
ABSTRACT
The gut microbiota is essential for maintenance and repair of the intestinal epithelial barrier. As shifts in both intestinal epithelial barrier function and microbiota composition are found in inflammatory bowel disease patients, it is critical to understand the role of distinct bacteria in regulating barrier repair. We identified a mouse commensal E. coli isolate, GDAR2-2, that protects mice from Citrobacter rodentium infection and dextran sulfate sodium-induced colitis. Colonization with GDAR2-2 in mice resulted in expansion of CX3CR1+ mononuclear phagocytes, including CX3CR1+ macrophages/dendritic cells and monocytes, along with IL-22-secreting type 3 innate lymphoid cells and improved epithelial barrier function. In vitro co-culture of macrophages with GDAR2-2 resulted in IL-1β production. In vivo, protection after GDAR2-2 colonization was lost after depletion of CX3CR1+ MNPs, or blockade of IL-1β or IL-22. We further identified human commensal E. coli isolates that similarly protect mice from C. rodentium infection through CX3CR1+ MNP and IL-1β production. Together, these findings demonstrate an unexpected role for commensal bacteria in promoting IL-1β secretion to support intestinal barrier repair.
Introduction
Commensal bacteria have coevolved with their hosts and support multiple host functions including digestion and absorption of nutrients, modulation of immune responses, maintenance of the epithelium and regulation of intestinal motility.Citation1 The greatest load of microbes resides within the large intestine, where a single layer of epithelial cells forms a barrier to separate intestinal contents from host internal tissues.Citation2,Citation3 Thus, protection and repair of this barrier are key to maintaining digestion as well as limiting entry of intestinal microbes into the gut tissue. Signals from the microbiota support epithelial functions with increased pathology in models of epithelial damage in germ-free or antibiotic-treated mice.Citation4–6 Further, altered interactions between host and microbiota can exacerbate intestinal inflammation and underlie chronic inflammatory disorders including inflammatory bowel diseases (IBD).Citation2,Citation7–9
Barrier repair is supported by the immune system. Mononuclear phagocytes (MNPs) expressing the chemokine receptor CX3CR1 are central regulators of this process,Citation10,Citation11 with multiple functions regulated by the microbiota.Citation10–13 CX3CR1+ MNPs arise from Ly6Chi monocyte precursors that differentiate within the tissue into dendritic cells (DCs) or macrophages.Citation14–16 In the steady state, lamina propria CX3CR1+ MNPs are highly phagocytic and clear apoptotic cells, cellular debris, and microbes.Citation17 After microbial exposure, CX3CR1+ MNPs produce limited pro-inflammatory cytokines such as IL-6 or TNF but produce high amounts of the anti-inflammatory cytokine IL-10.12,Citation18,Citation19 During intestinal injury, microbial signals drive IL-1β and IL-23 production from CX3CR1+ MNPs which activates type 3 innate lymphoid cells (ILC3s) to secret IL-22. This pathway supports barrier repair and anti-microbial defenses.Citation10,Citation20–23 IL-22 induces epithelial cell proliferation, survival,Citation24,Citation25 and production of anti-microbial peptides (AMPs), such as RegIIIβ and RegIIIγ.Citation21 IL-22 also regulates goblet cell regeneration and mucus secretion.Citation21,Citation26
Numerous studies describe changes in the intestinal microbiota composition, also known as dysbiosis, that are associated with inflammatory diseases. In IBD, decreased Bacteroidetes along with increased mucosa-associated bacteria including Proteobacteria,Citation7–9 a phylum including E. coli, are found. This increases opportunity for direct interaction between the microbes and host tissue potentially impacts disease severity. Similar microbial shifts are also observed in diseases such as arthritis, diabetes, and asthma.Citation27–29 As the microbiota orchestrates the development and functions of the immune system, changes in bacterial composition are thought to underlie inflammatory disorders. However, the role of specific microbes in such diseases are still being determined.
Here, we identified a mouse commensal E. coli isolate, GDAR2-2, that protects mice from increased damage after either Citrobacter rodentium infection or dextran sulfate sodium (DSS) treatment. After depleting the microbiota with antibiotics, colonization with GDAR2-2 promotes CX3CR1+ MNP expansion in the intestine, which then amplifies IL-22 secretion by type 3 innate lymphoid cells (ILC3s) and promotes transit-amplifying cell proliferation. Unexpectedly, this protection depends on IL-1β but not IL-23 production. Blockade of IL-22 also results in loss of protection. We further identified additional human intestinal E. coli that induce IL-1β production and offer similar intestinal protection. These findings highlight the pro-inflammatory pathways activated by intestinal microbes can be critical to limit intestinal damage and promote barrier repair.
Results
Commensal microbiota protects mice in models of colitis
We and others previously showed that the microbiota promotes barrier repair and is protective in models of colitis.Citation10 To better understand protective signals induced by the microbiota, we treated wild type (WT) mice with the broad-spectrum antibiotics ampicillin, vancomycin, metronidazole and neomycin (AVMN) cocktail to deplete the microbiota and infected mice with C. rodentium, a model of attaching and effacing (A/E) E. coli infection.Citation21,Citation30 Unlike previous reports,Citation4,Citation31 treatment with AVMN did not alter susceptibility to C. rodentium infection as demonstrated by no increase in weight loss, mortality, or decrease in colon length (). To test whether this regime depleted all intestinal microbes, we utilized qPCR to detect 16s rRNA and found no reduction in intestinal microbes ()). We then treated mice with AVMN in combination with streptomycin (AVMN+S) prior to administration of C. rodentium. As assessed by qPCR, this treatment was sufficient to deplete intestinal microbes below the level of detection ()). Upon infection with C. rodentium, AVMN+S-treated mice showed increased susceptibility to C. rodentium with increased weight loss and shorter colon length, indicating more severe intestinal inflammation ().
Figure 1. AVMN-resistant mouse commensal bacteria protects mice from C. rodentium infection. (a-c) WT C57BL/6 (B6) mice treated with AVMN, AVMN+S or left untreated were infected with C. rodentium. (a) weight change, (b) survival rate, and (c) colon length at day 12 post infection. (d-e) AVMN-resistant mouse commensal bacteria identified in feces. (d) Total fecal bacteria as detected by qPCR. (e) Bacteria colony forming units (CFU) as determined by plating on blood agar plates. Data are representative from at least 2 experimental repeats. (a,d,e) Data are shown as mean ± SEM compared by one-way ANOVA with Bonferroni correction or (b) log rank test. (c) Data points are individual mice with mean as compared by one-way ANOVA with Bonferroni correction. **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. none detected (n.d.). Dotted line represents limit of detection for assay.
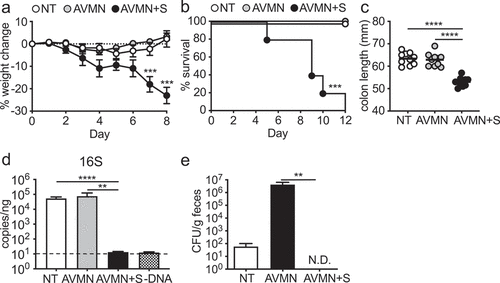
To identify the microbe(s) that offer protection after AVMN treatment, we cultured fecal microbes from AVMN-treated mice under aerobic and anaerobic conditions. As predicted by qPCR analysis, we found bacterial growth from AVMN but not AVMN+S-treated mice with equivalent growth under both aerobic and anaerobic conditions () and data not shown).
We isolated 20 colonies from the AVMN-treated group and identified all as E. coli by sequencing small subunit ribosomal 16S gene amplicons and serotyping as O112:H8. We further confirmed that all isolates were resistant to the AVMN antibiotic cocktail but remained susceptible to streptomycin and gentamicin. By PCR or culture of colonic contents from AVMN treated mice we were unable to identify outgrowth of any additional bacteria.
To further characterize the E. coli, we selected one isolate for genomic sequencing. The complete genome of the B1 phylotype, mouse derived E. coli isolate GDAR2-2 was sequenced using PacBio and assembled de novo into two fully closed contigs. This consisted of the bacterial genome of 4,928,781 base pairs (bp) in length with a 50.73% GC content. We also identified an associated 71,810 bp IncF low copy number plasmid with 94 predicted coding genes with mostly hypothetical functions. Sequence annotations of the genome predicted 4841 coding sequences (Figure S1a) including the presence of genes commonly found associated with commensal E. coli. We also found adhesion genes for long polar fimbriae and pilus assembly proteins.
To test whether treatment with individual antibiotics were sufficient to disrupt gut commensal bacteria colonization, we utilized ampicillin alone and ampicillin in combination with streptomycin. Treatment with ampicillin and streptomycin was sufficient to reduce intestinal bacteria load to a level below qPCR detection (Figure S2a). We utilized this antibiotic combination (ABX) in subsequent experiments.
