
ABSTRACT
The gut-lung axis has been implicated as a potential therapeutic target in lung disorders. While increasing evidence suggests that gut microbiota plays a critical role in regulating host immunity and contributing to tuberculosis (TB) development and progression, the underlying mechanisms whereby gut microbiota may impact TB outcomes are not fully understood. Here, we found that broad-spectrum antibiotics treatment increased susceptibility to Mycobacterium tuberculosis (M. tuberculosis) infection and modulated pulmonary inflammatory responses in mouse M. tuberculosis infection model. We then identified a commensal gut bacteria-regulated lncRNA, termed lncRNA-CGB, which was down-regulated by dysbiosis of gut microbiota during TB infection. Furthermore, we found that Bacteroides fragilis (B. fragilis) was a direct regulator of lncRNA-CGB, and oral administration of B. fragilis enhanced expression of lncRNA-CGB and promoted anti-TB immunity. Genomic knock-out of lncRNA-CGB led to reduced IFN-γ expression and impaired anti-TB immunity, therefore leading to detrimental effects on M. tuberculosis infection. Mechanistically, lncRNA-CGB interacted with EZH2 and negatively regulated H3K27 tri-methylation (H3K27Me3) epigenetic programming, leading to enhanced IFN-γ expression. Thus, this work not only uncovered previously unrecognized importance of gut bacteria-lncRNA-EZH2-H3K27Me3 axis in conferring immune protection against TB but also identified a potential new paradigm to develop a microbiota-based treatment against TB and potentially other diseases.
Introduction
The gastrointestinal tract represents a large ecosystem, housing millions of microbial cells, which are of critical importance for modulating systemic equilibrium of the immune system toward healthy homeostasis.1,Citation2 The concept of the gut-lung axis was born out of the observation that different lung diseases could be influenced by intestinal microenvironment changes and vice versa.Citation1,Citation2 Moreover, a higher prevalence of pulmonary diseases in those patients who also have chronic gastrointestinal diseases highlights the importance of crosstalk between the gut and lung.Citation3 Increasing evidence has highlighted that disturbing the gut-lung axis may contribute to impaired lung homeostasis and influencing the outcomes of lung diseases, such as asthma, chronic obstructive pulmonary disease, respiratory virus infection, and cystic fibrosis.Citation4–8
Mycobacterium tuberculosis (M. tuberculosis) infects one-third of the world population and leads to tuberculosis (TB), a deadly infectious disease with currently an estimated 1.4 million deaths annually.Citation9,Citation10 While the complex interplay between genetic factors, host- and pathogen-specific factors contribute to the development and progression of the TB infection, significant risk factors for developing active TB are associated with both structural and functional changes in the gut microbiota.Citation11–13 Emerging evidenceCitation14,Citation15 has suggested that altered microbiota is likely to have deleterious consequences for susceptibility to TB development and progression. Successfully addressing TB infection and disease may require targeting other modifiable risk factors, such as host microbiota. However, available research findings provide relatively limited evidence on the crosstalk between gut and lung during TB infection, and the underlying mechanism(s) whereby gut microbiota regulates the TB disease development and progression are largely not explained.
It has been suggested that the soluble microbial components and metabolites transported via the circulation are one means of communication between the gut and the lung.Citation1,Citation2 The gut microbiota therefore can directly and/or indirectly influence lung homeostasis via host-derived inflammatory mediators and determine the protective or pathological outcomes of microbial infection not only in gastrointestinal tract but also in lungs.Citation16–20 For instance, major mechanisms attributed to the protective effect of lactobacilli during respiratory infection are the induction of protective immunity via stimulation of the immunomodulatory mediators such as IFN-γ as well as the inhibition of lung immunopathology via the induction of IL-10.Citation21 However, the signals from the gut microbiota that affect TB infections in lungs are largely undetermined.
While the roles of protein-coding genes in host-microbiota interactions have been subjected to intensive investigations, emerging evidence has revealed that regulatory non-coding RNAs (ncRNAs) play a critical role in modulating host-microbe interactions, and ncRNAs have been proposed as potential modulators of the host response to microbiome-linked pathologies such as cancers and obesity.Citation22 An in-depth understanding of the roles and underlying mechanisms of ncRNAs in modulating host-microbe interactions involving gut-lung axis thus may provide more effective diagnostic tools and therapeutic agents for microbiome-linked diseases not only locally but also systemically. Recent studies have reported that compared with germ-free mice, those that were colonized with specific bacteria displayed a significantly different long non-coding RNA (lncRNA) profile, which suggest that crosstalk between host microbiota and noncoding RNAs may play a role in TB development, progression and prognosis.Citation22,Citation23 However, little is known as to whether and by which gut microbiota modulate circulating non-coding RNAs such as lncRNAs that contribute to M. tuberculosis infection outcomes.
Significant amounts of lncRNAs have been identified in the mammalian genomes,Citation24 of which only a minority, but increasingly more, have been functionally characterized in different processes, including infections.Citation25,Citation26 Some host lncRNAs have been reported to regulate the expression of inflammatory mediators or be regulated by interferon (IFN) in respiratory viral infection,Citation27 and our previous identification of lncRNA-CD244 suggested that lncRNA might have immunomodulatory properties for a more effective TB control.Citation28 However, whether a specific lncRNA is involved in gut-lung axis during TB infection in response to gut microbiota-derived cues is unclear. Thus, understanding how the intestinal microbiota primes the host inflammatory response through regulating the expression and functions of lncRNA to combat TB is vital for shaping future strategies of more effective prevention and treatment of TB.
Here, we found that dysbiosis of gut microbiota induced more severe TB infection and identified a series of commensal bacteria-associated lncRNAs, of which, a lncRNA (lncRNA-ENSMUSG00000086503, termed lncRNA-CGB) was significantly down-regulated in M. tuberculosis-infected mice with disruption of gut bacteria. Consistently, the human homologue of lncRNA-CGB was further characterized as the most significantly depressed lncRNA in active TB patients whose gut microbiome’s diversity was perturbed. Genomic knock-out of lncRNA-CGB developed much more severe TB disease after M. tuberculosis infection hinting that lncRNA-CGB played a beneficial role in developing immune resistance to M. tuberculosis infection and TB pathology. Moreover, we found that gut dysbiosis depleted bacterial species regulating proper immune functioning and therefore induced significantly repressed M. tuberculosis-killing cytokines, IFN-γ. Furthermore, oral transfer of the resident intestinal probiotic Bacteroides fragilis (B. fragilis) sustained expression of lncRNA-CGB, and promoted anti-TB immune protection via epigenetically modulating IFN-γ expression, indicating that a selected lncRNA signature modulated by commensal gut bacteria might be a key mediator regulating gut-lung axis homeostasis during M. tuberculosis infection.
Thus, this work not only uncovered previously unrecognized importance of gut microbiota-dictated lncRNA signature in conferring immune protection in gut-lung axis but also highlighted a potentially new paradigm to develop microbiota-based therapeutic interventions for lung diseases.
Results
Gut dysbiosis disrupts lung homeostasis and promotes M. tuberculosis infections in M. tuberculosis-infected mouse model
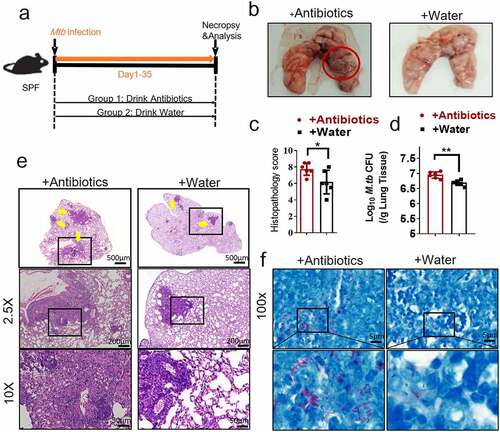
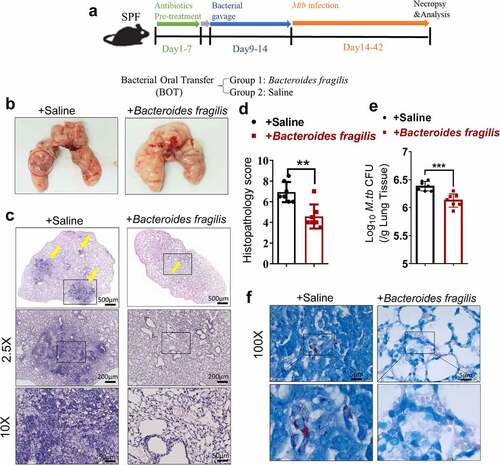
As an initial step to determine whether gut microbiota regulated the M. tuberculosis outcomes through modulating the expression and function of the lncRNAs, we first analyzed whether disruption of gut microbiota via antibiotics feeding could impact TB infection outcomes, thus we performed an antibiotic administration protocol during TB infection. Rapid loss of diversity of gut microbiota was observed after the broad-spectrum antibiotics (ampicillin, neomycin, vancomycin and metronidazole) feedings (Supplementary Figure S1). More importantly, oral treatment of these antibiotics was found to induce dysbiosis, but no significant effect on M. tuberculosis viability was observed even in mice with antibiotics administration for 42 days.Citation29 To this end, mice were fed daily with broad-spectrum antibiotics or saline as control during M. tuberculosis infection (). Therefore, the TB infection outcomes with or without the disruption of bacterial equilibrium by antibiotics treatment in mice can be determined. When compared with water controls, antibiotics feedings led to more severe lung pathology characterized by larger scale damage of lung structure and defined granuloma, more significant infiltration of inflammatory cells in the lungs (, c, e), much higher M. tuberculosis burdens in pulmonary compartments (), and more bacilli positive for acid-fast staining in granuloma lesion area, compared with water controls (). Thus, these results not only suggested that microbial diversity in the gastrointestinal tract was required to prime or sustain systemic immune resistance toward TB but also outlined the potential role of the gut microbiota in gut-lung crosstalk involved in TB infection.
