families other than Enterobacteriaceae (including Moraxellaceae, Lactobacillaceae, and Erysipelotrichaceae), which is likely the result of transferring the mice into non-sterile facilities after the administration of the probiotic (Figure S3(a), Supplementary file 2). A much greater portion of the putative mRNA reads remained unclassified (48.5% on average) but a small search of a random sampling of those reads showed that these reads mostly belong to the host (Mus musculus) and altogether suggests that the relative abundance of E. coli Nissle mapped reads is appropriate given its context.
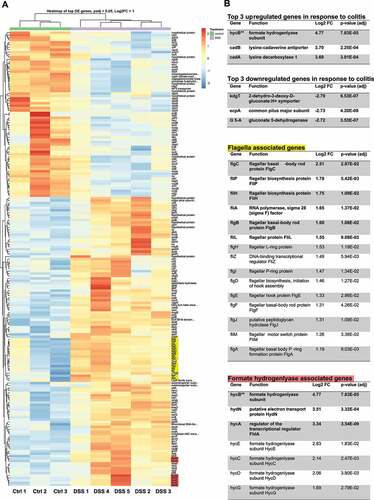
Differential expression analysis identified 187 differentially expressed genes (Log2 fold change >1.0, FDR adjusted p-value <0.05) from a total of 4996 total genes between the samples taken from DSS-treated mice and control mice: 119 upregulated genes and 68 downregulated genes (). Principal component analysis confirmed that samples from DSS-treated mice clustered separately from the control samples, while the DSS-treated samples clustered into two different groups (Figure S3(b)). We suspected that these differences would have been the result of cage effects: members of each cluster coincided precisely with the cages they were held in (Cage 1: DSS 1, 2, 3; Cage 2: DSS 4, 5). The distribution of all identified genes showed that most expression levels were not affected by DSS-induced inflammation, but only a small subset passed our thresholds for statistically significant differential expression (Figure S3(c)). The sequences retrieved from each of the differentially expressed genes in E. coli Nissle were aligned against the genome of the most studied E. coli strain, K-12, to match with its most closely related gene symbol. Eighty-seven percent of the upregulated genes and 85% of the downregulated genes were almost perfectly matched against a corresponding gene in E. coli K-12 (Supplementary file 3). Of the unmatched genes, three genes (3/25) shared over 50% homology with a corresponding fimbrial protein in E. coli K-12 (fimA, filM, fimD) and were therefore annotated as such. The remaining unmatchedgenes were either genes with predicted but unconfirmed protein functions or hypothetical proteins and were excluded from further analysis. Of the upregulated genes in response to colitis with characterized functions, we noted multiple genes involved in the flagellar biosynthesis pathway (fliA, flgC, fliP, fliH, flgB, fliL, flgH, fliZ, flgI, flgD, flgE. flgF, flgJ, flgA, fliM) suggesting a role for motility in the response to inflammation in the intestine. We also observed several genes belonging to the formate hydrogenlyase complex hyc operon (hycA, hycB, hycC, hycD, hycE, hycG), suggesting a role for formate metabolism in the response to colitis. To determine if any other groups of genes were enriched, we subjected our differentially expressed gene lists to pathway and biological process analysis (). As we suspected, flagellar assembly pathway was enriched in our differentially upregulated genes (p value = 4.41E-09) as well as genes involved in amino-acid metabolism (p value = 1.10E-02). Biological processes were enriched for several motility terms and amino acid metabolism but also showed enrichment for anaerobic respiration (p value = 1.41E-02) and nitrate assimilation (p value = 1.60E-02). However, this analysis did not confirm an enrichment for the formate hydrogenlyase pathway even though most of the members of its operon are present. The downregulated genes in response to colitis only showed enrichment for genes within the ribosome pathway (p value = 2.29E-05). The biological processes affected also indicated a negative effect on translation but also on several peptide transport terms.
