
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Over 90% of epidemic non-bacterial gastroenteritis are caused by human noroviruses (NoVs), which persist in a substantial subset of people allowing their spread worldwide. This has led to a significant number of endemic cases and up to 70,000 children deaths in developing countries. NoVs are primarily transmitted through the fecal-oral route. To date, studies have focused on the influence of the gut microbiota on enteric viral clearance by mucosal immunity. In this study, the use of mouse norovirus S99 (MNoV_S99) and CR6 (MNoV_CR6), two persistent strains, allowed us to provide evidence that the norovirus-induced exacerbation of colitis severity relied on bacterial sensing by nucleotide-binding oligomerization domain 2 (Nod2). Consequently, Nod2-deficient mice showed reduced levels of gravity of Dextran sodium sulfate (DSS)-induced colitis with both viral strains. And MNoV_CR6 viremia was heightened in Nod2-/- mice in comparison with animals hypomorphic for Atg16l1, which are prone to aggravated inflammation under DSS. Accordingly, the infection of macrophages derived from WT mice promoted the phosphorylation of Signal Transducer and Activator of Transcription 1 (STAT1) and NOD2’s expression levels. Higher secretion of Tumor Necrosis Factor alpha (TNF) following NOD2 activation and better viral clearance were measured in these cells. By contrast, reduced levels of pSTAT1 and blunted downstream secretion of TNF
were found in Nod2-deficient macrophages infected by MNoV_S99. Hence, our results uncover a previously unidentified virus-host-bacterial interplay that may represent a novel therapeutic target for treating noroviral origin gastroenteritis that may be linked with susceptibility to several common illnesses such as Crohn’s disease.
Introduction
The development of high throughput sequencing techniques progressively helped us gain a better comprehension of the composition of the enteric virome.Citation1 This is of particular importance for unraveling how some enteric viruses can establish persistent infections and subsequently influence our health.Citation2,Citation3 Among these, noroviruses (NoVs) have been studied for several years. These single stranded RNA (+) viruses, belonging to the family of Caliciviridae, are responsible for the majority of non-bacterial gastroenteritis worldwide. Until recent progress in cultivation methods of noroviruses from genogroups I, II, IV, VIII, and IXCitation4–6 that infect humans (HuNoVs), mechanistic details were mostly obtained from studies using the mouse norovirus (MNoV).Citation7 Indeed, MNoVs can be easily produced in vitro in macrophages or dendritic cells.Citation8 Several MNoV strains have been described based on their shedding time in the stool and classified as either persistent or acute (non-persistent) strains.Citation9 Owing to the variety of circulating NoV strains, strain-dependent effects are expected to occur and should be studied in more details. Even a single amino acid change in the NS1/2 viral protein is sufficient to switch the acute MNoV_CW3 strain to a persistent state,Citation10 showing the high degree of adaptability of NoV genomes and hence their versatility to counteract host defense mechanisms. Type III interferon (IFN-λ) response has been shown to control the in vivo propagation of the persistent MNoV_CR6 strain,Citation11 while type I and II interferons mediate innate immune responses against acute MNoV infections.Citation12,Citation13
Multiple intracellular pattern recognition receptors (PRRs) have been associated with indirect noroviral detection, especially during NoV genome replication, when double stranded RNA intermediates are generated.Citation14 As such, the Toll-like receptor 3 (TLR3) that recognizes any dsRNA in the endosomal compartment and the melanoma differentiation associated protein-5 (MDA-5), a member of the RIG-I-like receptor (RLR) family that recognizes long dsRNA, have been shown to participate in the detection of the MNoV_1 strain and its plaque isolate the MNoV_CW3.Citation15 Among the Nod-like receptor (NLR) family, it was shown that the loss of Nlrp6 impairs antiviral immunity to MNoV_1.Citation16 Besides its ability to sense muropeptides from Gram positive and negative bacteria,Citation17 the nucleotide-binding oligomerization domain 2 (NOD2) was also shown to directly bind with the genome (ssRNA-) of the respiratory syncytial virus (RSV) to induce antiviral responses.Citation18 The recruitment of MAVS by NOD2 was needed to activate the interferon-regulatory factor 3 (IRF3) dependent signaling and promote IFN-β production. In the same line, NOD2-dependent signaling was shown to disrupt intestinal homeostasis and increase lethality in an E. Coli and MNoV_1 co-infection mouse model.Citation19
Another molecular player that is essential to activate antiviral signaling is the cytosolic protein signal transducer and activator of transcription 1 (STAT1). MNoV_1 infection was shown to be lethal in STAT1-deficient mice.Citation12 Furthermore, STAT1-dependent interferon responses were shown to limit MNoV_1 replication and dissemination in vivo.Citation20
Interestingly, Cadwell and colleagues have demonstrated how infection with the MNoV persistent strain CR6 rendered Atg16l1HM mice more susceptible to DSS-induced colitis.Citation21 Interestingly, the interaction of NOD2 with ATG16L1 was shown to regulate the autophagy clearance of pathogenic bacteria.Citation22 Furthermore, it has been established that loss-of-function point mutations of either NOD2 or ATG16L1 predispose humans to Crohn’s disease.Citation23,Citation24 Insights into the mechanisms of how NOD2 signaling is regulated upon noroviral infection and whether it may subsequently modulate colitis susceptibility remained to be assessed.