To confirm colonization with GDAR2-2 was sufficient to protect mice from C. rodentium infection, we treated mice with ABX and then left them uncolonized or colonized with the lab adapted E. coli strain MG1655 (K-12) or GDAR2-2 before infection with C. rodentium. Uncolonized mice or mice colonized with K-12 were equally susceptible to C. rodentium infection with equivalent weight loss, decreased survival, shorter colon lengths, and enhanced severity of intestinal pathology (). In contrast, GDAR2-2 colonized mice were protected from C. rodentium infection with reduced weight loss, increased survival, longer colon length, and ameliorated pathology with lower histological colitis scores after C. rodentium infection (). Supporting improved barrier function in GDAR2-2 colonized mice, unlike K-12, we found lower fecal albumin and increased expression of the tight junction protein, Cldn1, in the intestine of GDAR2-2 colonized mice as compared to uncolonized mice ().
Figure 2. Mouse commensal E. coli isolate GDAR2-2 protects mice from C. rodentium infection. (a-i) Ampicillin and streptomycin (ABX)-treated B6 mice were colonized with lab strain E. coli K-12, GDAR2-2 or left uncolonized and further infected with C. rodentium. (a) weight change, (b) survival rate, (c) colon length 2 weeks after infection. (d-e) Representative H&E staining and colitis scores of colon. (f) albumin concentration in feces was measured at 4 days post infection. Expression of (g) mCldn1, (h) mAxin2 and (i) mLgr5 in colon 4 days after infection. Feces were collected at indicated time points for (j) E. coli and (k) C. rodentium quantification of bacterial load by qPCR. (l-m) 3 days after infection, colon tissues were collected and washed. Mucosa-associated (l) E. coli and (m) C. rodentium were determined by qPCR. Data are representative of at least 2 independent experiments. (a) Data are shown in mean ± SEM (a) or individual mice (c, e-m) and compared by one-way ANOVA with Bonferroni correction (a, c, e, h-k), Kruskal-Wallis with Dunn’s multiple comparison (f-g), log rank test (b), or Student’s t test (l-m). *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
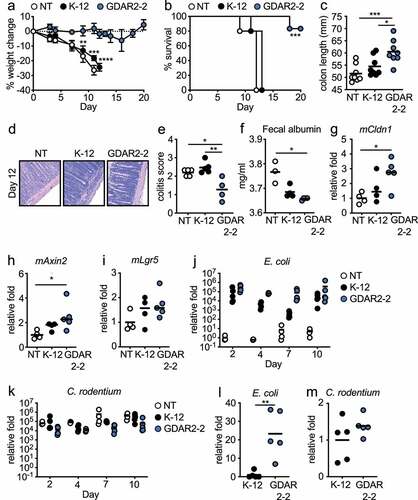
Regeneration of intestinal epithelium occurs by inducing proliferation of transit-amplifying cellsCitation32 or by induction of Wnt agonist that promotes stem cell proliferationCitation33,Citation34 with both pathways shown to play a role in protection from C. rodentium infection.Citation33,Citation34 To determine if either of these pathways was activated after GDAR2-2 colonization, we utilized qPCR and found increased expression of transit-amplifying cell marker, Axin2 in the colon of GDAR2-2-colonized C. rodentium infected mice ()). In contrast, we did not find activation of the Wnt pathway, as expression of the Wnt agonist, Rspo3, was similar between untreated, K-12, and GDAR2-2 colonized C. rodentium infected mice (Figure S3a). Further, the expression of Lgr5, a stem cell marker and Wnt target gene, was also not upregulated in GDAR2-2 colonized mice ()). We also found increased expression of Ascl2 in GDAR2-2-colonized mice which could indicate epithelial differentiation in GDAR2-2 mediated protectionCitation35 (Figure S3b). These data suggest that GDAR2-2 colonization is sufficient to protect mice from C. rodentium infection by increasing proliferation of transit-amplifying cells.
To test if GDAR2-2 inhibited C. rodentium intestinal colonization, we determined the levels of E. coli and C. rodentium in feces and intestinal mucosa of ABX-treated mice colonized with K-12 or GDAR2-2. By analyzing relative bacterial abundance in feces, we found both E. coli colonized equivalently at day of infection with C. rodentium ()). However, colonization with either E. coli did not affect C. rodentium colonization levels as we found comparable levels of C. rodentium in feces in all groups and timepoints ()). To test if there were differences in colonization at the intestinal mucosa, we performed qPCR of mucosa-associated bacteria in the intestine and found higher levels of GDAR2-2 compared to K-12 ()). Our findings supported genomic analyses indicating that GDAR2-2 carries adherence genes such as long polar fimbria and pilus assembly proteins, both of which could mediate epithelial adhesion.Citation36–38 However, we found no difference in mucosa C. rodentium levels between K-12 and GDAR2-2-colonized mice ()). Together this suggests that protection after GDAR2-2 colonization is not due to competition with C. rodentium as has been shown with other E. coli isolates.Citation39
To determine if protection after GDAR2-2 colonization was specific for C. rodentium infection, we treated mice with ABX before treatment with DSS which causes damage to the intestinal epithelium.Citation40 As with C. rodentium infection, mice treated with ABX succumbed to DSS-induced colitis with significant weight loss (Figure S4(a-b)). Pre-colonization with GDAR2-2 protected mice from increased weight loss with increased survival after DSS treatment (Figure S4(c-d)).
Mouse commensal E. coli induces ILC3 IL-22 secretion and epithelium regeneration
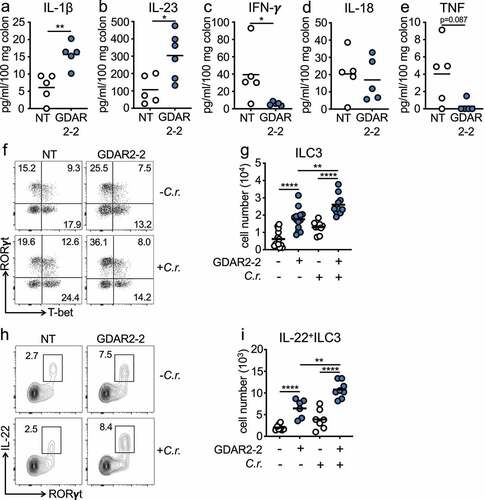
To understand how tissue immune responses were changed after GDAR2-2 colonization, we colonized ABX-treated mice with GDAR2-2 before infection with C. rodentium and measured cytokine secretion in colon explants 6 days post infection. In GDAR2-2-colonized mice, we found elevated colon IL-1β and IL-23 secretion () alongside decreased IFN-γ secretion ()). Similar IL-18 and TNF levels were detected in both groups (). IL-1β and IL-23 both induce type three innate lymphoid cell (ILC3) production of IL-22 which is required for protection from C. rodentium infection.Citation10,Citation21,Citation22 To determine if GDAR2-2 induced ILC3 IL-22 secretion, we inoculated ABX-treated WT mice with or without GDAR2-2 and left untreated or infected with C. rodentium and performed flow cytometry of lamina propria cells. In the large intestine lamina propria of GDAR2-2-colonized mice, we found higher numbers of ILC3 with an increased proportion making IL-22 as compared to uncolonized mice (). After C. rodentium infection, we saw limited expansion of ILC3 or induction of IL-22 after infection in the absence of GDAR2-2 (). In contrast, in C. rodentium-infected GDAR2-2-colonized mice, we saw expansion of ILC3 with a higher number producing IL-22 ().
Figure 3. GDAR2-2 induces ILC3 and IL-22+ILC3 expansion. (a-e) ABX-treated B6 mice were colonized with GDAR2-2 or left un-colonized and infected with C. rodentium. 6 days after infection, colon secretion of (a) IL-1β, (b) IL-23, (c) IFN-γ, (d) IL-18 and (e) TNF was measured by LegendPlex. (f-i) ABX-treated B6 mice were colonized with GDAR2-2 or left un-colonized and infected with or without C. rodentium. Five days after infection, ILC3s and IL-22+ILC3s from large intestine laminar propria were isolated and analyzed by flow cytometry. (f) Representative dot plots of live lin−Eomes−CD90+ cells and (g) absolute number of live lin−Eomes−CD90+RORγt+ILC3. (h) Representative dot plots of live lin−Eomes−CD90+ cells and (i) absolute number of live lin−Eomes−CD90+RORγt+IL-22+ILC3. Data are representative of at least 2 independent experiments. (a-e, g, i) Data points are single mouse with mean. (a-c, e) Student’s t test, (d) Mann–Whitney test and (g, i) one-way ANOVA with Bonferroni correction. *p ≤ 0.05, **p ≤ 0.01, **** p ≤ 0.0001.