Figure 1. Gut microbiota is required for the control of M. tuberculosis infection.
The gut microbiota regulates the expression of the lncRNA-CGB, and such gut commensal-modulated lncRNA involves in active TB infection
To systematically identify gut microbiota-regulated lncRNAs, we then determined whether alteration of gut commensal bacteria would modulate lncRNA expression profiles via destruction/aberration of gut microbiota by antibiotic feedings during M. tuberculosis infection. RNA sequencing-based transcriptomic analysis was performed to comparatively measure lncRNAs in gut tissues from antibiotics-fed mice and controls during M. tuberculosis infection. ~7592 lncRNAs were deregulated in the gut tissues of antibiotics-fed mice during TB infection and among them, lncRNA-ENSMUSG00000086503 was one of the most significantly down-regulated lncRNA signatures when gut microbiota was depleted (). We therefore temporarily named it as lncRNA-CGB (commensal gut bacteria-associated lncRNA) and focused on this lncRNA-CGB for in-depth functional and mechanistic studies.
Figure 2. LncRNA-CGB expression was regulated by gut microbiota and involved in TB infection.
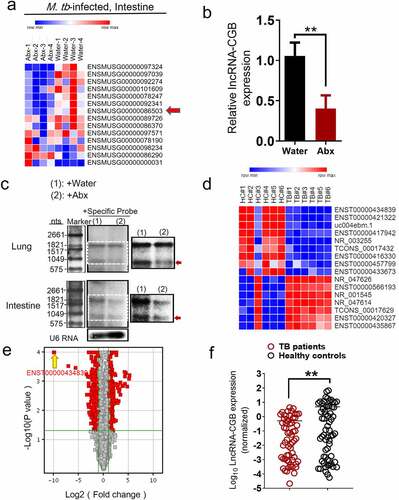
Localization and protein-coding capability analyses of lncRNA-CGB suggested that lncRNA-CGB was located on chromosome X, predominantly localized in the nuclear fractions, was highly conserved in different species including Homo sapiens and Mus musculus and did not show detectable coding ability or translation events (Supplementary Figure S2a-f).Moreover, qRT-PCR – and northern blot-based quantitative assays demonstrated that antibiotics feeding of M. tuberculosis-infected mice while destroying microbiota, significantly reduced expression levels of lncRNA-CGB in lungs and intestines (, c and Supplementary Figure S3). These data implicated that lncRNA-CGB might indeed somehow be involved in regulating TB pathogenesis induced by destruction/aberration of gut microbiota.
To further investigate whether lncRNAs were involved in the gut-lung immune axis in response to M. tuberculosis infection, we then evaluated whether TB infections were associated with any gut microbiota-regulated lncRNA signatures using human lncRNAs microarray and hierarchical clustering analyses. The comparative analysis between active TB patients and healthy controls (HC) allowed us to display distinct lncRNAs expression profiles during TB infection. The supervised hierarchical clustering segregation analysis then identified dominant groups of lncRNAs differentially expressed in TB patients (). From 15,683 denoted lncRNAs, 13 lncRNAs were identified to be differentially expressed (TB/HC > 2 or TB/HC< 0.5) in peripheral blood mononuclear cells (PBMCs) between TB and healthy subjects (). Remarkably, among them, lncRNA-ENST00000434839, the human homologue of lncRNA-CGB (Supplementary Figure S2a), was identified as one of the three mostly down-regulated lncRNAs in patients with active TB disease, displaying mean 4.7-fold down-regulation in differentially expressed lncRNAs. High consistency of qRT-PCR validation of lncRNA-ENST00000434839 in a larger cohort of TB patients (N = 50) and healthy controls (HC) (N = 43) further supported that gut microbiota-regulated lncRNA-CGB might somehow involve in active TB disease progression (). To further identify whether this specific lncRNA-CGB was associated with active TB infection, we then explored the expression of lncRNA-CGB in PBMCs derived from M. tuberculosis-infected mice by qPCR. Significantly decreased expression of lncRNA-CGB was detected in M. tuberculosis-infected mice when compared with those in mock-infected mice (Supplementary Figure S4).
These findings strongly suggested alteration of lncRNA-CGB expression might indeed somehow be involved in immunomodulating gut-lung crosstalk during M. tuberculosis infection.
LncRNA-CGB is required for host resistance against M. tuberculosis infection
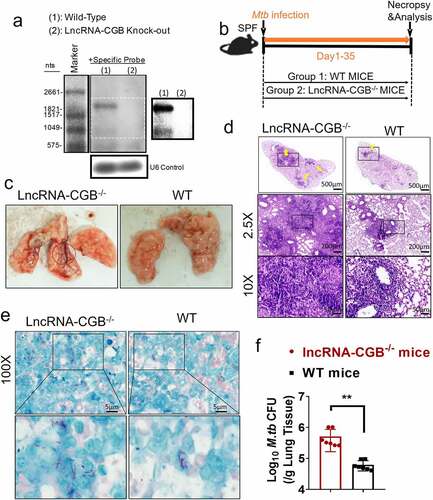
Since gut microbiota was required for developing resistance to TB, and depressed lncRNA-CGB expression patterns correlated with bad outcomes of M. tuberculosis infection in mouse model and active TB disease in humans, we then determined the exact role of lncRNA-CGB in M. tuberculosis infection. We developed a lncRNA-CGB genomic knock-out (KO) mouse strain using the CRISPR-Cas9 system deletion of the lncRNA regions (Supplementary Figure S5a). Genomic deletion and loss of lncRNA-CGB transcripts in the lymphocytes from the lncRNA knock-out (KO) mice were confirmed by northern blotting () and PCR analysis (Supplementary Figure S5b). The genomic knock-out (KO) did not interfere with the expression of flanking genes, and there was a lack of any abnormal findings for the lncRNA knock-out (KO) mice. We then examined whether in vivo deficiency of lncRNA-CGB would result in a beneficial or detrimental effect on M. tuberculosis infection (). Importantly, lncRNA-CGB knock-out (KO) mice developed much more severe TB disease after M. tuberculosis infection than did wild-type animals. Virtually, gross pathology and hematoxylin and eosin (H&E) staining of lung sections revealed that lncRNA-CGB knock-out (KO) mice developed more severe disruption of lung structure and larger scale granuloma lections than did control mice (, ). In addition, higher bacillus burdens in lungs derived from lncRNA-CGB knock-out (KO) mice were observed than did control mice (, ).
Figure 3. LncRNA-CGB knock-out (KO) mice developed more severe TB than did control mice.
To confirm whether human lncRNA-CGB also played a role in mediating anti-TB resistance as mouse lncRNA-CGB did, we developed a human T cell adoptive transfer-based M. tuberculosis infection therapeutics study model with lncRNA-CGB knock-down in CD3 + T cells, as we previously did.Citation28 Briefly, CD3 + T cells and CD14+ monocytes purified from PBMCs of active TB patients were firstly transduced with lentiviral (LV) vector encoding shRNA targeting lncRNA-CGB or LV vector only (LV-Ctrl). LV-lncRNA–transduced (lncRNA-CGB–depressed) and LV-Ctrl–transduced CD3 + T cells were then transferred into each of recipient M. tuberculosis-infected mice which had also received autologous monocytes from TB patients to facilitate M. tuberculosis infection. We found that lncRNA-CGB knock-down impaired the ability of human CD3 + T cells to limit M. tuberculosis replications and infections in this T cell adoptive transfer-based M. tuberculosis infection therapeutics study model (Supplementary Figure S6a, b). Thus, these results suggested that both mouse and human lncRNA-CGB played a critical role in limiting M. tuberculosis infection and TB pathology.
Taken together, we found that gut bacteria might mediate host resistance against TB through sustaining or enhancing the expression of lncRNA-CGB.
B. fragilis, a significantly altered microbiota during TB, is a key mediator of the lncRNA-CGB expression profile
We then identified which component of gut microbiota mediated the expression and function of lncRNA-CGB. We firstly colonized antibiotic-treated mice with microbiota from healthy participants and then measured the expression of lncRNA expression in the lungs. Fecal microbiota restoration substantially increased lncRNA-CGB expression in lung tissues (). This indicated that human gut microbiota indeed could modulate lncRNA-CGB expression.
Figure 4. B. fragilis was identified as a significantly altered gut microbiota to orchestrate the expression of lncRNA-CGB during TB infection.