In this study, we have investigated how NOD2 influences the pathogenesis of the two persistent murine norovirus strains MNoV_S99 (Berlin)Citation25 and MNoV_CR6 in vivo in DSS-induced colitis models. At the cellular level, the Nod2-dependent pro-inflammatory signaling pathway activation was further examined in response to MNoV_S99 and MNoV_CR6 infection in myeloid cells to determine how this in turn subsequently affects viral production.
Results
MNoV_S99 exacerbates the severity of DSS-induced inflammation
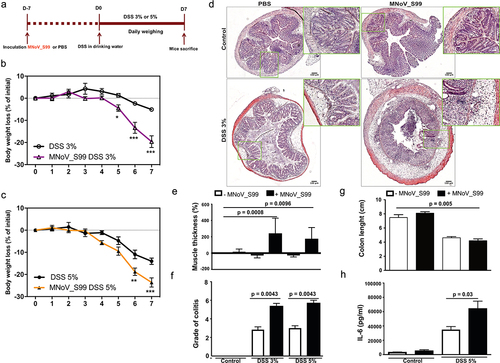
Since MNoV_S99 is a natural strain commonly found in animal facilities that is able to establish persistent infections,Citation25 we assessed the implication of MNoV_S99 in the exacerbation of colitis. Hence, we examined MNoV_S99’s impact in a widely used chemically induced colitis model that is induced upon the administration of Dextran Sodium Sulfate (DSS). Specifically, C57BL/6J mice were first infected with a single dose of 5 × 107 TCID50/mL of MNoV_S99 or mock-treated with PBS (200 µL). One week later, drinking water was replaced with either a 3% DSS or 5% DSS solution for a period of seven consecutive days (). Body weight-loss was monitored on a daily basis together with signs of diarrhea and rectal bleeding. With both concentrations of DSS, the pre-treatment with MNoV_S99 had induced a more significant body weight loss (purple and orange curves ). After 7 d of DSS treatment, mice were euthanized and the disease severity was evaluated. Histological analysis of colon sections revealed an increased infiltration of inflammatory cells and more pronounced epithelial damages in infected mice treated with DSS when compared to similarly challenged animals that were not infected (). Accordingly, increased muscle thickness was measured in colon sections from mice treated with the virus in the presence of both 3% and 5% DSS in comparison with mock-treated control mice (). Similarly, histological scoring of the hematoxylin and eosin (H&E) stained colonic sections expectedly showed significantly more severity in MNoV_S99-infected mice treated with either 3% or 5% DSS than DSS alone (). Upon mice sacrifice, resected colons were measured. A significantly reduced length was obtained in mice infected with MNoV_S99 and treated with 5% DSS in comparison with non-infected control animals (PBS) (). The increased level of colitis was also evidenced by a significantly augmented secretion of the pro-inflammatory cytokine interleukin-6 (IL-6) in the supernatants of overnight in vitro cultured colon explants from MNoV_S99 infected +5% DSS treated mice versus those from DSS only mice (). Taken together, these data clearly show that MNoV_S99 infection is able to lower the resistance of mice to chemically induced colitis.