CD4+ T cell responses are also critical for C. rodentium clearance.Citation41,Citation42 However, we found no changes in total T cell numbers or in the frequency of T cell subsets after C. rodentium infection in GDAR2-2-colonized mice as compared to uncolonized controls (Figure S5(a-e)). To further confirm that protection after GDAR2-2 colonization was independent of adaptive immunity, we treated Rag2−/− mice with ABX and left uncolonized or colonized with GDAR2-2 before infection with C. rodentium. As in WT mice, GDAR2-2 colonization was sufficient to protect Rag2−/− mice from C. rodentium infection. Compared to uncolonized mice, Rag2−/− mice colonized with GDAR2-2 showed less weight loss and longer colon length than control Rag2−/–infected mice ()), suggesting T cells were dispensable for GDAR2-2-induced protection against C. rodentium infection. Together, these data support that the expansion of IL-22+ILC3s is required for the GDAR2-2-induced protection,
Colonic CX3CR1 MNPs promote barrier protection
Since we found increased mucosa colonization by GDAR2-2 as compared to K-12, we hypothesized that interactions with the epithelium could initiate the observed protective effects. To test if GDAR2-2 induced epithelial responses, we left untreated or co-cultured human intestinal cell line Caco-2 with K-12 or GDAR2-2 and measured gene expression by qPCR. We found increased expression of monocyte recruiting chemokines, hCcl2, hCx3cl1, hCcl4, Ccl5 and Ccl20, after GDAR2-2 stimulation (, Figure S6(a-c). Additionally, increased expressions of hIl8 and pro-inflammatory cytokines such as hIl23a, hIl6, hTnf, hIl1a, hIl1b were found with no change in hIl18 and hIl1rn expression after GDAR2-2 stimulation (Figure S6(d-k).
Figure 4. GDAR2-2 induced protection depends on CX3CR1+ MNPs. (a-b) Caco-2 cells were co-cultured with K-12, GDAR2-2 or left untreated. mRNA expression of (a) hCcl2 and (b) hCx3cl1 determined by qPCR are shown. (c-f) ABX-treated CX3CR1-GFP mice were colonized with GDAR2-2 or left un-colonized. 2 days later, CX3CR1+ MNPs from large intestine laminar propria were analyzed by flow cytometry. (c) Representative dot plots of live (left) and CD11b+CX3CR1int population (right). Absolute numbers of (d) CX3CR1int macrophages (Mp)/dendritic cells (DCs), (e) CX3CR1+ monocytes and (f) CX3CR1int transitional (trans)-monocytes are shown. (g-k) DT-treated littermate control (CX3CR1+, CX3CR1+ MNP sufficient) and CX3CR1-DTR (CX3CR1-, CX3CR1+ MNP deficient) mice were treated with ABX and colonized with GDAR2-2 before infection with C. rodentium. (g) Weight change, (h) survival, (i) colon length 12 days post infection was shown. (j-k) Absolute number of (j) ILC3s and (k) IL-22+ILC3s 5 days post infection are shown. See also Figure S5. Data are representative of at least 2 independent experiments. (a-b) Data are shown as mean with SEM and K-12 and GDAR2-2 group are compared by student’s t test. (d-f) Data points are single mouse with mean and compared by Student’s t test. (g) Data are shown as mean ± SEM and compared by one-way ANOVA with Bonferroni correction for each time point. (h) log rank test. (i-k) Data points are single mouse with mean. One-way ANOVA with Bonferroni correction. *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
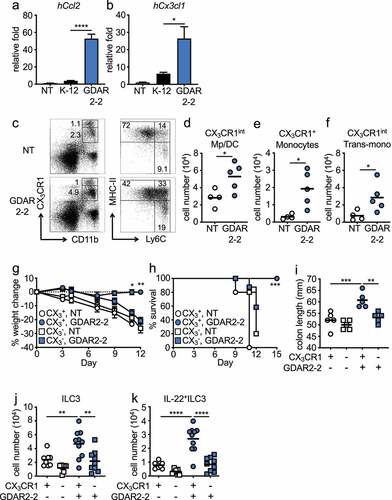
Previously, we demonstrated that CX3CR1+ MNPs are required to protect against C. rodentium infection through the induction of IL-22 production by ILC3sCitation10. We hypothesized that the upregulated monocyte recruiting chemokines resulted in an influx of monocytes into the intestine that differentiated into CX3CR1+ MNPs to provide protection. In the intestine, CX3CR1+ MNPs can be divided into two major populations based on CX3CR1 expression, CX3CR1hi and CX3CR1int MNPs. CX3CR1hi MNPs are considered macrophagesCitation13,Citation16,Citation43 whereas CX3CR1int MNPs are composed of three subgroups including MHC-II+Ly6C− cells that have characteristics of both macrophages and dendritic cells, MHC-II−Ly6C+ monocytes and the MHC-II+Ly6C+ transitional monocytes.Citation13,Citation16,Citation43 To determine if GDAR2-2 regulated CX3CR1+ MNPs, we colonized ABX-treated mice with GDAR2-2 and analyzed MNP populations by flow cytometry. After GDAR2-2 colonization, we found expansion of CX3CR1int macrophages and dendritic cells, CX3CR1+ monocytes and MHC-II+Ly6C+ transitional monocytes with no change in CX3CR1hi macrophages in the intestine (). These data suggest that GDAR2-2-induced CX3CR1+ MNP expansion is likely driven by intestinal epithelium recruitment of monocytes.
To determine whether CX3CR1+ MNPs were critical for GDAR2-2-mediated protection, we utilized a mouse model where the diphtheria toxin receptor (DTR) is driven by the CX3CR1 promoter (CX3CR1-DTR)Citation10 that allows for selective depletion of CX3CR1+ cells after diphtheria toxin (DT) administration.Citation12 We administered DT to ABX-treated CX3CR1-DTR mice and WT littermate controls and colonized them with GDAR2-2. Two days later, we infected mice with C. rodentium. Colonization with GDAR2-2 did not protect mice from C. rodentium infection in the absence of CX3CR1+ MNPs. Similar to uncolonized mice, mice lacking CX3CR1+ MNPs showed increased weight loss, decreased survival, and decreased colon length compared to mice with normal CX3CR1+ MNPs (). In the steady state or after C. rodentium infection, in the absence of CX3CR1+ MNPs, GDAR2-2 induced neither colonic ILC3 expansion nor IL-22 secretion (, Figure S7(a-b)). Together, these data indicate a critical role of CX3CR1+ MNPs in GDAR2-2-induced protection.
Live mouse commensal E. coli promotes IL-1β secretion
To determine if GDAR2-2 directly regulated macrophage cytokine secretion, we co-cultured WT bone marrow derived macrophages (BMDMs) with K-12 or GDAR2-2 and measured cytokine secretion by LegendPlex. As compared to K-12, co-culture with GDAR2-2 induced similar levels of TNF ()) and undetectable IL-23 production (data not shown). In contrast, GDAR2-2 induced higher IL-6 and IL-1β secretion (). We next sought to understand if live GDAR2-2 or GDAR2-2 metabolites were required for IL-1β secretion. To test this, we co-cultured immortalized BMDMs (iBMDMs) with live GDAR2-2, heat-killed GDAR2-2, or GDAR2-2 culture supernatant and found that only live GDAR2-2 induced IL-1β production from iBMDMs ()). To further understand if the inflammasome was required for GDAR2-2 induced IL-1β production, we utilized iBMDMs deficient for different inflammasome components and found that IL-1β production is abolished in Casp11−/−, Casp1/11−/−, Gsdmd−/− and Nlrp3−/− iBMDM ()). However, IL-1β secretion is not blunted in Nlrc4−/− BMDM ()). These data indicate that live GDAR2-2 is required to activate the inflammasome and results in IL-1β secretion from macrophages.