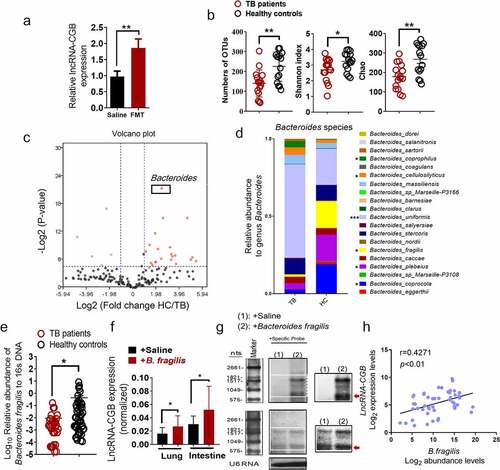
We therefore performed a detailed comparison of gut microbial profiles using 16S rRNA gene sequencing in fecal samples from active TB patients and healthy controls (HC). The baseline characteristics of the participants are shown in . Consistent with previous reports,Citation30–32 we observed significantly lower species richness in TB patients’ samples (TB) compared to healthy controls (HC) (Wilcoxon rank-sum test; ). Consistently, the Shannon index and the Chao1 Index, indicating within-sample diversity, were much lower for the TB patient group than that for the healthy controls (HC) (p-values<0.01, <0.01; Wilcoxon rank-sum test; ). These data suggested that microbial diversity was decreased in active TB patients and M. tuberculosis infection exhibited alterations of the discriminatory genus of gut microbiota.
Table 1. Demographic and clinical characteristics of active TB patients (TB) and healthy controls (HC)
To analyze the gut microbial profiles of the study participants and identify the exact gut bacteria driving lncRNA-CGB alteration, a comprehensive comparison between the patients with TB and controls in terms of the abundance of genera was performed. Of the 164 genera identified, 28 (17.1%) were found to have significant differential log abundances between both groups (). Specifically, we found that the relative abundance of the genus Bacteroides was visibly lower in TB patients compared with controls (). Considering that previous reports have shown a lower abundance of the genus Bacteroides in children with pulmonary tuberculosisCitation33 and Bacteroides has been implicated to play a critical role in mediating immune resistance against a variety of diseases such as cancers,Citation34,Citation35 we then focused on the Bacteroides species to investigate whether the altered abundance of Bacteroides species was associated with the development and progression of human TB infection. Notably, B. fragilis was identified as one of the most significantly differently and abundantly expressed species within the genus Bacteroides in 16S rRNA gene sequencing analysis, displaying mean 12.13-fold up-regulation in 20 species of gut microbiota from healthy controls over TB patients (). Furthermore, in order to verify the validity of 16S rDNA analysis, qRT-PCR analysis targeting B. fragilis was performed in a larger cohort of TB patients (TB) (N = 50) and healthy controls (HC) (N = 43). qRT-PCR analysis showed a much lower enrichment of B. fragilis in the feces from a large cohort of TB patients ().
To further validate the intestinal microbiota changes were due to M. tuberculosis infection, we next comparatively the compositions of the microbiome in M. tuberculosis-infected or uninfected mice by qPCR. In agreement with previous study,Citation36 we observed similarly decreased trends of gut microbiota diversity during murine M. tuberculosis infection. Interestingly, the genus that showed higher abundance in the feces of healthy controls (HC) also displayed higher abundance levels in uninfected mice, including genus Veillonella, Blautia, Neisseria Fusobacterium, Desulfovibrio, Bacteroides, Lactobacillus, Enterococcus and Campylobacter (Supplementary Figure S7). More importantly, the relative abundance of genus Bacteroides was significantly decreased in the M. tuberculosis-infected mice, which is consistent with those alterations in TB patients’ gut microbiome (Supplementary Figure S7). Moreover, such alterations of gut microbiome in response to M. tuberculosis infection did not induce observable histopathological changes of cecum in Hematoxylin and Eosin (H&E) staining (Supplementary Figure S8).
Thus, these data further suggested that B. fragilis was a gut microbial taxa that was the most significantly altered by active TB infection.
LncRNA-CGB is upregulated by B. fragilis and is clinicopathologically related to TB features and outcomes
Given the appreciated importance of B. fragilis in governing microbiota-host interaction,Citation37 shaping host immune responseCitation34,Citation37,Citation38 and regulating T cell immunity,Citation39 we further hypothesized that B. fragilis might play, or be at least one of, the most important roles in regulating lncRNA-CGB expression and gut-lung immune axis, which therefore determined the outcomes of M. tuberculosis infection. To test this hypothesis, we quantified the lncRNA-CGB expression after oral administration of nontoxic commensal gut bacteria, B. fragilis. Remarkably, analyses based on qRT-PCR and northern blotting revealed that B. fragilis treatment increased or sustained the lncRNA-CGB expression in M. tuberculosis-infected mice (, ). In consistent with mouse data showing the positive relationship between higher lncRNA-CGB expression and B. fragilis abundance, we also observed a significant correlation between higher abundance of B. fragilis in fecal samples and the lncRNA-CGB up-regulation in active TB patients (). Sputum smear positivity in patients with active TB are usually correlated with higher disease severity.Citation40 Thus, to provide further evidence showing the association between the relative abundance of B. fragilis and TB infection, we further examined the relationship between the levels of B. fragilis abundance in fecal samples and the grades of smear positivity. A trend toward increased levels of the B. fragilis with decreasing numbers of M. tuberculosis bacteria in the sputum was observed (Supplementary Figure S9). A significant negative correlation between the relative abundance of B. fragilis and sputum smear grades was found in smear-positive TB patients (r = −0.9531, p < .05) (Supplementary Figure S9).
Therefore, these data from mouse model and humans collectively indicated that lncRNA-CGB expression was dictated by gut bacteria, and such gut commensal-modulated, variable lncRNA-CGB expression patterns might be a mechanism for a selected gut commensal governing gut-lung immune interaction axis in response to M. tuberculosis infection.
B. fragilis enhanced host resistance against M. tuberculosis infection in mice
LncRNA-CGB, which was dictated by B. fragilis, conferred the beneficial effects on host resistance to M. tuberculosis infection, raised a critical question as to whether dominant or abundant bacteria B. fragilis was responsible for conferring anti-TB beneficial effects. To define the role of B. fragilis during M. tuberculosis infection, we assessed B. fragilis in oral transfer for its ability to function as an active anti-TB component in TB-associated or TB-altered gut bacteria (). Mice that received bacteria oral transfer (BOT) of B. fragilis displayed significantly attenuated tissue pathology and decreased M. tuberculosis burdens while saline-treated mice showed more severe lung pathology characterized by large-scale damage of lung structure and defined granuloma (). Moreover, B. fragilis-treated mice showed much lower M. tuberculosis CFU numbers in lungs than that of saline-treated mice (), and lung sections derived from B. fragilis-treated mice contain fewer acid-fast-staining-positive M. tuberculosis bacilli (). Interestingly, the structure of cecum exhibited no significant differences on M. tuberculosis-infected lncRNA-CGB knock-out/wild type mice with/without oral treatments of B. fragilis and their matched controls (Supplementary Figure S8). Taken together, B. fragilis acted as a beneficial regulator in conferring immune control of pulmonary M. tuberculosis infection and TB pathology.
Figure 5. Direct oral administration of B. fragilis protected mice against M. tuberculosis infection.
LncRNA-CGB mediates gut microbiota-reduced host susceptibility to TB by inducing IFN-γ production
We then examined how commensal bacteria was involved in mediating protective immunity against M. tuberculosis infection. The gene set enrichment analysis (GSEA) of the lungs from antibiotics-treated or control mice (GSEA false discovery rate [FDR] respectively q < 0.05; ) suggested that gut microbiota might modulate cytokines and inflammatory responses during M. tuberculosis infection. Furthermore, RNA sequencing-based transcriptomic analysis also demonstrated significantly down-regulated IFN-γ transcriptional responses in the lungs of antibiotics-fed mice during M. tuberculosis infection (). It is noteworthy that IFN-γ is absolutely required for immune resistance to TB,Citation41 and gut microbiota is critically important for the production of IFN-γ.Citation20,Citation42 We therefore postulated that IFN-γ might be a key mediator linking gut microbiota, lncRNA profiles and pulmonary immune homeostasis.
Figure 6. Gut microbiota and its regulated lncRNA-CGB modulate de novo IFN-γ responses during M. tuberculosis infection.
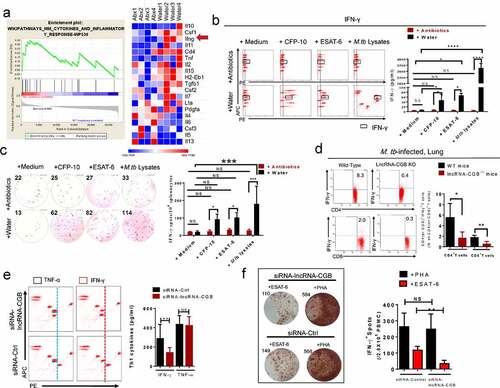
Concurrently, antibiotics feedings of M. tuberculosis-infected mice shut down or remarkably reduced the ability of lung lymphocytes to mount M. tuberculosis-specific IFN-γ responses as revealed by flow-cytometry-based quantitation () and IFN-γ-ELISPOT assays (). Moreover, antibiotics feedings significantly decreased de novo production of IFN-γ by intestinal CD4+ and CD8 + T cells derived from M. tuberculosis-infected mice without further ex vivo re-stimulation with M. tuberculosis antigens (Supplementary Figure S10).
In addition, lncRNA-CGB knock-out indeed reduced IFN-γ expression by CD4+ and CD8 + T cells during M. tuberculosis infection in mice (), implicating that commensal bacteria-dictated lncRNA-CGB was involved in mediating IFN-γ expression and therefore conferred beneficial effect against M. tuberculosis infection. In agreement with these mouse data, knock-down of lncRNA-CGB in CD3 + T cells derived from active TB patients also led to an inhibited expression of IFN-γ (). In parallel, oral administration of B. fragilis allowed increased induction of IFN-γ expression during M. tuberculosis infection in vivo and ex vivo (Supplementary Figure S11). These results suggested that IFN-γ was one of the key immune mediators linking gut-lncRNA-CGB-lung axis.