Table 1. Pathway and biological function enrichment analysis of differentially expressed genes
Figure 1. Differential expression analysis of Escherichia coli Nissle reveals upregulation of flagella and formate hydrogenlyase genes in response to colitis. (a) Heatmap of the differentially expressed genes of E. coli Nissle between DSS-treated mice and untreated (control) mice. There are 187 differentially expressed genes; 119 upregulated and 68 downregulated genes clustered based on similarity. The cutoff set for differential expression were log2 fold change over 1.0 and FDR-adjusted p-values under 0.05. Each column represents samples from either DSS treated mice (5 samples) or control mice (3 samples) clustered based on similarity. Red indicates relative overexpression and blue indicates relative repression. (b) Tables highlighting results of differential expression analysis on the top upregulated/downregulated genes in response to colitis as well as genes associated with flagella assembly and formate hydrogenlyase activity. Functions related to gene symbol was retrieved from the BioCyc online database.Citation23 Log2 fold change (Log2FC) and associated p value are listed for every gene listed. Genes in bold were also validated by qRT-PCR in the subsequent section. Flagella genes (yellow) and formate hydrogen lyase genes (red) are highlighted on the heatmap. “ Formate hydrogenlyase gene hycB is listed in the top three upregulated genes table instead of the formate hydrogenlyase genes table
Independent validation by qRT-PCR confirms upregulation of flagella and formate hydrogenlyase genes in response to colitis
To validate some of the transcriptional biomarkers, we repeated the experiment with a new set of mice (2 treatment groups, n = 4) and collected RNA from the stool samples for qRT-PCR. Of the differentially expressed genes, we chose the top three upregulated and downregulated genes, in terms of their calculated fold change compared to control mice, as well as an additional 11 target genes, to independently verify by qRT-PCR. Of the top three upregulated genes (hycB, cadA, and cadB), all three were confirmed to be upregulated in response to colitis by fold changes of 4.6X, 20.6X, and 18.0X, respectively ()). However, of the top three downregulated genes (gluconate-5-dehydrogenase, ecpA, and kgdT) only one gene (kdgT) was found to be significantly downregulated (−5.1X) in response to colitis ()). These discrepancies were expected given the dissimilarities between the two RNA relative quantification methods and the use of samples from independently replicated experiments. But, given the fact that we were still able to confirm differential expressions for at least some of the target genes (4 out of 6) strengthens the case that those cross-validated genes can be considered genuine transcriptional markers of colitis.
Figure 2. Confirming differential expression of E. coli Nissle genes associated with flagella assembly and formate hydrogenlyase activity in DSS-treated mouse by qRT-PCR. (a) Validation of differential expression of top three upregulated and downregulated genes, (b) genes associated with flagella assembly, (c) genes associated with formate hydrogenlyase activity, and (d) genes involved in dipeptide transportation. RNA was recovered from mouse stool samples colonized with E. coli Nissle of an independently replicated experiment (N = 4 per treatment group). Extracted RNA was converted into cDNA and relative expression levels of target genes was accessed by qRT-PCR with specific primers previously validated for specificity and amplification efficiency against genomic DNA of E. coli Nissle. Gene expression for each target was normalized to the geometric mean of two independent housekeeping genes: 16S rRNA and cysG. Fold increase (or fold decrease as a negative number) between DSS-treated and untreated (control) mice are indicated above each mean as well as the result of Student t-test (* = p value<.05, n.s. = not significant)
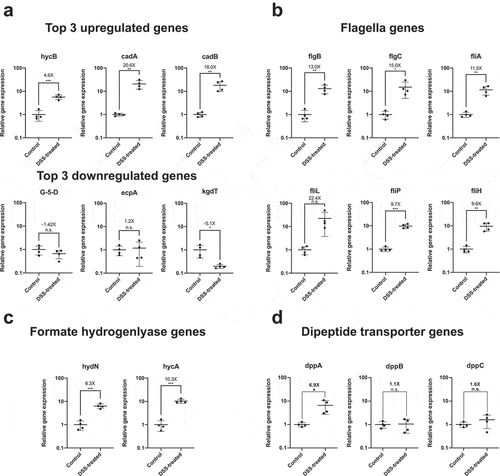
Next, we wanted to validate target genes that were of interest, including genes involved in the flagellar biosynthesis pathway (fliA, flgB, flgC, fliL, flip, fliH); genes involved in dipeptide transport (dppA, dppB, and dppC); and genes associated with the formate hydrogenlyase pathway (hydN and hycA). Of the flagellar genes, 5 out of 6 genes tested showed significantly increased expression (ranging from 9.6X to 15X) in DSS-treated mice compared to the control ()). Interestingly, the transcription factor fliA (sigma factor 28) that is responsible for the transcriptional regulation of flagellar genes is upregulated by 11.5 fold. This strongly suggests that E. coli Nissle is responding to colitis by increasing its flagella assembly pathway and presumably its motility. Two other genes of interest were also independently tested by qRT-PCR. hycA and hydN, together with hycB (one of the top upregulated genes from the RNA-seq analysis), are involved in formate hydrogenlyase activity and regulated by the transcriptional activator flhA in the response to anaerobic fermentation. Both these genes were confirmed to be upregulated in response to colitis (10.3X and 6.3X, respectively) ()). Finally, we were also interested in confirming the downregulation of the dipeptide transport genes; however, our independent validation of their expression levels in DSS-treated mice did not reveal a significant downregulation compared to control mice ()). In fact, dppA expression was relatively increased in DSS-treated mice compared to control mice (6.9X) as measured by qRT-PCR unlike in the RNA-seq analysis, again suggesting some variability between the RNA-seq experimental dataset and independent qRT-PCR experiments. What causes differential expression of these genes within E. coli Nissle appears to be dependent on the context of the inflamed colon environment as neither DSS alone nor the fecal supernatants recovered from DSS-treated mice were sufficient to induce the expression of the flagella and formate hydrogenlyase genes in E. coli Nissle grown in vitro (Figures S4 and S5). Overall, we were able to validate 12 out of the 17 E. coli Nissle genes tested for differential expression in response to colitis.