Figure 5. GDAR2-2-induced IL-1β secretion protects mice from C. rodentium infection. (a-c) WT bone marrow derived macrophages (BMDM) were co-cultured with K-12, GDAR2-2 or left untreated. Secretion of (a) IL-6, (b) TNF and (c) IL-1β were analyzed by LegendPlex. (d) Secretion of IL-1β from immortalized BMDM (iBMDM) co-cultured with live, heat-killed (HK), supernatant of GDAR2-2 or left untreated. (e-f) Secretion of IL-1β were detected from (e) WT, casp11−/−, casp1/11−/−, Gsdmd−/− and Nlrp3−/− iBMDM co-cultured with GDAR2-2 or from (f) WT and Nlrc4−/− BMDM co-cultured with GDAR2-2. (g-h) ABX-treated B6 mice were colonized with GDAR2-2, infected with C. rodentium and administered with α-IgG antibody (Ab) or α-IL-1β Ab. (g) Weight change and (h) colon length 6 days after infection were shown. (i-j) B6 mice were treated with ABX, colonized with GDAR2-2, infected with C. rodentium and treated with α-IgG Ab or α-IL-23 Ab. (i) Weight change and (j) colon length were shown. (k-l) ABX-treated B6 mice were colonized with GDAR2-2, infected with C. rodentium and treated with α-IgG Ab or α-IL-22 Ab. (k) Weight change and (l) colon length were shown. Data are pooled from (g,h,k,i) or are representative of (a-f, i, j) at least 2 independent experiments. (a-f) Data are shown as mean with SEM. (a-e) One-way ANOVA with Bonferroni correction. (f) Student’s t test. (g, i, k) Data are shown as mean ± SEM. Student’s t test at each time point. (h, j, l) Data points are single mouse with mean. Student’s t test. *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
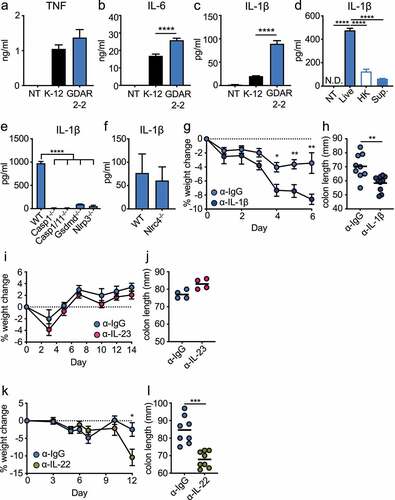
IL-1β is required for GDAR2-2 protection against infection
Secretion of either IL-1β or IL-23 by CX3CR1+ MNPs can activate ILC3 production of IL-22.Citation10,Citation44 To determine whether IL-1β induced by GDAR2-2 was required for protection after C. rodentium infection, we administered isotype control or an antagonistic IL-1β antibody (Ab) to ABX-treated, GDAR2-2-colonized, and C. rodentium-infected WT mice. Mice treated with anti-IL-1β Ab showed significant weight loss and shorter colon length (), suggesting a critical role for IL-1β in GDAR2-2-mediated protection. We similarly used an antagonistic anti-IL-23 Ab. In contrast to IL-1β blockade, blocking IL-23 did not result in changes of weight loss or colon length (), suggesting that IL-23 is dispensable for GDAR2-2-induced protection. These results suggest that GDAR2-2-induced IL-1β secretion from CX3CR1+ MNPs is crucial for protection against C. rodentium infection.
To determine the role of IL-22 in GDAR2-2-induced protection, we similarly blocked IL-22 with an IL-22 blocking Ab in GDAR2-2 colonized C. rodentium infected mice. We found increased weight loss with shorter colon length in mice treated with anti-IL-22 blocking Ab as compared to isotype control, suggesting that GDAR2-2-induced IL-22 production is critical for protection from C. rodentium infection ().
Select human commensal E. coli isolates promote barrier protection through IL-1β secretion
In patients with inflammatory bowel diseases (IBD), increased levels of mucosa-associated bacteria such as E. coli are found.Citation7,Citation8,Citation45,Citation46 These microbes are thought to amplify pathology. To test if they could also regulate protection similar to our identified mouse E. coli, we assembled a panel of human microbiome project (HMP)Citation47 mucosa-associated E. coli (). We treated mice with ABX as above before colonizing mice with individual human E. coli isolates. Mice were then infected with C. rodentium. While the majority did not protect from C. rodentium, we identified a subset of protective human E. coli as shown by reduced weight loss (), Figure S8(a), ). Similar to the mouse E. coli, GDAR2-2, we found comparable E. coli and C. rodentium fecal load in mice colonized with either protective or neutral E. coli isolates (. Supporting a similar mechanism of protection, we found increased colonic CX3CR1+ MNP and ILC3 numbers in mice colonized with protective as compared to neutral E. coli ().
Table 1. Characteristics of human E. coli isolates used in the study. Effect describes outcome in the C. rodentium infection model. HM339 is pathogenic when administered to mice in absence of C. rodentium infection. Crohn’s disease (CD) ulcerative colitis (UC), control = non IBD
Figure 6. Select human E. coli isolates protect mice from C. rodentium infection through induction of IL-1β. (a-e) ABX-treated B6 mice were colonized with individual human E. coli isolates or left un-colonized and infected with C. rodentium. (a) Weight change. (b) E. coli and (c) C. rodentium titer in feces 5 days after infection analyzed by plating. Absolute number of (d) live CX3CR1+ MNPs and (e) lin−Eomes−CD90+RORγt+ILC3 of C. rodentium infected mice colonized with indicated E. coli isolate. (f) IL-1β secretion from iBMDM co-cultured with indicated E. coli isolate. (g) ABX-treated B6 mice were colonized with indicated E. coli isolates, infected with C. rodentium and administered with α-IgG Ab or α-IL-1β blocking Ab. Colon length is shown. Data are representative of at least 2 independent experiments. (a, f) Data are shown as mean ± SEM. (b-e, g) Data points are single mouse with mean. (d, e, g) One-way ANOVA with Bonferroni correction. *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
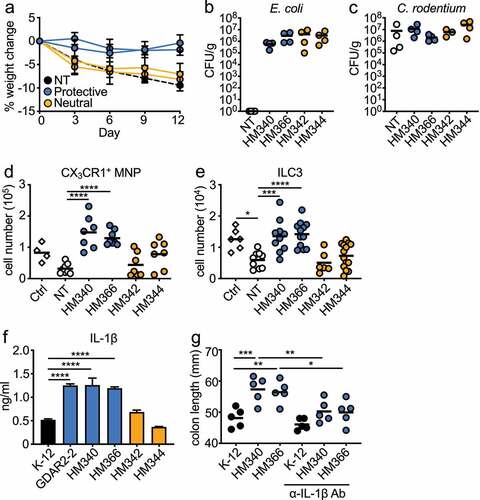
To determine whether the protective human commensal E. coli also induced macrophage IL-1β production, we co-cultured individual human E. coli with iBMDM and analyzed supernatants for cytokine production by ELISA and LegendPlex. All E. coli induced similar levels of IL-6, TNF and IL-23 (Figure S8(c-e)). Similar to GDAR2-2, protective human E. coli induced higher IL-1β levels () and Figure S8(b)). To determine whether IL-1β induced by the protective human E. coli also contributed to protection from C. rodentium, we colonized ABX-treated mice with protective strains and treated them with isotype control or anti-IL-1β Ab. The protection was lost as shown by decreased colon length ()) in the anti-IL-1β Ab-treated animals. These results indicate that, similar to the mouse commensal E. coli GDAR2-2, select human mucosa-adherent E. coli can protect against C. rodentium-induced colitis by enhancing macrophage IL-1β production.
Discussion
Within the intestine, host-microbe interactions regulate many aspects of host physiology. We and others previously demonstrated that one of these functions is to promote barrier repair.Citation10,Citation48,Citation49 However, the mechanisms by which individual bacterium and microbial signals regulate intestinal epithelial integrity remain unclear. Using antibiotic selection, we identified a multidrug resistant mouse commensal E. coli, GDAR2-2, that protects against C. rodentium infection and DSS-induced colitis in mice. We found that this protection is not due to direct competition with C. rodentium but depends on the regulation of host innate immune responses.
Colonization with GDAR2-2 induced IL-22+ILC3 expansion in mice. IL-22 is required for survival from C. rodentium infectionCitation21 by promoting intestinal epithelium proliferation and survival,Citation25 including enhancing transit-amplifying cell proliferation,Citation32 facilitating goblet cell regeneration and mucin secretion,Citation24 and promoting anti-microbial peptide production.Citation21,Citation26,Citation50 We found colonic upregulation of Axin2 and Ascl2, markers of transit-amplifying cells, after GDAR2-2 colonization. We also found loss of protection when IL-22 was blocked in GDAR2-2-colonized mice, indicating its requirement for GDAR2-2-induced protection against C. rodentium infection.
Attachment of microbes to the gut epithelium is an important mechanism to regulate host immunity.Citation12,Citation51 In vivo, we found enriched GDAR2-2 at the intestinal epithelium, suggesting that protection might be downstream of epithelial attachment. Using the Caco-2 human colon cell line, we found up-regulation of monocyte chemoattractants upon GDAR2-2 stimulation. In line with this finding, GDAR2-2 colonization led to expansion of CX3CR1+ monocytes, CX3CR1int transitional monocytes and CX3CR1int macrophage/DC in the intestine. Previous studies showed that increased intestinal monocytes are pathogenic in colitis modelsCitation14,Citation52,Citation53 while macrophages are found to be protective.Citation12,Citation54,Citation55 Using mice that can be selectively depleted of CX3CR1+ cells,Citation12,Citation13 we demonstrated that the GDAR2-2-induced protection against C. rodentium infection is CX3CR1+ MNP dependent. We previously demonstrated that CX3CR1+ MNPs are required for ILC3 production of IL-22Citation10. Depletion of CX3CR1+ MNPs also abolished GDAR2-2-induced IL-22+ILC3 expansion. Collectively, the commensal E. coli GDAR2-2 promotes barrier repair functions of CX3CR1+ MNPs that results in IL-22+ILC3 expansion.