Taken together, the above findings allowed us to hypothesize a potential biological sequence or pathway: gut microbiota →lncRNA-CGB→IFN-γ expression →resistance to TB.
LncRNA-CGB directly interacted with EZH2 and negatively regulated H3K27Me3, leading to increased TB-specific IFN-γ expression and immunity against M. tuberculosis infection
We next sought to investigate the molecular mechanisms by which antibiotics’ destruction of microbiota depressed lncRNA-CGB and IFN-γ responses leading to a loss of immune resistance to M. tuberculosis infection. Given that emerging evidence has highlighted the critical roles for lncRNAs in mediating immune response genesCitation43 or T cell immune responsesCitation28,Citation44–47 by directly or indirectly interacting with epigenetic modifiers,Citation28,Citation44,Citation47–55 we then performed global histone methylation analyses in lymphocytes isolated from the lungs of M. tuberculosis-infected lncRNA-CGB knock-out (KO) mice or wild-type mice. We found that genomic knock-out of lncRNA-CGB increased expression of H3K27Me3 but reduced H3K9Me1 and H3K4Me3 (), implicating that lncRNA-CGB knock-out (KO) led to a globally more repressive chromatin state during M. tuberculosis infection. Since tri-methylation (Me3) at H3K27 could be catalyzed by the EZH2 subunit of histone methyltransferase Polycomb Repressive Complex 2 (PRC2),Citation56 we therefore presumed that lncRNA-CGB might directly interact with EZH2/H3K27Me3 epigenetic programming of IFN-γ transcription. To test this, we first predicted the interaction potential between lncRNA-CGB and EZH2. We identified a 51-nt motif with a predicted secondary structure in lncRNA-CGB similar to a previously validated 89-mer EZH2 interacting motif in lncRNA-HotairCitation56 (Supplementary Figure S12a), but the interaction potential of EZH2/lncRNA-CGB appeared to be stronger than (or at least as strong as) that of EZH2/lncRNA-Hotair (Supplementary Figure S12b,c).
Figure 7. LncRNA-CGB negatively regulates H3K27Me3 in IFN-γ (ifng) promoter via physically interacting with EZH2.
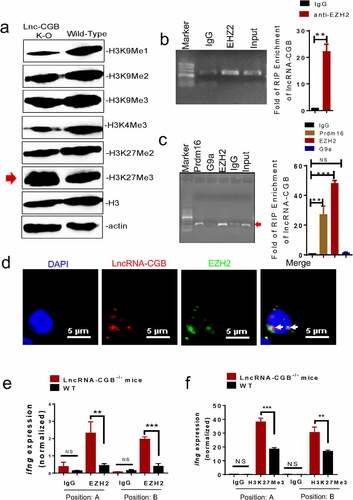
We then performed RNA immunoprecipitation (RIP) retreat assay using CD3 + T cells isolated from active TB patients. Indeed, we found that lncRNA-CGB was immunoprecipitated with both EZH2 and Prdm16 in lysates from CD3 + T cells of TB patients, with no or very little in G9a or IgG controls (). Similar amounts of lncRNA-CGB/EZH2 interactions were also found in lymphocytes derived from the lungs of M. tuberculosis-infected mice (). Consistently, confocal microscope-based fluorescence in situ hybridization (FISH) analysis of lncRNA-CGB and immunofluorescent staining of EZH2 showed that significant amounts of EZH2 co-localized with lncRNA-CGB in the nucleus of CD3 + T cells from patients with active TB (). Concurrently, weak interaction events between lncRNA-CGB and EZH2 were observed in confocal imaging and RIP in PBMCs derived from healthy controls (Supplementary Figure S13), further supporting the view that lncRNA-CGB interacted with the EZH2 subunit of histone methyltransferase PRC2 and thereafter regulated EZH2 and H3K27Me3 epigenetic programming in the ifng promoter. Besides, ChIP-qPCR analysis showed that lncRNA-CGB knock-out increased EZH2 abundance and enhanced tri-methylation (Me3) of H3K27 in the ifng promoter during M. tuberculosis infection (, f), suggesting that lncRNA-CGB was required to inhibit EZH2 translocation to the ifng promoter where they might mediate H3K27Me3 for inhibition of IFN-γ expression.
The studies presented here identified that lncRNA-CGB governed by B. fragilis and lncRNA-dictated IFN-γ expression were key mediators of gut-lung axis (Supplementary Figure S14). Thus, this work may shed new light on developing a microbiota-based treatment against lung diseases such as M. tuberculosis infection.
Discussion
While an emerging area of intense interest is the role of gut-lung axis in the pathogenesis of lung diseases,Citation2,Citation57 accumulating evidence has demonstrated that gut microbiota plays a critical role in mediating gut-lung axis.Citation58,Citation59 However, due to the complex crosstalk, the exact mechanisms through which the gut impacts lung health or disease and vice versa are only starting to be uncovered. The gut-lung axis allows for the passage of endotoxins, microbial metabolites, cytokines, or hormones into the bloodstream connecting the gut niche with that one of the lungs. However, mechanistic insights into the pathways and mediators involved in gut-lung axis are largely unexplored. For the first time, this work has characterized the previously unrecognized importance of a commensal gut bacteria-modulated lncRNA signature in mediating host defense mechanisms against deadly lung diseases, such as TB, via epigenetic programming of expression of an immune mediator.
Although it has been firmly established that the intestinal microbiota may exert a tremendous impact on the development and homeostasis of the immune system locally and systemically, our understanding of the exact mechanisms by which lung health is determined by intestinal microbiota is still at the beginning. While signals delivered by gut microbes were documented to influence the functions of innate immune cells,Citation4,Citation18,Citation60 adaptive T cells, such as CD4+ TCRαβ T helper (Th) cells, were also influenced by the intestinal epithelium-associated microbiota.Citation61 Pulmonary TB disease is a classic example of how a successful control of M. tuberculosis infection relies on delicate orchestration between adequate immune responses and inhibited activation and functional exertion of inflammatory networks.Citation62,Citation63 However, there is limited evidence regarding potential mechanistic pathways on gut-lung interactions that may orchestrate immune balance in both gut-lung axis that may further affect TB susceptibility, manifestation and progression. Our results provide the first line of evidence suggesting that the commensal gut bacteria-related lncRNA-CGB is linked to the regulation of the gut-lung axis for protective immunity against lung pathogenesis.
This work highlighted the importance of lncRNAs in shaping and promoting IFN-γ expression and function to support pulmonary homeostasis. The IFN-γ-deficient mice displayed significantly higher pulmonary M. tuberculosis burdens,Citation41 and regulation events from a gut microbiota-associated lncRNA in considerable responses by CD4 + T and CD8 + T effector subpopulations in naïve settings were observed in this work. Thus, it is not surprising to see changes in IFN-γ-driven anti-TB immunity in lncRNA-CGB knock-out mice. Also, relatively smaller alterations in M. tuberculosis CFU numbers were observed in lncRNA-CGB knock-out mice in this study than that of ifng knock-out mice.Citation41 This difference may be explained by the notion that lncRNA-CGB-enhanced anti-TB immunity may be partially compromised by other gut bacteria-regulated lncRNAs which are also involved in the immune network regulation during M. tuberculosis infection. Furthermore, since increased IFN-γ productions may lead to over-reacted immune response and immunopathology and significant fractions of M. tuberculosis-infected individuals with active TB disease express quite appreciable amounts of IFN-γ, enhanced expression of IFN-γ driven by lncRNA-CGB may not mean full immune protection against M. tuberculosis infection.Citation64 Thus, full investigations of more comprehensive lncRNAs-gut microbiota-immune network should be required for uncovering exact TB pathogenesis mechanisms.
Although it was anticipated that TB can alter abundance, composition and diversity in gut microbiota, exact TB-induced alterations of gut microbial components and how such TB-altered gut microbial species orchestrate pulmonary or systemic immunity against TB infection are unclear. We have identified that B. fragilis was a most significantly altered gut microbiota taxa that discriminated active TB patients from healthy controls. A recent study has reported that the relative abundance of B. fragilis might be increased after anti-TB treatment,Citation65 suggesting the recovery in the functional ability of B. fragilis after anti-TB treatment. The importance of B. fragilis in mediating immune protection or homeostasis against TB further gained support from our study wherein oral administration of B. fragilis conferred homeostatic benefits and immune resistance against M. tuberculosis infection. Since the structure and function of gut microbiota are dynamically regulated by genetics, environment, and food, the pre-treatment of antibiotics may help the mice orally treated with human gut microbiota or a single strain bacteria tend to be at the same baseline in terms of structure and function of their gut microbiota in our study. However, the true impacts and underlying mechanisms of the baseline of gut microbiome on treatments of FMT or gut microbiota interference during TB infection need further study.
Moreover, our study revealed that a significant shift of gut bacteria toward beneficial microbial consortium could be highly associated with the sustaining of antibiotics-depressed lncRNA-CGB expression and production of protective IFN-γ response against TB by B. fragilis. The data therefore consistently point to the notion that lncRNA is a decisive mediator for B. fragilis to control TB.