Figure 1. MNoV_S99 aggravates inflammation in DSS-induced colitis. a)- in vivo experimental design, C57BL/6J mice were orally gavaged with PBS (200 µL) or MNoV_S99 (5×107 TCID50/mL), 7 d later, all the mice were given DSS in drinking water for a week. Mice were weighed daily for weight loss comparison. b)- 3% DSS-induced colitis ± MNoV_S99 (n = 6). c)- 5% DSS-induced colitis ± MNoV_S99 (n = 6). In b) and c), statistical differences were determined by two-way ANOVA test, *p < 0.05, **p < 0.01, ***p < 0.001. d)- Representative images from H&E staining of colon sections from mock-treated (control) or 3% DSS ± MNoV_S99 treated mice. Insets showing higher magnifications from each image are presented next to each image. e)- muscle wall thickness was determined with image J from colon section images. Percentages of increase are plotted after baseline subtraction relative to the values obtained from control mice (n = 6). f)- histological intestinal epithelial inflammation scores comparison between control, 3% or 5% DSS ± MNoV_S99 treated mice (n = 6). g)- colon length comparison between control and 5% DSS ± MNoV_S99 treated mice h)- IL6 secretion levels from colon explants from mock or 5% DSS ± MNoV_S99 treated mice (n = 6). Statistical differences were determined with Student’s t-test in E, F, G and H.
MNoV_S99-associated inflammation is abrogated in Nod2−/− mice
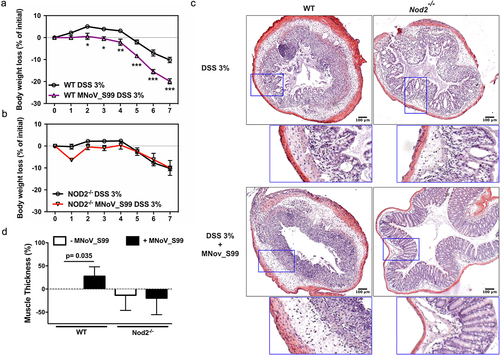
In cells from the myeloid lineage, NOD2 was shown to participate in the sensing of Citrobacter rodentium and of commensal bacteria that are translocating in response to DSS-induced epithelial injuryCitation26. To determine whether NOD2 is also implicated in the immunopathology associated with MNoV in our preclinical model of colitis, we compared the severity of DSS-induced colitis in the presence or absence of MNoV_S99 between WT and Nod2-deficient mice. In contrast to what was observed in WT mice (), the sensitizing effect of the virus on the daily weight-loss was absent upon MNoV_S99 infection of Nod2-/- mice (). These data suggest that the pro-inflammatory effect of MNoV_S99 observed in WT mice may depend on the degree of bacterial sensing through NOD2. Indeed, DSS induces an abrasion of the epithelial barrier allowing commensal bacteria to cross the mucosa and unleash a NOD2-dependent pro-inflammatory response. This was further confirmed after mice were autopsied. Colon tissue section staining showed decreased levels of colonic inflammation from MNoV_S99 infected Nod2-/- vs WT mice (). In agreement, muscle thickness quantification from these colon sections showed significantly increased thickness in MNoV_S99-infected WT mice under 3% DSS treatment vs 3% DSS alone (). The variations of colon length measurements that were induced upon MNoV_S99 infection in WT mice were absent by contrast in Nod2-/- mice (Fig. S1A). Similarly, differences in histological scores and mucosal wall thickness measured from MNoV_S99-infected WT mice vs DSS alone were also blunted in Nod2-/- infected mice (Fig. S1B). And a clear trend was observed regarding the increased levels of secretion of tumor necrosis factor α (TNFα) in the supernatants of in vitro cultured colon explants from WT mice infected with MNoV_S99 and treated with 3% DSS vs those with DSS only. This effect was absent in the supernatants from Nod2-/- mice infected and treated similarly (Fig. S1C).
Figure 2. MNoV_S99-associated inflammation is NOD2 dependent weight loss comparison in a)- 3% DSS-induced colitis ± MNoV_S99 in WT mice (n = 6) and b)- in Nod2-/- mice (n = 6), statistical differences were determined by two-way ANOVA test, *p < 0,05, **p < 0,01, ***p < 0,001. c)- Representative images of H&E staining of colon sections from mice in a) and b). Insets showing higher magnifications are presented below each image. d)- Percentages of increase of muscle wall thickness from colon section images are plotted after baseline subtraction relatively to the values obtained from WT mice treated only with DSS (n = 6), statistical differences were determined with Student’s t-test.