Previous studies showed that secretion of IL-1β and IL-23 from CX3CR1+ MNPs activate ILC3s and induce IL-22 production from ILC3s.Citation10,Citation23,Citation44,Citation56 We found macrophage stimulation with live GDAR2-2 resulted in enhanced IL-1β secretion as compared to live K-12. In addition to activating IL-22 production, previous studies showed that IL-1β also supports Th17 cell responses with IL-1β required for early Th17 differentiation and synergizes with IL-6 and IL-23 to promote Th17 cell survival and effector functions.Citation57 Accumulation of Th17 cells can lead to chronic inflammation.Citation58 Although we found elevated IL-1β secretion from GDAR2-2 stimulated macrophages, we did not see expansion of Th17 cells nor other T cell populations. Further, neutralization of IL-1β but not IL-23 abrogated the protective effect of GDAR2-2 in ABX-treated, GDAR2-2-colonized mice. These findings indicate the importance of IL-1β activation of IL-22 production by ILC3 in the GDAR2-2-induced protection from C. rodentium infection.
In IBD patients, a shift in the microbiota composition has been identified with increased mucosa-associated bacteria including Proteobacteria.Citation7,Citation8,Citation45,Citation46 Using human E. coli isolated as part of the NIH human microbiome projectCitation47 we identified several human E. coli isolates that also protect from C. rodentium infection. Similar to GDAR2-2, these E.coli isolates increase intestinal ILC3 numbers and induce macrophage IL-1β secretion. As with GDAR2-2, blocking IL-1β ameliorates their protection. Although increased Proteobacteria is associated with IBD and certain human E. coli isolates were isolated from IBD patients, these results highlight that distinct commensals can play distinct roles depending on tissue context.
IL-1β is a pro-inflammatory cytokine that promotes various inflammatory disorders, including rheumatoid arthritis (RA) and type 2 diabetes (T2D) where neutralization of IL-1β effectively represses inflammation and reduces disease severity.Citation59,Citation60 Similar to RA and T2D, increased IL-1β is found in IBD patients.Citation61,Citation62 IL-1β can increase intestinal tight junction permeability,Citation63–65 which can increase intestinal bacteria penetration from the lumen to lamina propria and results in colitis in mouse models.Citation66 However, blocking IL-1β does not ameliorate symptoms in IBD patients.Citation61,Citation67 Protective effects of IL-1β are found in the skin where bacteria induced IL-1β secretion results in skin regeneration after injury.Citation68 Further, additional studies support an anti-inflammatory role for IL-1β which promotes Treg functions and induces IgA production.Citation69–71
Work by Seo et al. shows that Proteus mirabilis activates monocyte secretion of IL-1β.Citation52 Unlike GDAR2-2, P. mirabilis induction of IL-1β is pathogenic in DSS-induced colitis in WT mice. P. mirabilis is a pathobiontCitation52,Citation72 that carries virulence factors such as Proteus toxic agglutinin and type III secretion systems.Citation73 In addition, Viladomiu and Metz et al. demonstrate that the large subunit of propanediol dehydratase (PduC) encoded by Adherent-invasive E. coli (AIEC) triggers IL-1β secretion from CX3CR1+ MNPs.Citation56 Similar to P. mirabilis, IL-1β induced by PduC-encoding AIEC also causes severe pathology in DSS-induced colitis in Il10-deficient mice. The metabolic activity of PduC also exacerbates T cell-dependent colitis.Citation56 On its own, GDAR2-2 does not induce intestinal pathology and these data highlight that immune activation in combination with microbial characteristics together contribute to the disease state of the intestine.
In addition to the ability of IL-1β to induce IL-22 secretion by ILC3, a recent study demonstrated that IL-1-signaling on mesenchymal cells results in increased production of Wnt agonists that promote intestinal stem cell proliferation and can protect in DSS-induced colitis and after C. rodentium infection.Citation33 However, we did not see up-regulation of Wnt agonist, Rspo3, and the Wnt target stem cell gene, Lgr5, suggesting that GDAR2-2 does not protect mice from C. rodentium infection through Wnt signaling to induce intestinal stem cell regeneration. Instead, we found up-regulation of Axin2 and Ascl2, which are the transit-amplifying cell markers induced by IL-22 signaling, suggesting that GDAR2-2 provides protection likely through IL-22-induced transit-amplifying cell proliferation. The loss of protection in the GDAR2-2-colonized mice given anti-IL-22 blocking Ab also strengthens this finding. Together, these results support the protective role of GDAR2-2 against C. rodentium infection through inducing barrier repair by activating the IL-1β-IL-22 pathway. This may indicate spatial interactions between CX3CR1+ MNPs and ILC3s instead of activating mesenchymal cells that secret Wnt agonist after GDAR2-2 colonization to protect from C. rodentium infection.
As microbiota composition changes are often found in IBD and autoimmune diseases,Citation9 understanding how individual commensal bacterium shapes the host immune responses will offer important clues for developing strategies to ameliorate disease. Here we describe the identification of a subset of intestinal mucosa-associated E. coli that induce CX3CR1+ MNP IL-1β production to protect the intestinal barrier. Together, these findings provide potential therapeutic interventions for IBD by regulating microbial composition and signals as well as revisiting the role of IL-β.
Materials and methods
Mice
CX3CR1-DTR mice were previously describedCitation10,12 . C57BL/6 J (JAX # 000664) and CX3CR1-GFP (JAX # 005582) mice are from Jackson Laboratories and Rag2−/− (RAGN12) are from Taconic. All mice are bred in-house under standard SPF conditions at the animal facilities of Baylor College of Medicine or Memorial Sloan Kettering Cancer Center. All mouse experiments were performed with mice between 8 and 12 weeks of age with males and females at similar ratios, unless otherwise specified. Littermate controls were used for each experiment and mice were randomly assigned to experimental groups with a minimum of 3 mice per group. All experiments were performed in accordance with approved protocols by the Institutional Animal Care and Usage Committee at Baylor College of Medicine or Memorial Sloan Kettering Cancer Center.
Depletion of gut microbiota
Animals were provided with 1 g/L ampicillin (A; Sigma, A0166), 500 mg/L vancomycin (V; Sigma, V2002), 1 g/L neomycin sulfate (N; Sigma, N1876), and 1 g/L metronidazole (M; Sigma, M3761) in drinking water for four weeksCitation4,Citation13 or treated with single dose of 20 mg/ml streptomycin (Sigma, S9137-25 G) in 100 μl water by per os (P.O.) and with 1 g/1 L ampicillin (Fisher BioReagent, BP176025) in drinking water for 1–2 weeks. Ampicillin water was replaced with regular drinking water 2 days before E. coli colonization as described below. Microbiota depletion was confirmed by bacterial DNA in feces as detected by real time qPCR.
GDAR2-2 isolation and sequencing
Fecal pellets from AVMN-treated mice were resuspended in PBS to 100 mg/ml and dilutions were plated on blood agar plates (Fisher) and cultured overnight at 37°C under normal or anaerobic conditions using BD GasPak (Fisher). For sequencing, genomic DNA was extracted with phenol chloroform, and DNA was sheared to 15kb using Covaris g-TUBE® devices, allowing for sizes 5 kb and larger. The library preparation was carried out using SMRTbell Template kit 1.0 Exo VII protocol and the sample was barcoded with PacBio Adaptor. Genome sequencing was performed using the Pacific Biosciences Sequel sequencing platform. Long reads were assembled de novo into two contigs (main chromosome and 1 plasmids) using Canu (v. 1.6).Citation73 Gene prediction and annotation were carried out using the webservice PATRIC.Citation74 Genomic visualization was performed using Circos v0.69–9.Citation75 Genomic comparison was done using the PATRIC webservice and phylogenomic reconstructions were done using the GToTree pipeline and its associated dependencies.Citation76–80 Sequencing reads and the genome assembly were submitted to NCBI under the bioproject PRJNA725420.
Bacteria
K-12 (ATCC® PTA-7555) and Citrobacter rodentium (ATCC® 51,459) are from ATCC. Human Microbiome ProjectCitation46 Escherichia coli isolates are from ATCC with details in . For heat-killed GDAR2-2, GDAR2-2 was grown to OD600 = 1 and was left at 85°C for 15 min. For GDAR GDAR2-2 supernatant, GDAR2-2 was grown to OD600 = 1, spun down at 3000 g, RT for 10 min. The supernatant was then filtered through a 0.22 μm filter.