To date, commensal probiotic B. fragilis or short chain fatty acids (SCFAs) fermented from B. fragilis have been shown to inhibit inflammation and infection by pathogenic bacteria and support cancer therapy via microbe-host immune interactions.Citation34,Citation60 Other views also suggest that B. fragilis may control pathogenesis by readjusting the composition of the gut microbiota or by enhancing the intestinal barrier defense system.Citation66 Notably, the roles and mechanisms of B. fragilis in the regulation of any lncRNA in the case of TB have not been reported elsewhere. Our current study elucidated that gut microbiota was important for the control of M. tuberculosis infection through sustaining expression of lncRNA-CGB and underscored the concept that gut microbiota not only directly mediated immune responses but also regulated non-coding genes to induce the immunomodulatory effects in response to M. tuberculosis infections.
While it is still at the beginning to understand exact molecular mechanisms regulating the gut-lung axis, our epigenetic mechanism study reveals that lncRNA-CGB inhibits H3K27Me3 programming in IFN-γ promoter and allows for IFN-γ expression and subsequently protective homeostasis and outcomes during M. tuberculosis infection. Notably, lncRNA-CGB binding to EZH2 did not seem to directly repress PRC2 enzymatic activity of EZH2. Rather, such binding probably interferes with EZH2 genomic targeting in the ifng promoter through antagonizing activities of EZH2, leading to relieving the suppressive function of EZH2 in the ifng promoter. We have observed a reduced expression of H3K9Me1 in M. tuberculosis-infected lncRNA-CGB KO mice and revealed interactions between lncRNA-CGB and Prdm16, an H3K9Me1 modifier. It will be interesting to further determine whether lncRNA-CGB action may also require other PRC2-binding lncRNAs or other involved histone modifiers.
As systemic activation via translocation of gut microbial products from the intestine into the blood circulation has been well-documented,Citation2 we thus postulate that there are several possible scenarios for how gut microbial products may impact the anti-TB immunity: firstly, gut microbial products such as SCFAs may be released into the bloodstream and reach lungs and thereby influencing the expression of lncRNA-CGB in resident T subsets in lungs and triggering them to further exert effector functions and thus restrict intracellular M. tuberculosis; secondly, naïve T cells in gut-associated lymphoid tissues may be directly activated by gut microbial products via sustaining or enhancing their lncRNA-CGB expression and then migrate into lung tissues, where they facilitate anti-M. tuberculosis immune functions; thirdly, gut microbial products may systemically protect T cell viability from entering into exhaustion via regulating expressions of inhibitory markers such as TIM-3, PD-1 and maintain their anti-M. tuberculosis immune functions in either lung or gut compartments. However, these postulations need to be further investigated. Recent studies suggest that the microbiota and its metabolites influence genomic reprogramming such as DNA methylation and histone modification.Citation67 Previous work on the correlation of the gut microbiota dominant group Bacteroidetes and differential methylation status of gene promoters in cardiovascular diseasesCitation68 hint that the control of lncRNA-CGB expression during TB infection evolved as one potential mechanism for impaired anti-TB immunity.
It remains extremely difficult to control TB due to the broad emergence of drug-resistant strains of M. tuberculosis, insufficient BCG vaccine efficacy against pulmonary TB and HIV-1 co-infection.Citation69 Developing new strategies for effective control of TB, such as better vaccines or new therapeutics, can be facilitated by identifying a new paradigm of protective immunityCitation62and uncovering previously unknown pathogenesis mechanisms during M. tuberculosis infection.Citation70Citation71 Our gut microbiota approach therefore might potentially serve as adjunctive therapeutics against TB although in-depth studies in animals and humans are required to confirm that lung protections and homeostasis could really be achieved via oral administrations of a single-strain gut bacteria or a rationally-designed gut bacteria consortium.
Overall, the current study defines gut microbiota functions and mechanisms for maintaining lung immune homeostasis through modulating lncRNA expression. Gut dysbiosis can dictate expression patterns of lncRNA in both gut and lung and induce immune dysfunction at distant organs. We also made useful observations implicating microbiota-mediated regulatory pathways: gut microbiota →lncRNA-CGB →EZH2/H3K27Me3 epigenetics →IFN-γ expression → anti-TB immunity. Thus, this work not only provides a novel framework and in-depth understanding of significances and underlying mechanisms in homeostatic control of M. tuberculosis infection of gut-lung immune interaction axis but also reveals a previously unidentified strategy for the development of a protective immune paradigm against deadly TB and other lung diseases.
Materials and methods
Methods
Study subjects: Subjects with newly diagnosed active TB infection but did not receive any anti-TB treatment were recruited by the Guangdong Provincial Center for TB Control in this study. All subjects were ≥18 years of age and were sero-negative for HIV-1/2, other infectious diseases and major diseases, such as cancer and diabetes. The sex ratios (female/male) of each group are healthy controls (HC) (28/15) and TB patients (TB)(29/26). The ages of each group (median [interquartile range]) were HC, 27 (24–35); TB, 34 (26–40). The healthy controls (HCs) came to the same hospital clinics for annual routine health examinations while providing fecal samples, with final reports indicating healthy statuses. Diagnosis of pulmonary TB was based on symptoms, roentgen graphic findings (chest X-ray and/or HRCT) and sputum bacillus examination. The numbers of bacilli in sputum smears were counted according to World Health Organization (WHO) guidelines. Of 55 TB patients, 48 had sputum positive culture for M. tuberculosis and were divided into four groups as per grading of the sputum AFB smear: group I (sputum1+, n = 9): 1 to 9 AFB per 100 fields; group II (sputum 2+, n = 8): 10 to 99 AFB per 100 fields; group III (sputum 3+, n = 8): 1 to 10 AFB per field in 50 fields; and group IV (sputum 4+, n = 5): more than 10 AFB per field in 20 fields. Sputum samples were classified according to the highest number of AFB per specimen. Fecal samples and blood samples from TB subjects were collected in sterile containers within 7 days prior to initiating standard anti-TB treatment. Informed consent from all participants included in the study was obtained according to protocols approved by the Internal Review and the Ethics Boards of Zhongshan School of Medicine of Sun Yat-sen University (SYSU).
Mice: Specific-pathogen-free (SPF) C57BL/6(3 ~ 4 weeks) were obtained from SYSU Experimental Animal Center. LncRNA-CGB KO mice were generated using a CRISPR genome-editing system in the C57BL/6 background. Single guide RNAs (sgRNAs) flanking region flanking exon 1 and exon 2 to exon 7 of lncRNA – CGB were designed using a CRISPR design tool (genome-engineering.org). A Cas9 expression plasmid (Addgene) was linearized with PmeI and used as a template for in vitro transcription. Purified Cas9 mRNA and sgRNAs were mixed and injected into the cytoplasm of fertilized eggs of C57BL/6 mice in the M2 medium (Sigma-Aldrich). Successful KO mice were validated by PCR with the following primers: “Pair1-F: GGCCTCTGATTTAGCCAGCACTG”; “Pair1-R: GTGTACCCCAGCATAATGGCGAT;” “Pair2-F: CAATGGCTTGACCCAGACTTAGGAG;” and“Pair2-R: GACATTCCCTGGCATTCATATCAGG”. The detected mutated allele was selected to mate with the wild-type C57BL/6 strain to obtain F1-generation mice. Heterozygous F1 offspring were interbred to establish lncRNA-CGB-/ – Strain. All procedures were carried out under approval by the SYSU Institutional Animal Care and Use Committee (Approval number SYSU-IACUC-2019-B527).
M. tuberculosis infection: Mice were infected with M. tuberculosis (strain H37Rv) via aerosol challenge. To determine the M. tuberculosis burden at indicated times, the lung of each mouse was homogenized in PBS, and ten-fold serial dilutions were made in PBS and plated on 7H10 plates. Colonies were counted after 21d of incubation at 37°C, and CFUs per lung were determined. Approximately 100 ~ 150 CFUs of M. tuberculosis were deposited in the lungs of each mouse upon initial infection.
Histopathological, bacterial and immune analyses of M. tuberculosis-infected mice: At the end of the M. tuberculosis infection period, animals were sacrificed. The right lung and cecum were excised and carefully homogenized. For H&E and acid-fast Kinyoun’s analysis, lung or gut tissues of mice were fixed in 10% formalin and processed for paraffin embedding. Paraffin sections of 5 µm were counterstained with H&E or Kinyoun’s. Images of acid-fast staining were obtained using a microscope (Olympus BX63). H&E staining was taken by Digital Slide Scanning System AxioScan.Z1. All sections were interpreted by the same person and scored semiquantitatively, blinded to the variables of the experiment. The histopathological parameters inflammatory lesions and granuloma formation were scored as our previous study.Citation14
Antibiotics treatment: A mix of ampicillin (1 mg/ml), neomycin (1 mg/ml), metronidazole (1 mg/ml) and vancomycin (0.5 mg/ml) were added in sterile drinking water of mice. Solutions and bottles were changed 2 ~ 3 times a week. The antibiotic activity was confirmed by macroscopic changes observed at the level of the cecum (dilatation) and by cultivating the fecal pellets resuspended in BHI+15% glycerol at 0.1 g/ml on blood agar plates for 48 h at 37°C within aerobic or anaerobic conditions.