Nitric oxide is not solely responsible for the transcriptional response of E. coli Nissle toward colitis
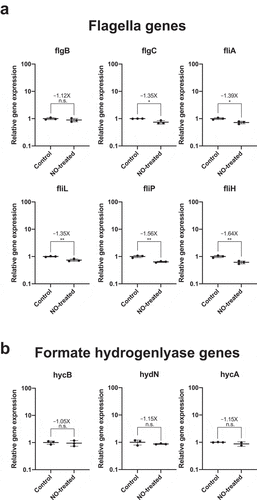
Both the genes involved in motility and the formate hydrogenlyase complex have been shown to have binding sites for the nitric -oxide-dependent NsrR transcription factor upstream of their operons in E. coli, and nitric oxide stress has been shown to increase the transcription of flagellin genes increasing its motility.Citation24 Interestingly, nitric oxide is a known biomarker for IBD.Citation25 Nitric oxide, along with reactive oxygen species, is believed to be one of the main precursors for many other reactive oxidants including peroxynitrite, released in the gut by the innate immune system as a response to inflammation and invading pathogens.Citation26 To find that genes involved in flagella assembly and in the formate hydrogen lyase complex were both upregulated in response to colitis led us to propose that this might be the result of increased nitric oxide levels in the gut. In order to test this hypothesis, we compared gene expression of motility genes and formate hydrogenlyase genes in E. coli Nissle cultures grown either with or without a nitric oxide donor, NONOate. We initially determined an appropriate concentration and induction needed to initiate stress response E. coli Nissle. We determined that a concentration of 100 µM of NONOate and 1 h induction was optimal for our experiment as it caused a 50% growth defect compared to untreated cells, indicating that the levels of nitric oxide are sufficiently high enough to activate a cellular response in E. coli Nissle’s without completely arresting growth (Figure S6). However, transcription levels of the flagella and formate hydrogenlyase genes between nitric oxide-treated and untreated cells did not increase; in fact, most of the motility genes slightly decreased in response to nitric oxide stress (). This suggests that nitric oxide cannot by itself be responsible for the transcriptional activation of motility and formate hydrogenlyase genes in our mouse model of colitis and that there are likely other factors that help regulate the transcriptional response of E. coli Nissle upon induction of inflammation. Repetition of the nitric oxide experiment under anaerobic conditions yielded more substantial transcriptional decreases in all genes except fliA (results not shown), again supporting the notion that the in vivo results do not arise solely from nitric oxide stress.
Figure 3. Nitric oxide stress alone is not sufficient to increase the transcription of flagella biosynthesis genes and formate hydrogenlyase genes in E. coli Nissle. Cultures of E. coli Nissle were grown in triplicate to their logarithmic growth phase (LB media, 37°C, 220 rpm) and subjected to nitric oxide stress by addition of 100 µM of a nitric oxide donor, spermine NONOate, for 1 h. Pellets were harvested and RNA was extracted for qRT-PCR. Gene expression for each target was normalized to the geometric mean of two independent housekeeping genes: 16S rRNA and cysG. Fold increase (or fold decrease as a negative number) between NONOate-treated and untreated (control) cells are indicated above each mean as well as the result of Student t-test (* = p value<.05, n.s. = not significant)
DISCUSSION
Summary of results
In this paper, we determined the transcriptional response of E. coli Nissle within a DSS-mouse model of colitis. RNA-seq analysis identified 187 differentially expressed genes within this context, with several genes belonging to the flagella assembly pathway and formate hydrogenlyase complex. A subset of these genes were also independently verified to be differentially expressed by subsequent qRT-PCR experiments on a completely new set of mice. In total, we were able to confirm 12 transcriptional markers of inflammation within E. coli Nissle.