Colonization of mice with E. coli
Mice were treated with ABX to disrupt microbiota before colonizing with E. coli. E. coli was incubated at 37°C overnight with shaking at 220 rpm in LB broth. ABX-treated animals were colonized with E. coli isolates: K-12, GDAR2-2 or HMP E. coli at 1 × 108 colony forming unit (CFU) in 100 μl PBS. Colonization was confirmed by real time qPCR with E. coli-specific qPCR primers before infecting with C. rodentium as described below.
C. rodentium infection
ABX-treated mice were left uncolonized or colonized with E. coli 2 days before C. rodentium infection as described above. C. rodentium was grown overnight in LB broth at 37°C with shaking at 220 rpm. Stationary phase C. rodentium was diluted 1:100 in LB broth and grown at 37°C with shaking until log phase. Mice were infected with 1 × 1010 colony forming units (CFU) of C. rodentium in 100 μl PBS. Infection was confirmed by real time qPCR with C. rodentium-specific qPCR primers.
DSS-induced colitis
Mice were given 2% dextran sodium sulfate (DSS) (Alfa Aesar, J63606) in drinking water for 5 days after which they were switched to regular drinking water.
Diphtheria toxin (DT) and blocking Ab administration
Starting 2 days before E. coli colonization, mice were intraperitoneally (I.P.) injected every other day with 200ng DT (Sigma, D0564) in PBS. For IL-1β blocking, mice were injected I.P. with 200 μg anti-IgG control Ab (BioCell, BE0290) or anti-IL-1β blocking Ab (BioCell, BE0246) a day before E. coli inoculation. Mice were then injected i.p. every other day starting at day of E. coli colonization with 50 μg anti-IgG control Ab or anti-IL-1β blocking Ab. For IL-23 blocking experiment, mice were injected twice a week by i.p. with 50 μg anti-IgG control Ab (BioCell, BE0290) or anti-IL-23 blocking Ab (BioCell, BE0313) starting from 2 days before E. coli colonization. For IL-22 blocking, mice were i.p. injected with 100 μg anti-IgG control Ab or anti-IL-22 blocking Ab (ThermoFisher, 16–7222-38) every other day from the day of E. coli inoculation.
Fecal albumin detection
Feces were freshly collected at day 4 after C. rodentium infection from ABX-treated mice colonized with K-12, GDAR2-2 or left untreated. Feces albumin was detected by Bromocresol Green Albumin Assay Kit (Sigma-Aldrich, MAK124).
Colon explant
Colon explant was performed as described elsewhere.Citation81 In brief, 6 days after C. rodentium infection, same area of the colons were isolated from each mouse, opened and washed. Colons were homogenized in lysis buffer supplemented with protease inhibitor. After centrifuging down the debris, supernatant was collected for cytokine detection using a customized LegendPlex kit (BioLegend).
Lamina propria cell isolation
Isolation of cells from large intestine laminar propria has been described previously.Citation13 Briefly, large intestines were opened longitudinally and washed in cold PBS to remove luminal contents. Tissue was cut into 1 cm sections and incubated while shaking in PBS with 1 mM DTT (Sigma, D9779), 30 mM EDTA (ThermoFisher, AM9621) and 10 mM HEPES (HyClone, SH30237.01) for 10 minutes at 37°C followed by PBS with 30 mM EDTA and 10 mM HEPES for 10 minutes at 37°C. Tissue was then digested at 37°C in a humidified chamber in RPMI/10% FBS with 200 U/ml type 8 collagenase (Sigma, C2139) and 150ug/ml DnaseI (Sigma, DN25) followed by separation on a 40% /80% Percoll (GE Health, 17–0891-01) gradient.
Antibody, staining, and flow cytometry
Flow cytometry analysis was performed with BD LSRII or Cytek Aurora and analyzed with FlowJo (Tree Star Inc.). Antibodies were from BD, eBiosciences, and BioLegend. For live/dead cell staining, DAPI (Sigma, D9542) or live/dead fixable blue dead cell stain (ThermoFisher, L34962) were used. To prevent nonspecific binding, CD16/32 Ab were used on all samples. For cell counts, 123 count ebeads (ThermoFisher, 01–1234) were used to determine absolute cell numbers. For cytokine staining, cells were activated with 50 ng/ml phorbol myristate acetate and 1 μM ionomycin for 4 hours. Together with intracellular staining, cells were fixed, permeabilized and stained with anti-mouse IL-22, T-bet, GATA-3, RORγt, and Foxp3 according to the Foxp3/Transcription Factor Staining Buffer Set instructions (ThermoFisher, 00–5523-00). For ILCs, lineage (CD3, CD5, CD8, Ly6G, CD19) negative (lin−) CD90+ cells were analyzed for transcription factors and IL-22. For CX3CR1+ MNP staining, CX3CR1-GFP, CD11b, MHC-II and Ly6C were used. For T cell staining, CD3, TCR-β and CD4 were used. Surface staining was stained in FACS buffer (PBS with 2 mM EDTA and 2% FBS) for 15 min, 4°C in the dark. All antibody information is listed below.
Bacterial DNA isolation
Bacterial DNA was isolated from feces or intestines using DNeasy PowerSoil Kit (QIAGEN, 12,888–100) according to manufacturer’s protocol.
RNA extraction and real time RT-PCR
RNA extraction from Caco-2 cells were performed with Trizol (ThermoFisher, 15,596,018) following manufacturer’s protocol. RNA was reverse transcribed to cDNA using iScript Reverse Transcription Supermix (BIO-RAD, 1,708,841). qPCR was performed with iTaq Universal SYBR Green Supermix (BIO-RAD, 1,725,125), 5 μmol of forward and reverse primers listed below, 100 ng cDNA using a QuantStudio 6 Pro (ThermoFisher Scientific). The qPCR program was 95°C for 2 min to initiate the reaction followed by 95°C for 15s, 60°C for 30s, and 72°C for 30s for a total of 40 cycles. Relative expression of target genes expression was determined by calculating delta Ct compare to reference gene.
Cell lines and micorbial co-culture
For Caco-2 cells (ATCC® HTB-37), 2.5 × 104 cells were seeded in 24-well tissue culture (TC) plate in 1 ml DMEM (Corning, MT10017CV) supplemented with 10% FBS (Gibco, 26,140–079), 10 mM HEPES (HyClone, SH30237.01), 2 mM glutamine (HyClone, SH30034.01), 1 mM sodium pyruvate (HyClone, SH30239.01), 1X MEM non-essential amino acids (ThermoFisher, 11,140–050), 100 unit/ml penicillin-streptomycin (HyClone, SV30010). Media was changed every other day until cells were confluent. For coculture with Caco-2 cells, one day prior to E. coli co-culture, cells were washed twice with PBS and media was replaced with complete DMEM without antibiotics. The next day, cells were spin infected at RT, 1000 x g for 10 min and co-cultured with indicated E. coli at MOI = 20 for 3 hours. Cells were washed once with PBS and resuspend in Trizol for RNA extraction. Immortalized bone marrow derived macrophages (iBMDM) were gifts from Dr. Jonathan C. Kagan.Citation82 2 × 105 iBMDM cells were seeded in 96-well TC plate in 200 μl complete DMEM without antibiotics and incubated overnight. iBMDM cells were then spin infected at RT, 1000 x g for 10 min with indicated E. coli at MOI = 20 and incubated for 10 min. Cells were washed twice with sterile PBS and media was replaced with complete DMEM containing 100 μg/ml gentamycin (Sigma, G1272). After 1 hour incubation, cells were washed once with PBS, and media was replaced with complete DMEM with 20 μg/ml gentamycin. After 10 or 24 hour incubation, supernatant was collected for cytokine detection by IL-1β ELISA kit (ThermoFisher, 88–7013-88) or customized LegendPlex kit (BioLegend).
Bone marrow derived macrophage (BMDM) culture
Femurs and tibias from WT and Nlrc4−/− (from Dr Isabella Rauch) mice were used for BMDM culture. BM was isolated from the indicated mice, treated with RBC lysis buffer and plated at a density of 1x106/ml in a 15 cm TC dish in bone marrow macrophage (BMM) medium (complete DMEM supplemented with 30% L-929 cell-conditioned medium). At day 3, same amount of the BMM media was added to the cell culture. Day 6 cell culture was harvested for microbial stimulation as described for the iBMDM.