Mouse lncRNA and mRNA sequencing, data analysis and statistics: Total RNA of lungs in M. tuberculosis-infected mice with or without the treatment of broad-spectrum antibiotics were isolated with TRIzol reagent (Thermo Fisher). After RNA cleanup and quality analysis, total RNA was used for mRNA and lncRNA high throughput sequencing analysis. Briefly, a total amount of 2 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (#E7530L, NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. The details of library construction showed as follows: Firstly, ribosomal RNA was removed by kits, RNA fragmentation and short RNA strands were carried out by NEB Next First Strand Synthesis Reaction Buffer under elevated temperature. Subsequently, the first cDNA strand was synthesized using random hexamer primers and RNA fragments as a template. Second strand cDNA synthesis was subsequently performed using buffer, dNTPs, DNA polymerase I and RNase H. The library fragments were purified with QiaQuick PCR kits and elution with EB buffer, then terminal repair adds poly (A) and adapter were implemented. In order to select cDNA fragments of preferentially 300bp in length, the library fragments were purified and the UNG enzyme was used to digest the second strand of cDNA. PCR was performed, aimed products were to retrieve, and the library was completed. Afterward, the clustering of the index-coded samples was performed on a cBot cluster generation system using TruSeq PE Cluster Kit v4-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the libraries were sequenced on an Illumina platform and 150 bp paired-end reads were generated.
Gene Set Enrichment Analysis (GSEA): GSEA was performed using the GSEA software (http://www.broadinstitute.org/gsea) with permutation type = gene set, number of permutations = 1000 enrichment; statistic = weighted, and metric for ranking genes = Singal2Noise. GSEA for analyzing the pathway for antibiotics treatment was performed using the gene set of mouse pathway target gene sets. The heat map was generated and visualized by using the Morpheus software (https://software.broadinstitute.org/morpheus/).
Northern blot: Total RNA was extracted from indicated samples by Trizol (Roche) according to the manufacturer’s manual. A total of 10 μg of RNA was electrophoresed on a 10% formaldehyde gel and transferred to a Biodyne Nylon membrane (Pall). The membrane was further hybridized with a digoxin-labeled lncRNA-CGB probe (5ʹDIG – AGATGATGGTAGGATGTGCTT-3ʹDIG) (Exiqon) at 68°C overnight in ULTR AhybTM-Oligo buffer. U6 probe and Scramble probe were used as a positive and negative control, respectively. The membrane washes were performed as described in the Northern Max Kit protocol (Roche) and the signals were visualized using an Odyssey infrared scanner (Li-Cor).
DNA extraction, 16S rDNA gene amplification, and pyrosequencing (human fecal samples): Fresh fecal stool samples were collected from patients with active TB disease and healthy controls, respectively. DNA was isolated with the DNA STOOL Kit (QIAGEN) followed the manufacturer’s instructions. The purity, concentration of DNA was detected by using the NanoPhotometer spectrophotometer and Qubit2.0 Fluorometer. DNA was then amplified by the corresponding primer: the 338 F-533 R for the V3 Regions, the 341 F-805 R for V3+ V4 regions and the 967 F-1046 R for the V6 regions. The index sequences were added and enriched after the extraction finished. The Qubit2.0, Agilent 2100 and Bio-rad CFX 96 were used to quantify the concentration and purity of the library to ensure the quality. After all had finished, the library was sequenced on an Illumina HiSeq2500 by using the 250 paired-end protocol.
Alpha diversity analysis: Alpha diversity was implemented to display the diversity of bacteria among the different samples, which mainly included 4 indices: Observed-species, Chao1, Shannon and Simpson. All these indices in our samples were calculated with QIIME (qiime-1.8.0) and displayed by R software.
OTU abundances analysis: To investigate differences in OTUs, genera, species abundances between both groups, raw counts were normalized then log-transformed using the normalization method below, as performed by a previous study:70
Normalized count = log10 ((raw count/ number of sequences in that sample) × average number of sequences per sample+1).
Wilcoxon tests were used to compare mean differences between two groups for genera and species log abundances. Considering t = total number of taxa tested, p = raw p-value and R = sorted rank of the taxon, P-values were corrected for multiple testing 70 using:
Adjusted p-value = t × p R Fold changes for each genus/OTU were calculated using:
Log2 FC = log2 (TB average + 1) − log2 (HC average + 1)
Monoclonal antibodies (mAbs) for intracellular cytokine staining (ICS): The following antibodies were used for flow cytometric assay: mouse anti-CD4 APC-eFluor 780(GK1.5, eBio), mouse anti – CD8-FITC (53–6.7, eBio), mouse anti-IFN-γ APC (XMG1.2, eBio).
Flow cytometric analysis: Experiments were conducted as described in our previous publication.Citation28 Briefly, cells were first stained with cell-surface markers and then fixed and permeabilized for 30 min before staining the intracellular molecules for another 45 min. Finally, cells were fixed with 2% formalin/PBS and processed for polychromatic flow cytometry. Isotype matched IgG staining was served as a negative control. Data were obtained from Beckman Coulter Gallios and analyzed by Kaluza 1.5 software.
Bacterial culture: Bacteroides fragilis (purchased from CCTCC, Wuhan, China) were grown on blood agar plates or selective agar plates for 48 hours at 37°C in anaerobic conditions. Bacteria were harvested from the agar plates, suspended in sterile saline with 10% glycerol, centrifuged and washed once with saline, then resuspended in sterile saline at an optical density (600 nm) of 1, which corresponded approximately to 1 × 109 colony-forming units (CFU)/ml. The bacterial culture was aliquoted and stored at −80°C for future use.
Bacteria oral transfer: SPF mice receiving antibiotics for 1 week were orally gavaged daily with 1 × 109 CFU of B. fragilis or saline until death. Colonized mice with dedicated bacteria were subjected to M. tuberculosis infection 1-week post bacteria oral transfer.
Fecal microbial transplantation (FMT): Fecal materials from healthy controls used for 16s sequencing were pooled by sterile saline supplemented with 15% glycerol and homogenized. The homogenate was centrifuged at 1000 g for 10 min at 4°C. The supernatant was aliquoted and stored at −80°C for future use. Antibiotics-treated mice were used for the colonization with fecal microbiota. FMT was performed by thawing the fecal materials and 0.2 ml of the pooled suspension containing fecal microbiota obtained from healthy controls was transferred by oral gavage into each recipient.
Quantification of bacteria by qRT-PCR: Genomic DNA was isolated from fecal samples using the QIAamp DNA Stool Mini Kit (Qiagen) following the manufacturer’s instructions. Targeted qRT-PCR systems were applied using either Taqman technology targeting the All Bacteria domain or SybrGreen for Bacteroides fragilis species. The primers used were summarized in .
Table 2. The list of qPCR primer couples
Adoptive transfer. CD3 + T cells and CD14+ monocytes were purified from PBMC of active TB patients using positive selection using a MACS Pro Separator (Miltenyi Biotech). Cell purity was consistently ≥95%. Three days before transfer, four-week-old SCID mice were infected with M. tuberculosis (H37RV). 5 × 105 CD3 + T cells were transduced with LV-lncRNA-CGB or LV-Ctrl for five days. Culture supernatants for CD3 + T cells, and CD3 + T cells transduced with indicated LV vector and 4 × 104 autologous monocytes were transferred into each of recipient mice via i.p. injections.
Western blotting: Cells isolated from indicated mice were seeded. Total protein was extracted from cells using RIPA lysis buffer. Extracted proteins were mixed with loading buffer, subject to 10% SDS-PAGE and transferred to PVDF membranes, which were subsequently blocked in a 5% solution of nonfat milk for 1 hour. The membranes were subsequently incubated with the following antibodies overnight at 4°C: rabbit anti-H3K9Me1(ab9045,Abcam), mouse anti-H3K27Me3(ab6002,Abcam), rabbit anti – H3K4Me3(ab8580,Abcam), rabbit anti-H3(ab1791,Abcam),mouse anti-β actin (ab6276,Abcam), rabbit anti-H3K27Me2(ab24684,Abcam), mouse anti – H3k9Me2(ab1220, Abcam), rabbit anti-H3K9Me3(ab8898,Abcam).The membranes were further incubated with goat anti-rabbit or goat anti-mouse secondary antibody (Thermo Fisher) for 1 hour at room temperature. Signals were detected with Western Lightning Plus-ECL Enhanced Chemiluminescence reagent. Images were captured by the BIO-RAD ChemiDoc Touch machine.
Isolation of peripheral blood mononuclear cells (PBMCs): PBMCs were obtained by density centrifugation of diluted blood (1-part blood to 1-part PBS) over Ficoll-Paque. Cells were washed twice in PBS and suspended in the medium (RPMI 1640) supplemented with 10%FBS and were cultured for future use in a 96 well plate.
RNA interference: Control siRNA and siRNA target lncRNA-CGB were synthesized by Exiqon. siRNA transfections were performed using Lipofectamine RNAi MAX (Thermo Fisher) following the manufacturer’s instructions. The final concentration of the siRNA molecules is 20 nM and cells were stimulated with or without M. tuberculosis lysates for 48 hours according to the purposes of the experiments. Transfected cells were analyzed by qRT-PCR to analyze the expression of lncRNA-CGB post-transfection to determine transfection efficiency.
FISH and immunofluorescence of lncRNA-CGB, EZH2 in PBMCs: PBMCs were fixed 4% paraformaldehyde for 20 min at RT, and further permeabilized by 70% ethanol for another 2 hours. PBMCs were then incubated with rabbit anti-EZH2 with digoxin labeled lncRNA-CGB in hybridization buffer overnight at 37°C, respectively. Nonspecific binding of probes was removed by a subsequent wash with washing buffer. Cells were further stained with goat anti-rabbit-FITC and anti-digoxin-Rhodamine at 37°C for another 30 min. ProLong Gold Antifade with DAPI was used for nuclear counterstain and cells were air-dried for further microscopic analysis by using a confocal microscope (Zeiss 780).