Potential explanation for the upregulation of flagella and formate hydrogenlyase genes within E. coli Nissle as a response to colitis
There is evidence that flagellar biosynthesis pathways in the gut microbiota are affected by colitis, but the specific response differs based on the mouse models used for investigation. In a Tbet−/− Rag2−/− ulcerative colitis (TRUC) and the Helicobacter hepaticus models of colitis, flagella genes were found to be enriched in the gut microbiota;Citation10,Citation27 whereas in the DSS model of colitis, the presence of flagella genes decreased.Citation28 However, these studies focussed on the community response of the gut microbiota toward colitis making it hard to determine its effect of specific bacterial species. In our isolated context, a DSS-induced colitis causes an increase in flagella associated gene transcripts in E. coli Nissle. These molecules were discovered to be the main pathogen-associated-molecular pattern that stimulates the activity of the toll-like receptor 5 (TLR-5) found on the surface of intestinal epithelial cells.Citation29 Therefore, an increased abundance of flagella transcripts in E. coli Nissle might not only be an adaptation toward its new environment but might also have a significant effect on the innate immune response of the host. Increased flagella biosynthesis could also help E. coli Nissle maintain its niche within the intestine. Bacterial flagellum is considered an important factor for adherence and virulence.Citation30 The flagellin gene in E. coli Nissle (fliC) has been found to be responsible for efficient adhesion of the bacteria toward tissues.Citation31 Thus, increased flagella component expression could lead to increased adherence within the intestine. In this particular mouse model of colitis (DSS), the treatment leads to damaged endothelial cell wall, loss of crypt formations, mucosal edema, and infiltration of immune cells in the mucosa.Citation32 More flagella might allow E. coli Nissle to maintain its niche but also increase its motility to help it escape the host immune response. Furthermore, the components of the flagella organelles of E. coli Nissle are important factors for host recognition as well as its anti-inflammatory effects. Deletion of the hypervariable region of the flagellin gene fliC in E. coli Nissle inhibits its anti-inflammatory properties in a DSS-mouse model of colitis.Citation33 This effect is mediated and dependent on an interaction with the TLR-5 recognition domain on epithelial cells. Interestingly, administration of recombinant flagellin proteins from E. coli Nissle but not from other non-probiotic E. coli strains can recapitulate this anti-inflammatory effect in mice, providing an explanation for its probiotic properties.
Changes in formate metabolism in E. coli Nissle has also been previously linked with adaptation to the colitic environment. Increased levels of formate originating from the gut microbiota has been detected in the gut lumen of a DSS-mouse model of colitis, and formate dehydrogenase genes improve the colonization and growth of E. coli Nissle within the inflamed gut environment.Citation34 There are three major formate dehydrogenases in E. coli responsible for the oxidation of formate, each competing for the same pool of substrate and each activated under specific conditions: FDH-N, FDH-O, and FDH-H. FDH-N is activated under anaerobic conditions and when nitrate is present,Citation35 FDH-O is active under aerobic conditions or when nitrate is present,Citation35 and FDH-F, together with the formate hydrogenlyase complex (FHL) is activated in anaerobic conditions and in the absence of nitrate.Citation36 Our data show that most of the genes encoded by hyc operon hycABCDEFGHI, which forms part of the FHL complex, is transcriptionally activated in E. coli Nissle within a DSS-mouse model of colitis. We independently verified the transcriptional activation of hycA and hycB as well as another gene thought to be involved in FHL activity, hydN, by qRT-PCR. However, it was previously demonstrated that FDH-N, not FHL, was mostly responsible for formate oxidation in a DSS-mouse model of colitis and was also necessary to confer a fitness advantage for E. coli Nissle within the inflamed gut.Citation34 Nonetheless, these two conclusions do not necessarily contradict each other. Expression of the hyc operon is normally repressed by nitrate and oxygen levels but can be relieved by increased formate concentrations.Citation36 This means that both formate metabolic pathways could be simultaneously activated in response to increased formate levels. Additionally, this same study showed that FDH-N no longer confers the same fitness benefit to E. coli Nissle administered to germ-free mice.Citation34 This may suggest that different formate oxidation pathway may be taken by E. coli Nissle when it is the only microbe present in the gut compared to conventionally raised mice. Activation of the FHL complex under these conditions could potentially be used by E. coli Nissle to adapt to its environment, helping deplete excess intracellular formate and maintaining viability in the inflamed gut environment.