Histopathology
Tissues were fixed in 10% buffered neutral buffered formalin (Fisher Scientific, SF100-4). Fixed tissues were paraffin embedded, processed, and stained with hematoxylin and eosin (H&E) using standard protocols. Samples were scored from 0 to 4 using established criteriaCitation12- Grade 0: Histology is normal. Grade 1: Mild inflammatory infiltrate in submucosa with edema. Minor erosion and muscularis remains intact. Grade 2: Grade 1 histology appear over 50% of the tissues. Grade 3: Severe inflammatory infiltration (predominantly neutrophil infiltrate) with edema. Ulceration in submucosa to muscularis layer. Grade 4: Grade3 histology shows in over 50% of the tissues.
Statistics
Normality was determined by Shapiro Wilk test. If the data was normally distributed, for two groups, Student’s t test was used; for more than two groups, one-way ANOVA with Bonferroni correction was used. If the data was not normally distributed, for two groups, Mann–Whitney test was used; for more than 2 groups, Kruskal–Wallis test with Dunn’s multiple comparison was used. For survival, log rank test was used. All statistics are un-paired, 2-tailed with 95% confidence interval. Data are shown in mean or mean ± SEM. Significance is determined as ns:p > 0.05, *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Data was plotted with GraphPad Prism 8.4.3.
Contributions
G.E.D. and W-J.H.W. designed experiments and wrote the manuscript with the input from all co-authors. G.E.D., W-J.H.W., and M.K., performed, designed and analyzed the experiments. L-C.C., F.B.S., and K.N. performed experiments. A.A. and B.S.S. performed E. coli genomic assembly and analysis.
Data availability
Sequencing reads and the genome assembly are openly available in NCBI under bioproject PRJNA725420
Supplemental Material
Download PDF (10 MB)Acknowledgments
We thank all members of the Diehl lab as well as Drs Katherine King, David Corry, Sasirekha Ramani, and Tor Savidge for critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Saldana-Morales FB, Kim DV, Tsai M-T, Diehl GE. 2021. Healthy intestinal function relies on coordinated enteric nervous system, immune system, and epithelium responses. Gut Microbes. 13(1):1–20. doi:10.1080/19490976.2021.1916376.
- Caruso R, Lo BC, Nuñez G. 2020. Host-microbiota interactions in inflammatory bowel disease. Nat Rev Immunol. 20(7):411–426. doi:10.1038/s41577-019-0268-7.
- Sommer F, Bäckhed F. 2013. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. 11(4):227–238. doi:10.1038/nrmicro2974.
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 118(2):229–241. doi:10.1016/j.cell.2004.07.002.
- Brasseit J, Althaus-Steiner E, Faderl M, Dickgreber N, Saurer L, Genitsch V, Dolowschiak T, Li H, Finke D, Hardt W-D, et al. 2016. CD4 T cells are required for both development and maintenance of disease in a new mouse model of reversible colitis. Mucosal Immunol. 9(3):689–701. doi:10.1038/mi.2015.93.
- Kang E, Zhou G, Yousefi M, Cayrol R, Xia J, Gruenheid S. 2018. Loss of disease tolerance during citrobacter rodentium infection is associated with impaired epithelial differentiation and hyperactivation of T cell responses. Sci Rep-uk. 8(1):847. doi:10.1038/s41598-017-17386-y.
- Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc of the Natl Acad of Sci. 104(34):13780–13785. doi:10.1073/pnas.0706625104.
- Peterson DA, Frank DN, Pace NR, Gordon JI. 2008. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 3(6):417–427. doi:10.1016/j.chom.2008.05.001.
- Wu W-JH, Zegarra-Ruiz DF, Diehl GE. 2020. Intestinal microbes in autoimmune and inflammatory disease. Front Immunol. 11:597966. doi:10.3389/fimmu.2020.597966.
- Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR. 2014. CX₃CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 211(8):1571–1583. doi:10.1084/jem.20140678.
- Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, Marderstein AR, Gandara J, Perez AR, Withers DR, et al. 2018. Microbiota-Induced TNF-like ligand 1a drives group 3 innate lymphoid cell-mediated barrier protection and intestinal t cell activation during colitis. Immunity. 49(6):1077–1089. e5.
- Kim M, Galan C, Hill AA, Wu W-J, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, et al. 2018. Critical role for the microbiota in CX3CR1+ intestinal mononuclear phagocyte regulation of intestinal t cell responses. Immunity. 49(1):151–163. e5.
- Diehl GE, Longman RS, Zhang J-X, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. 2013. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature. 494(7435):116–120. doi:10.1038/nature11809.
- Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, et al. 2012. Ly6Chi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 37(6):1076–1090. doi:10.1016/j.immuni.2012.08.026.
- Welty NE, Staley C, Ghilardi N, Sadowsky MJ, Igyártó BZ, Kaplan DH. 2013. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalismLP DCs maintain T cell homeostasis. J Exp Med. 210(10):2011–2024. doi:10.1084/jem.20130728.
- Varol C, Zigmond E, Jung S. 2010. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol. 10(6):415–426. doi:10.1038/nri2778.
- Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, et al. 2008. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 112(13):5026–5036. doi:10.1182/blood-2008-06-162404.
- Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. 2005. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 115(1):66–75. doi:10.1172/JCI200519229.
- Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Müller W, Sparwasser T, Förster R, Pabst O. 2011. Intestinal tolerance requires gut homing and expansion of foxp3+ regulatory t cells in the lamina propria. Immunity. 34(2):237–246. doi:10.1016/j.immuni.2011.01.016.
- Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. 2011. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 34(1):122–134. doi:10.1016/j.immuni.2010.12.009.
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 14(3):282–289. doi:10.1038/nm1720.
- Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, Niess JH. 2012. CX3CR1+ macrophages support IL-22 production by innate lymphoid cells during infection with citrobacter rodentium. Mucosal Immunol. 6(1):177–188. doi:10.1038/mi.2012.61.
- Wang B, Lim J-H, Kajikawa T, Li X, Vallance BA, Moutsopoulos NM, Chavakis T, Hajishengallis G. 2019. Macrophage β2-integrins regulate il-22 by ilc3s and protect from lethal citrobacter rodentium-induced colitis. Cell Rep. e5;26(6):1614–1626. doi:10.1016/j.celrep.2019.01.054.
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 118:534–544.
- Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, Butler JA, Boon L, Coles MC, Samsom JN, et al. 2015. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med. 212(11):1783–1791. doi:10.1084/jem.20150318.
- Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention -J-J, Thiam K, Cerf-Bensussan N, Mandelboim O, et al. 2008. Microbial flora drives interleukin 22 production in intestinal nkp46+ cells that provide innate mucosal immune defense. Immunity. 29(6):958–970. doi:10.1016/j.immuni.2008.11.001.
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. 2012. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 490(7418):55–60. doi:10.1038/nature11450.
- Davis KL. 2015. Low gut microbiota diversity in early infancy precedes asthma at school age. Pediatrics. 136(Supplement):S232–S232. doi:10.1542/peds.2015-2776T.
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. 2013. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2:e01202. doi:10.7554/eLife.01202.
- Collins JW, Keeney KM, Crepin VF, Rathinam VAK, Fitzgerald KA, Finlay BB, Frankel G. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Publ Group. 12:612–623.
- Kennedy EA, King KY, Baldridge MT. 2018. Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front Physiol. 9:1534. doi:10.3389/fphys.2018.01534.
- Zha J-M, Li H-S, Lin Q, Kuo W-T, Jiang Z-H, Tsai P-Y, Ding N, Wu J, Xu S-F, Wang Y-T, et al. 2019. Interleukin 22 expands transit-amplifying cells while depleting lgr5+ stem cells via inhibition of wnt and notch signaling. Cell Mol Gastroenterol Hepatol. 7(2):255–274. doi:10.1016/j.jcmgh.2018.09.006.
- Cox CB, Storm EE, Kapoor VN, Chavarria-Smith J, Lin DL, Wang L, Li Y, Kljavin N, Ota N, Bainbridge TW, et al. 2021. IL-1R1–dependent signaling coordinates epithelial regeneration in response to intestinal damage. Sci Immunol. 6(59):eabe8856. doi:10.1126/sciimmunol.abe8856.
- Harnack C, Berger H, Antanaviciute A, Vidal R, Sauer S, Simmons A, Meyer TF, Sigal M. 2019. R-spondin 3 promotes stem cell recovery and epithelial regeneration in the colon. Nat Commun. 10(1):4368. doi:10.1038/s41467-019-12349-5.
- Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, Wucherpfennig K, Michor F, Clevers H, Shivdasani RA. 2020. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell. e6;26(3):377–390. doi:10.1016/j.stem.2019.12.011.
- Bäumler AJ, Heffron F. 1995. Identification and sequence analysis of lpfABCDE, a putative fimbrial operon of salmonella typhimurium. J Bacteriol. 177(8):2087–2097. doi:10.1128/jb.177.8.2087-2097.1995.