Chromatin Immunoprecipitation and ChIP-qPCR: 1 × 106 cells were prepared for each round of ChIP. ChIP was performed using EZ MAGNA ChIP assay kit (millipore) according to the manufacturer’s manual. Briefly, after fixation of cells with formaldehyde (1%), Chromatin DNA was immunoprecipitated with the following antibodies: rabbit anti-H3K9Me1 (ab9045, Abcam), mouse anti-H3K27Me3 (ab6002, Abcam), rabbit anti-H3K4Me3 (ab8580, Abcam), rabbit anti-H3 (ab1791, Abcam), rabbit anti-H3K27Me2 (ab24684, Abcam), mouse anti-H3k9Me2 (ab1220, Abcam), rabbit anti-H3K9Me3 (ab8898, Abcam). PCRs were performed in 20-μl reaction mixtures containing 2 μl of DNA, 10 μl of SYBR green master mix and 1 μl of each primer. The following gene-specific ChIP-qPCR primers were used:
The PCR amplimers were confirmed to be a single band of the correct size by agarose gel electrophoresis. ChIP-qPCR data were normalized by the % input method. Results are expressed as means ± SEM of the indicated number of independent determinations.
RNA Immunoprecipitation: RIP was performed using the Magna RIP-Binding Protein Immunoprecipitation Kit (Millipore). Briefly, anti-EZH2 (AB3748; Abcam), anti-PRDM16 (ab106410, Abcam), anti-G9a (ab40542; Abcam) or control IgG were first incubated with Protein G dynabeads for 30 min, isolated PBMCs of TB patients or lymphocytes of lungs of M. tuberculosis-infected mice were lysed followed by incubation with antibody-dynabeads complexes overnight, immunoprecipitated RNA was then purified according to the manufacturer’s protocol. LncRNA-CGB and control RNAs were then analyzed by PCR and gel electrophoresis.
Reverse transcription qRT-PCR (qPCR): Total RNA was prepared from tissues or PBMCs using Trizol reagent (Takara). The concentration of total RNA was determined by a Nanodrop spectrometer. First-strand cDNA was synthesized using the PrimeScriptone step Strand cDNA Synthesis Kit (Takara) following the manufacturer’s instructions; qPCR was performed in technical triplicate using the SYBR Green to determine the expression of IFN-gamma and lncRNA-CGB. GADPH was used as an endogenous control to normalize gene expression. Relative mRNA expression levels were presented as means ± SEM. Statistical differences were analyzed by the Student’s t-test.
IFN-γ-ELISPOT analyses: ELISPOT plates (Millipore, MAIP S4510) were coated with purified IFN-γ (BD) antibody overnight at 4°C followed by blocking with 10% FBS in RMPI1640 for 2 hours at room temperature. Cells derived from indicated mice were plated at 1 × 106 cells per well and stimulated with ESAT-6 (6 kDa Early Secretory Antigenic Target) peptides, CFP-10 (10-kDa Culture Filtrate Protein) peptides and M. tuberculosis lysates. Spots were developed using the BD mouse IFN-γ kit (Cat. No. 551083), and the number of spots was measured using an Immunospot Series 3 Analyzer and analyzed using ImmunoSpot software.
Cytometric bead assay (CBA) analysis of Th1/Th2/Th17 cytokine expression: Cells derived from mice or isolated PBMC of TB patients with indicated treatments were seeded at a density of 1 × 106 cells per well and stimulated with ESAT-6 peptides, CFP-10 peptides and M. tuberculosis lysates at 37°C, respectively. Culture supernatants of cells were analyzed for the production of the following cytokines: IL-2, IL-4, IL-6, IFN-γ, TNF-α, IL-17A and IL-10 using the cytometric bead assay (CBA) mouse Th1/Th2/Th1 cytokine kit (BD) according to the manufacturer’s instructions. Data were acquired on the Beckman Coulter Gallios (Beckman). The concentrations of each cytokine were revealed by the fluorescence intensity. Cytokine concentrations were calculated relative to the standard dilution curve.
Bioinformatics analyses of evolutionary conservation and coding potential of lncRNA-CGB and plasmid constructions: Evolutionary conservation of lncRNA-CGB was analyzed using the University of California Santa Cruz (UCSC) Genome Browser (genome.ucsc.edu). The coding potential of lncRNA-CGB was first analyzed using the tools provided by the Peking University Center for Bioinformatics (cpc.cbi.ku.edu.cn/programs/run_cpc.jsp) and further constructed into the ORF of eukaryotic expression vector pReceiver-M98 with EGFP tag. Vector pReceiver-M98R with only EGFP tag or and vector pReceiver-M98Rwith EZH2 and EGFP tag were served as controls. All constructs were confirmed by DNA sequencing.
In silicon prediction for RNA structure and protein-RNA interaction: RNA secondary structure was predicted by RNA fold Webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) based on minimum free energy (MFE) and partition function. The binding propensity between EZH2 and RNA fragments was predicted by catRAPID (http://service.tartaglialab.com/page/catrapid_group).
Statistical analysis: Statistical analysis was performed using GraphPad Prism. Unless otherwise indicated, the non-parametric Mann-Whitney U-test was often used for two groups comparisons to avoid the assumption of a normal distribution. For samples more than 20, statistical analyses were performed with the unpaired two-tailed Student t-test as indicated in the figures. p-value <0.05 were considered significant. *p < .05, **p < .01, ***p < .001 and **** p < .0001. NS, no statistical significance. All experiments were replicated in the laboratory at least two times. In figure legends, N represents the number of samples.
Supplemental Material
Download Zip (15.8 MB)Disclosure statement
G.C.Z. is a founder of Revaissant Bioscience.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. The datasets generated for this study can be found in the GSE125870.https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125870
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–26. doi:10.1126/science.1241165.
- Budden KF, Gellatly SL, Wood DLA, Cooper MA, Morrison M, Hugenholtz P, Hansbro PM. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi:10.1038/nrmicro.2016.142.
- Wang H, Liu JS, Peng SH, Deng XY, Zhu DM, Javidiparsijani S, Wang GR, Li DQ, Li LX, and Wang YC, 2013 Gut-lung crosstalk in pulmonary involvement with inflammatory bowel diseases. World Journal of Gastroenterology 19, 6794–804, doi:10.3748/wjg.v19.i40.6794.
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, and Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza a virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5354–5359. doi:10.1073/pnas.1019378108.
- Russell SL, Gold MJ, Hartmann M, Willing BP, Thorson L, Wlodarska M, Gill N, Blanchet MR, Mohn WW, and McNagny KM, et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012; 13:440–447. doi:10.1038/embor.2012.32.
- Rutten EPA, Lenaerts K, Buurman WA, Wouters EFM. Disturbed intestinal integrity in patients with COPD: Effects of activities of daily living. Chest. 2014;145(2):245–252. doi:10.1378/chest.13-0584.
- Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, Mazmanian SK. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15(3):374–381. doi:10.1016/j.chom.2014.02.006.
- Dumas A, Bernard L, Poquet Y, Lugo-Villarino G, Neyrolles O. The role of the lung microbiota and the gut–lung axis in respiratory infectious diseases. Cell Microbiol. 2018;20(12). doi:10.1111/cmi.12966.
- World Health Organization 2020 Global tuberculosis report 2020 Geneva: World Health Organization; 2020. Licence: CC BY-NC-SA 3.0 IGO
- Rubin EJ. Troubles with tuberculosis prevention. N Engl J Med. 2014;370(4):375–376. doi:10.1056/NEJMe1312301.
- Lozupone CA, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ, Knight R, Fontenot AP, Palmer BE. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14(3):329–339. doi:10.1016/j.chom.2013.08.006.
- Gehrig JL, Venkatesh S, Chang HW, Hibberd MC, Kung VL, Cheng J, Chen RY, Subramanian S, Cowardin CA, and Meier MF, et al. Effects of microbiota-directed foods in gnotobiotic animals and undernourished children. Science. 2019;365. doi:10.1126/science.aau4732.
- Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut. 2014;63(9):1513–1521. doi:10.1136/gutjnl-2014-306928.
- Yang F, Yang Y, Chen Y, Li G, Zhang G, Chen L, Zhang Z, Mai Q, and Zeng G. MiR-21 is remotely governed by the commensal bacteria and impairs anti-TB immunity by down-regulating IFN-γ. 2021;11. doi:10.3389/fmicb.2020.512581.
- Khan N, Vidyarthi A, Nadeem S, Negi S, Nair G, and Agrewala JN. Alteration in the gut microbiota provokes susceptibility to tuberculosis. Front Immunol. 2016;7. doi:10.3389/fimmu.2016.00529.
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37(1):158–170. doi:10.1016/j.immuni.2012.04.011.
- Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535(7610):75–84. doi:10.1038/nature18848.
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi:10.1126/science.1223490.
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–323. doi:10.1038/nri2515.
- Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Ter Horst R, Jansen T, Jacobs L, Bonder MJ, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167(4):1125–36 e8. doi:10.1016/j.cell.2016.10.020.
- Zelaya H, Villena J, Lopez AG, Alvarez S, Agüero G. Modulation of the inflammation-coagulation interaction during pneumococcal pneumonia by immunobiotic Lactobacillus rhamnosus CRL1505: role of Toll-like receptor 2. Microbiol Immunol. 2014;58(7):416–426. doi:10.1111/1348-0421.12163.