Nitric oxide effect on the expression of flagella assembly and formate hydrogenlyase genes in vitro
From in vitro studies, both the flagella assembly genes and the anaerobic formate oxidation genes are regulated by the nitric oxide sensing repressor in E. coli. The fliAZY and fliLMNOPQR operons have a binding site for the nitric oxide-sensing transcriptional repressor NsrR and are transcriptionally activated by addition of nitric oxide.Citation24 Furthermore, it has been demonstrated that nitric oxide increases the motility and surface adherence of E. coli.Citation24 These phenotypes could be a mechanism for E. coli to establish a niche within the intestine. The hycA gene belonging to the hyc operon also contains binding sites for the NsrR repressor and is transcriptionally derepressed by the addition of nitrite.Citation24,Citation37 Although seemingly unrelated, this fact caught our attention because nitric oxide is a well-known biomarker for IBD and is found in elevated concentrations in the gut lumen of patients with ulcerative colitis.Citation38 Detection of nitric oxide and its derivatives like nitrate, tetrathionate, and thiosulfate have been used to create whole-cell biosensors to detect inflammation in the intestine.Citation39–42 It made sense to try and see if the transcriptional response of E. coli Nissle in the context of colitis was caused by increased levels of nitric oxide in the gut. However, our in vitro data suggest that nitric oxide stress by itself does not lead to the same transcriptional response within our probiotic. Neither the flagella genes nor the formate hydrogenlyase genes were derepressed by the addition of nitric oxide. Although it does not negate the potential effect of nitric oxide on E. coli Nissle, this does suggest that there are other factors required for the upregulation of flagella and formate hydrogenlyase genes.
Potential caveats and concluding remarks
There are some limitations to our experimental design, primarily because of the use of germ-free mice prior to E. coli Nissle administration. Presumably, in conventionally raised mice, there are interactions between the resident gut microbes and E. coli Nissle that would affect the transcriptional behavior of the probiotic differently than when E. coli Nissle is the only bacterium present. However, using fully colonized mice would have proven troublesome for RNA-seq analysis. Already in the mice used in this study, which are predominantly colonized with E. coli Nissle, only between 48.6% and 2.9% of the sequencing reads from our eight different samples belonged to E. coli Nissle with a large portion of the rest of the reads belonging to the host. It becomes a challenge to achieve enough sequencing coverage for our specific bacterium when RNA from other species is also present in the stool sample. We believe that the information we gathered within this specific context would still be useful for other researchers as this is a well-used model for studying the therapeutic potential of probiotics. It should also be noted that the DSS-treated mice did not significantly lose weight compared to the control mice (Figure S2(d)). Given the fact that E. coli Nissle confers protection against DSS-treatment in mice,Citation8 it is possible that colonization of E. coli Nissle prevented the DSS-treated mice from losing weight. However, we still detected elevated Lcn-2 levels in the stool of DSS-treated mice (Figure S2(e)) and found the presence of blood in the stool that confirmed that DSS-treated mice had inflammation in the intestine.
In conclusion, we present the transcriptional response of E. coli Nissle to colitis in a DSS-model of colitis. Through an independent verification experiment, we confirmed that part of its response involves increasing expression of flagella and formate hydrogenlyase genes for reasons that remain unclear but likely reflect an adaptive response to the prevailing inflammatory environment within the intestine. We have confirmed the differential transcription of 12 genes, offering the potential for these genes to be used as biomarkers for the presence of inflammation. Further verification of these transcriptional biomarkers should be validated in conventionally raised mice to determine if these effects are dependent on the presence of other microbes. Future experiments could also look at how increasing flagella synthesis and formate metabolism in E. coli Nissle, might be helping its survival in the inflamed gut environment and conferring health benefits to the host whether by increasing the motile capabilities of the bacterium, increasing host recognition and stimulation of cell surface receptors, or shifting metabolic pathways to respond to increased formate levels.