- Torres AG, Kanack KJ, Tutt CB, Popov V, Kaper JB. 2004. Characterization of the second long polar (LP) fimbriae of Escherichia coli O157:H7 and distribution of LP fimbriae in other pathogenic E. coli strains. FEMS Microbiol Lett. 238(2):333–344. doi:10.1016/j.femsle.2004.07.053.
- Sajjan US, Xie H, Lefebre MD, Valvano MA, Forstner JF. 2003. Identification and molecular analysis of cable pilus biosynthesis genes in Burkholderia cepacia. Microbiology+. 149(Pt 4):961–971. doi:10.1099/mic.0.26176-0.
- Kamada N, Kim Y-G, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. 2012. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 336(6086):1325–1329. doi:10.1126/science.1222195.
- Kiesler P, Fuss IJ, Strober W. 2015. Experimental models of inflammatory bowel diseases. Cell and Mol Gastroenterol and Hepatol. 1(2):154–170. doi:10.1016/j.jcmgh.2015.01.006.
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. 2006. Transforming growth factor-β induces development of the TH17 lineage. Nature. 441(7090):231–234. doi:10.1038/nature04754.
- Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. 2002. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, citrobacter rodentium, in mice lacking IL-12 or IFN-γ. J Immunol. 168(4):1804–1812. doi:10.4049/jimmunol.168.4.1804.
- Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, Fehling HJ, Hardt W-D, Shakhar G, Jung S. 2009. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 31(3):502–512. doi:10.1016/j.immuni.2009.06.025.
- Seo S-U, Kuffa P, Kitamoto S, Nagao-Kitamoto H, Rousseau J, Kim Y-G, Núñez G, Kamada N. 2015. Intestinal macrophages arising from CCR2+ monocytes control pathogen infection by activating innate lymphoid cells. Nat Commun. 6(1):8010. doi:10.1038/ncomms9010.
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. 2004. Enhanced adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 127(1):80–93. doi:10.1053/j.gastro.2004.03.054.
- Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 127(2):412–421. doi:10.1053/j.gastro.2004.04.061.
- Group TNHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, et al. 2009. The NIH human microbiome project. Genome Res. 19(12):2317–2323. doi:10.1101/gr.096651.109.
- Wrzosek L, Miquel S, Noordine M-L, Bouet S, Chevalier-Curt MJ, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C, et al. 2013. Bacteroides thetaiotaomicron and faecalibacterium prausnitziiinfluence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 11(1):61. doi:10.1186/1741-7007-11-61.
- Jinnohara T, Kanaya T, Hase K, Sakakibara S, Kato T, Tachibana N, Sasaki T, Hashimoto Y, Sato T, Watarai H, et al. 2017. IL-22BP dictates characteristics of Peyer’s patch follicle-associated epithelium for antigen uptake. J Exp Med. 214(6):1607–1618. doi:10.1084/jem.20160770.
- Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. 2018. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. 53(4):465–474. doi:10.1007/s00535-017-1401-7.
- Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, et al. 2017. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med. 9(376):1–12. doi:10.1126/scitranslmed.aaf9655.
- Seo S-U, Kamada N, Muñoz-Planillo R, Kim Y-G, Kim D, Koizumi Y, Hasegawa M, Himpsl SD, Browne HP, Lawley TD, et al. 2015. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity. 42(4):744–755. doi:10.1016/j.immuni.2015.03.004.
- Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, Mowat AM. 2013. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 6(3):498–510. doi:10.1038/mi.2012.89.
- Denning TL, Wang Y, Patel SR, Williams IR, Pulendran B. 2007. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17–producing T cell responses. Nat Immunol. 8(10):1086–1094. doi:10.1038/ni1511.
- Wynn TA, Vannella KM. 2016. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 44(3):450–462. doi:10.1016/j.immuni.2016.02.015.
- Viladomiu M, Metz ML, Lima SF, Jin W-B, Chou L, Bank JLC, Guo C-J, Diehl GE, Simpson KW, Scherl EJ, et al. 2021. Adherent-invasive E coli metabolism of propanediol in Crohn’s disease regulates phagocytes to drive intestinal inflammation. Cell Host Microbe. 29(4):607–619. e8.
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, et al. 2009. Critical regulation of early th17 cell differentiation by interleukin-1 signaling. Immunity. 30(4):576–587. doi:10.1016/j.immuni.2009.02.007.
- Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJIL. 2012. 1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. J Exp Med. 209(9):1595–1609. doi:10.1084/jem.20111453.
- Dinarello CA, Simon A, van der Meer JWM. 2012. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 11(8):633–652. doi:10.1038/nrd3800.
- Donath MY, Shoelson SE. 2011. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 11(2):98–107. doi:10.1038/nri2925.
- Mao L, Kitani A, Strober W, Fuss IJ. 2018. The role of NLRP3 and IL-1β in the pathogenesis of inflammatory bowel disease. Front Immunol. 9:2566. doi:10.3389/fimmu.2018.02566.
- Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. 1990. Role of interleukin 1 in inflammatory bowel disease–enhanced production during active disease. Gut. 31(6):686. doi:10.1136/gut.31.6.686.
- Al-Sadi R, Guo S, Ye D, Dokladny K, Alhmoud T, Ereifej L, Said HM, Ma TY. 2013. Mechanism of IL-1β modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation. J Immunol. 190(12):6596–6606. doi:10.4049/jimmunol.1201876.
- Al-Sadi R, Guo S, Dokladny K, Smith MA, Ye D, Kaza A, Watterson DM, Ma TY. 2012. Mechanism of interleukin-1β induced-increase in mouse intestinal permeability in vivo. J Interf Cytokine Res. 32(10):474–484. doi:10.1089/jir.2012.0031.
- Al-Sadi R, Ye D, Said HM, Ma TYIL. 2010. 1β-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-κB pathway. Am J Pathol. 177(5):2310–2322. doi:10.2353/ajpath.2010.100371.
- Rawat M, Nighot M, Al-Sadi R, Gupta Y, Viszwapriya D, Yochum G, Koltun W, Ma TY. 2020. IL1B increases intestinal tight junction permeability by up-regulation of MIR200C-3p, which degrades occludin mRNA. Gastroenterology. 159(4):1375–1389. doi:10.1053/j.gastro.2020.06.038.
- Carter JD, Valeriano J, Vasey FB. 2003. Crohn disease worsened by anakinra administration. Jcr J Clin Rheumatol. 9(4):276–277. doi:10.1097/01.RHU.0000081265.06408.e4.
- Wang G, Sweren E, Liu H, Wier E, Alphonse MP, Chen R, Islam N, Li A, Xue Y, Chen J, et al. 2021. Bacteria induce skin regeneration via IL-1β signaling. Cell Host Microbe. 29(5):777–791. e6.
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. 2010. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 464(7293):1371–1375. doi:10.1038/nature08949.
- Jung Y, Wen T, Mingler MK, Caldwell JM, Wang YH, Chaplin DD, Lee EH, Jang MH, Woo SY, Seoh JY, et al. 2015. IL-1β in eosinophil-mediated small intestinal homeostasis and IgA production. Mucosal Immunol. 8(4):930–942. doi:10.1038/mi.2014.123.
- Ganesh BB, Bhattacharya P, Gopisetty A, Sheng J, Vasu C, Prabhakar BS, Unutmaz D. 2011. IL-1β promotes tgf-β1 and il-2 dependent foxp3 expression in regulatory t cells. PLoS One. 6(7):e21949. doi:10.1371/journal.pone.0021949.
- Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, et al. 2010. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 8(3):292–300. doi:10.1016/j.chom.2010.08.004.
- Armbruster CE, Mobley HLT. 2012. Merging mythology and morphology: the multifaceted lifestyle of proteus mirabilis. Nat Rev Microbiol. 10(11):743–754. doi:10.1038/nrmicro2890.
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 27(5):722–736. doi:10.1101/gr.215087.116.
- Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, et al. 2015. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep-uk. 5(1):8365. doi:10.1038/srep08365.
- Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19(9):1639–1645. doi:10.1101/gr.092759.109.
- Lee MD, Ponty Y. 2019. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics. 35(20):btz 188. doi:10.1093/bioinformatics/btz188.
- Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11(1):119. doi:10.1186/1471-2105-11-119.
- Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P. 2020. A complete domain-to-species taxonomy for bacteria and archaea. Nat Biotechnol. 38(9):1079–1086. doi:10.1038/s41587-020-0501-8.
- Nguyen L-T, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi:10.1093/molbev/msu300.
- Shen W, Ren H. 2021. TaxonKit: a practical and efficient NCBI taxonomy toolkit. J Genet Genomics. doi:10.1016/j.jgg.2021.03.006.
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. 2018. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity. e6. 48(1):35–44. doi:10.1016/j.immuni.2017.11.013.