- Virtue AT, McCright SJ, Wright JM, Jimenez MT, Mowel WK, Kotzin JJ, Joannas L, Basavappa MG, Spencer SP, Clark ML, et al. The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs. Sci Transl Med. 2019;11(496). doi:10.1126/scitranslmed.aav1892.
- Dempsy J. Coordinate regulation of long non-coding RNAs and protein-coding genes in germ-free mice. BMC Genomics. 2018;19(1):834. doi:10.1186/s12864-018-5235-3.
- Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15(6):423–437. doi:10.1038/nrg3722.
- Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, Brahic M, Kirkegaard K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. 2013;152(4):743–754. doi:10.1016/j.cell.2013.01.015.
- Fortes P, Morris KV. Long noncoding RNAs in viral infections. Virus Res. 2016;212:1–11. doi:10.1016/j.virusres.2015.10.002.
- Wang P, Xu J, Wang Y, Cao X. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science. 2017;358(6366):1051–1055. doi:10.1126/science.aao0409.
- Wang Y, Zhong H, Xie X, Chen CY, Huang D, Shen L, Zhang H, Chen ZW, Zeng G. Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+T-cell immune responses in tuberculosis infection. Proc Natl Acad Sci U S A. 2015;112(29):E3883–92. doi:10.1073/pnas.1501662112.
- Chang CL, Park TS, Oh SH, Kim HH, Lee EY, Son HC, Kim CM. Reduction of contamination of mycobacterial growth indicator tubes with a modified antimicrobial combination. J Clin Microbiol. 2002;40(10):3845–3847. doi:10.1128/JCM.40.10.3845-3847.2002.
- Hu Y, Feng Y, Wu J, Liu F, Zhang Z, Hao Y, Liang S, Li B, Li J, and Lv N, et al. The gut microbiome signatures discriminate healthy from pulmonary tuberculosis patients. Front Cell Infect Microbiol. 2019;9:90. doi:10.3389/fcimb.2019.00090.
- Maji A, Misra R, Dhakan DB, Gupta V, Mahato NK, Saxena R, Mittal P, Thukral N, Sharma E, Singh A, et al. Gut microbiome contributes to impairment of immunity in pulmonary tuberculosis patients by alteration of butyrate and propionate producers. Environ Microbiol. 2018;20(1):402–419. doi:10.1111/1462-2920.14015.
- Winglee K, Eloe-Fadrosh E, Gupta S, Guo H, Fraser C, Bishai W. Aerosol mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PLoS ONE. 2014;9(5). doi:10.1371/journal.pone.0097048.
- Li W, Zhu Y, Liao Q, Wang Z, and Wan C. Characterization of gut microbiota in children with pulmonary tuberculosis. BMC Pediatr. 2019;19. doi:10.1186/s12887-019-1782-2.
- Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–1084. doi:10.1126/science.aad1329.
- Chan JL, Wu S, Geis AL, Chan GV, Gomes TAM, Beck SE, Wu X, Fan H, Tam AJ, Chung L, et al. Non-toxigenic Bacteroides fragilis (NTBF) administration reduces bacteria-driven chronic colitis and tumor development independent of polysaccharide A. Mucosal Immunol. 2019;12(1):164–177. doi:10.1038/s41385-018-0085-5.
- Winglee K, Eloe-Fadrosh E, Gupta S, Guo H, Fraser C, Bishai W. Aerosol Mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PLoS One. 2014;9(5):e97048. doi:10.1371/journal.pone.0097048.
- Wexler AG, Goodman AL. An insider’s perspective: bacteroides as a window into the microbiome. Nat Microbiol. 2017;2(5):17026. doi:10.1038/nmicrobiol.2017.26.
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–118. doi:10.1016/j.cell.2005.05.007.
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science. 2015;350(6264):1084–1089. doi:10.1126/science.aac4255.
- Qiu Z, Zhang M, Zhu Y, Zheng F, Lu P, Liu H, Graner MW, Zhou B, Multifunctional CX. CD4 T cell responses in patients with active tuberculosis. Sci Rep. 2012;2(1):216. doi:10.1038/srep00216.
- Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178(6):2249–2254. doi:10.1084/jem.178.6.2249.
- Thiemann S, Smit N, Roy U, Lesker TR, Galvez EJC, Helmecke J, Basic M, Bleich A, Goodman AL, Kalinke U, et al. Enhancement of IFNgamma production by distinct commensals ameliorates salmonella-induced disease. Cell Host Microbe. 2017;21(6):682–94 e5. doi:10.1016/j.chom.2017.05.005.
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341(6147):789–792. doi:10.1126/science.1240925.
- Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, Brahic M, Kirkegaard K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell. 2013;152(4):743–754. doi:10.1016/j.cell.2013.01.015.
- Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen BS, Melum E, Pertel T, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(7534):386–390. doi:10.1038/nature13848.
- Pang KC, Dinger ME, Mercer TR, Malquori L, Grimmond SM, Chen WS, Mattick JS. Genome-wide identification of long noncoding RNAs in CD8+T cells. Journal of Immunology. 2009;182(12):7738–7748. doi:10.4049/jimmunol.0900603.
- Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol. 2014;15(6):484–491. doi:10.1038/ni.2887.
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Morales DR, Thomas K, Presser A, Bernstein BE, and van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11667–11672. doi:10.1073/pnas.0904715106.
- Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding HM, Butty VL, Torrey L, Haas S, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152(3):570–583. doi:10.1016/j.cell.2013.01.003.
- Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338(6113):1435–1439. doi:10.1126/science.1231776.
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521(7551):232-+. doi:10.1038/nature14443.
- Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20(3):300–307. doi:10.1038/nsmb.2480.
- Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145(2):178–181. doi:10.1016/j.cell.2011.03.014.
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–U58. doi:10.1038/nature09819.
- Wang ZH, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, Ren SX, Wang XH, Chen I, Wang H, et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22(10):1131–1139. doi:10.1038/nm.4179.
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44(4):667–678. doi:10.1016/j.molcel.2011.08.027.
- Cervantes J, and Hong BY. The gut-lung axis in tuberculosis. Pathog Dis. 2017;75. doi:10.1093/femspd/ftx097.
- Bradley CP, Teng F, Felix KM, Sano T, Naskar D, Block KE, Huang H, Knox KS, Littman DR, Wu HJJ. Segmented filamentous bacteria provoke lung autoimmunity by inducing gut-lung axis Th17 cells expressing dual TCRs. Cell Host Microbe. 2017;22(5):697–704.e4. doi:10.1016/j.chom.2017.10.007.
- Negi S, Pahari S, Bashir H, and Agrewala JN. Gut microbiota regulates mincle mediated activation of lung dendritic cells to protect against mycobacterium tuberculosis. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.01142.
- Ramakrishna C, Kujawski M, Chu H, Li L, Mazmanian SK, Cantin EM. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat Commun. 2019;10(1):2153. doi:10.1038/s41467-019-09884-6.
- Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, Giry A, Serino M, Stenman L, Lahtinen S, et al. The gut microbiota regulates intestinal CD4 T cells expressing RORγt and controls metabolic disease. Cell Metab. 2015;22(1):100–112. doi:10.1016/j.cmet.2015.06.001.
- Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol. 2014;12(4):289–299. doi:10.1038/nrmicro3230.
- Orme IM, Robinson RT, Cooper AM. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol. 2015;16(1):57–63. doi:10.1038/ni.3048.
- Zeng G, Zhang G, Chen X. Th1 cytokines, true functional signatures for protective immunity against TB? Cell Mol Immunol. 2018;15(3):206–215. doi:10.1038/cmi.2017.113.
- Hu Y, Yang Q, Liu B, Dong J, Sun L, Zhu Y, Su H, Yang J, Yang F, Chen X, et al. Gut microbiota associated with pulmonary tuberculosis and dysbiosis caused by anti-tuberculosis drugs. Journal of Infection. 2019;78(4):317–322. doi:10.1016/j.jinf.2018.08.006.
- Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, Conner ME, Earl AM, Knight R, Bjorkman PJ, et al. Gut microbiota utilize immunoglobulin a for mucosal colonization. Science. 2018;360(6390):795–800. doi:10.1126/science.aaq0926.
- Romano KA, Martinez-Del Campo A, Kasahara K, Chittim CL, Vivas EI, Amador-Noguez D, Balskus EP, Rey FE. Metabolic, epigenetic, and transgenerational effects of gut bacterial choline consumption. Cell Host Microbe. 2017;22(3):279–90 e7. doi:10.1016/j.chom.2017.07.021.
- Kumar H, Lund R, Laiho A, Lundelin K, Ley Ruth E, Isolauri E, Salminen S, Ravel J. Gut microbiota as an epigenetic regulator: pilot study based on whole-genome methylation analysis. mBio. 2014;5(6):e02113–14. doi:10.1128/mBio.02113-14.
- Dheda K, Gumbo T, Gandhi NR, Murray M, Theron G, Udwadia Z, Migliori GB, Warren R. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. The Lancet Respiratory Medicine. 2014;2(4):321–338. doi:10.1016/S2213-2600(14)70031-1.
- Kaufmann SHE. Novel tuberculosis vaccination strategies based on understanding the immune response. J Intern Med. 2010;267(4):337–353. doi:10.1111/j.1365-2796.2010.02216.x.
- Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. Isme Journal. 2012;6(10):1858–1868. doi:10.1038/ismej.2012.43.