Supplemental Material
Download Zip (3.6 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307. doi:10.1038/nature10209.
- Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–14.
- Kaur N, Chen -C-C, Luther J, Kao JY. Intestinal dysbiosis in inflammatory bowel disease. Gut Microbes. 2011;2(4):211–216. doi:10.4161/gmic.2.4.17863.
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. doi:10.1186/gb-2012-13-9-r79.
- Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, D’Amato M, Bonfiglio F, McDonald D, Gonzalez A, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017;2(5):17004. doi:10.1038/nmicrobiol.2017.4.
- Sonnenborn U, Robertson L. Escherichia coli strain Nissle 1917—from bench to bedside and back: history of a special Escherichia coli strain with probiotic properties. FEMS Microbiol. Lett. 2016;363(19):fnw212. doi:10.1093/femsle/fnw212.
- Scaldaferri F, Gerardi V, Mangiola F, Lopetuso LR, Pizzoferrato M, Petito V, Papa A, Stojanovic J, Poscia A, Cammarota G, et al. Role and mechanisms of action of Escherichia coli Nissle 1917 in the maintenance of remission in ulcerative colitis patients: an update. World J. Gastroenterol. 2016;22(24):5505–5511. doi:10.3748/wjg.v22.i24.5505.
- Rodríguez-Nogales A, Algieri F, Garrido-Mesa J, Vezza T, Utrilla MP, Chueca N, Fernández-Caballero JA, García F, Rodríguez-Cabezas ME, Gálvez J, et al. The administration of Escherichia coli Nissle 1917 ameliorates development of DSS-induced colitis in mice. Front. Pharmacol. 2018;9:1–12. doi:10.3389/fphar.2018.00468.
- Schirmer M, Huttenhower C. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018;3(7351). doi:10.1038/s41564-017-0089-z.
- Ilott NE, Bollrath J, Danne C, Schiering C, Shale M, Adelmann K, Krausgruber T, Heger A, Sims D, Powrie F, et al. Defining the microbial transcriptional response to colitis through integrated host and microbiome profiling. ISME J. 2016;10(10):2389–2404. doi:10.1038/ismej.2016.40.
- Riglar DT, Silver PA. Engineering bacteria for diagnostic and therapeutic applications. Nat. Rev. Microbiol. 2018;16(4):214–225. doi:10.1038/nrmicro.2017.172.
- Landry BP, Tabor JJ, Allen Britton R, Cani PD. Engineering diagnostic and therapeutic gut bacteria. Microbiol. Spectr. 2017;5(5):333–361. doi:10.1128/microbiolspec.BAD-0020-2017.
- Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4(2):206–223. doi:10.1038/nprot.2008.227.
- Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci. Transl. Med. 2014;6(220):220ra11. doi:10.1126/scitranslmed.3008051.
- Viennois E, Chen F, Laroui H, Baker MT, Merlin D. Dextran sodium sulfate inhibits the activities of both polymerase and reverse transcriptase: lithium chloride purification, a rapid and efficient technique to purify RNA. BMC Res Notes. 2013;6(1):360. doi:10.1186/1756-0500-6-360.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
- Taj B, Adeolu M, Xiong X, Ang J, Nursimulu N, Parkinson J. MetaPro: a scalable and reproducible data processing and analysis pipeline for metatranscriptomic investigation of microbial communities. bioRxiv. 2021;02.23.432558. doi:10.1101/2021.02.23.432558.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):45e– 45. doi:10.1093/nar/29.9.e45.
- Robertson SJ, Zhou JY, Geddes K, Rubino SJ, Cho JH, Girardin SE, Philpott DJ. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes. 2013;4(3):222–231. doi:10.4161/gmic.24373.
- Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One. 2012;7(9):e44328. doi:10.1371/journal.pone.0044328.
- Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, Keseler IM, Krummenacker M, Midford PE, Ong Q, et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019;20(4):1085–1093. doi:10.1093/bib/bbx085.
- Partridge JD, Bodenmiller DM, Humphrys MS, Spiro S. NsrR targets in the Escherichia coli genome: new insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol. Microbiol. 2009;73(4):680–694. doi:10.1111/j.1365-2958.2009.06799.x.
- Kolios G, Valatas V, Ward SG. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113(4):427–437. doi:10.1111/j.1365-2567.2004.01984.x.
- Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet (London, Eng). 1994;344(8926):859–861. doi:10.1016/S0140-6736(94)92831-2.
- Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, Gallini CA, Beal C, van Hylckama-Vlieg JE, Ballal SA, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 2014;8(7):1403–1417. doi:10.1038/ismej.2014.3.
- Schwab C, Berry D, Rauch I, Rennisch I, Ramesmayer J, Hainzl E, Heider S, Decker T, Kenner L, Müller M, et al. Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J. 2014;8(5):1101–1114. doi:10.1038/ismej.2013.223.
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410(6832):1099–1103. doi:10.1038/35074106.
- Haiko J, Westerlund-Wikström B. The role of the bacterial flagellum in adhesion and virulence. Biol (Basel). 2013;2:1242–1267.
- Troge A, Scheppach W, Schroeder BO, Rund SA, Heuner K, Wehkamp J, Stange EF, Oelschlaeger TA. More than a marine propeller – the flagellum of the probiotic Escherichia coli strain Nissle 1917 is the major adhesin mediating binding to human mucus. Int. J. Med. Microbiol. 2012;302(7–8):304–314. doi:10.1016/j.ijmm.2012.09.004.
- Solomon L, Mansor S, Mallon P, Donnelly E, Hoper M, Loughrey M, Kirk S, Gardiner K. The dextran sulphate sodium (DSS) model of colitis: an overview. Comp. Clin. Path. 2010;19(3):235–239. doi:10.1007/s00580-010-0979-4.
- Steimle A, Menz S, Bender A, Ball B, Weber ANR, Hagemann T, Lange A, Maerz JK, Parusel R, Michaelis L, et al. Flagellin hypervariable region determines symbiotic properties of commensal Escherichia coli strains. PLoS Biol. 2019;17(6):e3000334. doi:10.1371/journal.pbio.3000334.
- Hughes ER, Winter MG, Duerkop BA, Spiga L, Furtado de Carvalho T, Zhu W, Gillis CC, Büttner L, Smoot MP, Behrendt CL, et al. Microbial respiration and formate oxidation as metabolic signatures of inflammation-associated dysbiosis. Cell Host Microbe. 2017;21(2):208–219. doi:10.1016/j.chom.2017.01.005.
- Sawers G, Heider J, Zehelein E, Böck A. Expression and operon structure of the sel genes of Escherichia coli and identification of a third selenium-containing formate dehydrogenase isoenzyme. J Bacteriol. 1991;173(16):4983–4993. doi:10.1128/jb.173.16.4983-4993.1991.
- Rossmann R, Sawers G, Böck A. Mechanism of regulation of the formate-hydrogenlyase pathway by oxygen, nitrate, and pH: definition of the formate regulon. Mol. Microbiol. 1991;5(11):2807–2814. doi:10.1111/j.1365-2958.1991.tb01989.x.
- Constantinidou C, Hobman JL, Griffiths L, Patel MD, Penn CW, Cole JA, Overton TW. A reassessment of the FNR regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J. Biol. Chem. 2006;281(8):4802–4815. doi:10.1074/jbc.M512312200.
- Lundberg JON, Lundberg JM, Alving K, Hellström PM. Greatly increased luminal nitric oxide in ulcerative colitis. Lancet. 1994;344(8938):1673–1674. doi:10.1016/S0140-6736(94)90460-X.
- Woo S-G, Moon S-J, Kim SK, Kim TH, Lim HS, Yeon G-H, Sung BH, Lee C-H, Lee S-G, Hwang JH, et al. A designed whole-cell biosensor for live diagnosis of gut inflammation through nitrate sensing. Biosens. Bioelectron. 2020;168:112523. doi:10.1016/j.bios.2020.112523.
- Archer EJ, Robinson AB, Süel GM. Engineered E. coli that detect and respond to gut inflammation through nitric oxide sensing. ACS Synth. Biol. 2012;1(10):451–457. doi:10.1021/sb3000595.
- Daeffler KN, Galley JD, Sheth RU, Ortiz‐Velez LC, Bibb CO, Shroyer NF, Britton RA, Tabor JJ. Engineering bacterial thiosulfate and tetrathionate sensors for detecting gut inflammation. Mol. Syst. Biol. 2017;13(4):923. doi:10.15252/msb.20167416.
- Riglar DT, Giessen TW, Baym M, Kerns SJ, Niederhuber MJ, Bronson RT, Kotula JW, Gerber GK, Way JC, Silver PA, et al. Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol. 2017;35(7):653–658. doi:10.1038/nbt.3879.