Overall, these data demonstrate that the excessive inflammatory response measured in the presence of MNoV_S99 depends on NOD2-dependent bacterial sensing in our DSS-induced colitis mouse model.
Mouse noroviruses infections trigger NOD2-dependent pro-inflammatory signaling
Mouse noroviruses are known to have a tropism for the myeloid cell lineages, namely macrophages and dendritic cells. Recently, it was shown that tuft cells belonging to the gut epithelial cellular lineages, that possess the CD300lf MNoV receptor can also be targeted by the MNoV_CR6 strain.Citation27
Making use of the GFP-Nod2 reporter mice line, we first examined in which cellular lineage Nod2 is expressed within the intestinal mucosa. We failed to observe any GFP-Nod2 expression in epithelial cells. By contrast, GFP signal was detectable in the bulk of the cells below the lamina propria where intestinal mononuclear phagocytes are primarily located (). Hence, the NOD2-dependent response to MNoV_S99 infection was further studied in macrophages, monocytes, and dendritic cells in our in vitro cellular models.
Figure 3. MNoV_S99 associated pro-inflammatory signaling is NOD2 and MAVS-dependent in myeloid lineage cells. a)- Representative image showing an intestinal villus from a Nod2GFP mouse. NOD2-GFP is shown in green, actin is stained with Phalloidin in gray and nuclei were stained with DAPI in blue; scale bare represents 50 µm. b)- Representative western blot showing STAT1 signaling pathway activation after 1 h infection (Moi 1) with the mentioned MNoV strains in BMDM. Below are shown quantification of pSTAT1 bands intensities normalized to β-actin and relative to mock-infected cells (n = 3, mean ± SEM), statistical differences were determined by one-way ANOVA test *p < 0,05, **p < 0,005. c)- STAT1 activation in response to MNoV_S99 (Moi 1 or 5) in BMDM from WT vs Nod2-/- infected for 30 or 60 minu. The lower panel shows quantification of pSTAT1, bands intensities normalized to β- actin and relative to mock-infected cells (n = 3, mean ± SEM). Statistical differences were determined by two-way ANOVA test *p < 0,05. d)- STAT1 activation in response to MNoV_S99 (Moi 0.1) in WT or Nod2-/- BMDM infected for 24 h. Relative quantification of pSTAT1 (n = 3, mean ± SEM) is shown in the lower panel, statistical differences were determined by two-way ANOVA test ***p < 0,0005. e)- STAT1 activation in response to MNoV_S99 (Moi 0.1) in BMDM from WT vs Mavs-/- infected for 60 min. Relative quantification of pSTAT1 (n = 4, mean ± SEM) are shown below. Statistical differences were determined by two-way ANOVA test *p < 0,05.
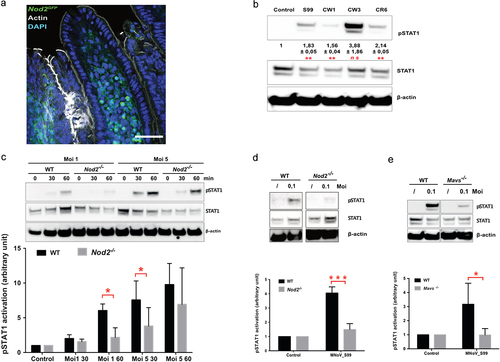
Interferon production being one of the most common responses to the intracellular presence of RNA genomes of foreign origin, we next examined if the STAT1 signaling pathway that lays upstream of ATG16l1-mediated interferon production was activated in response to murine norovirus infection. To this end, bone marrow-derived macrophages (BMDM) from WT mice were infected with several persistent and non-persistent MNoV strains at a multiplicity of infection (Moi) of 1, for 1 h and the levels of oexpression of pSTAT1 from cell lysates were measured by western blot (). In contrast to the non-persistent strain MNoV_CW3 that caused a striking increase in pSTAT1 levels, infections with the persistent strains S99 and CR6 induced a much milder STAT1 signaling pathway activation. The CW1 strain was associated with the lowest level of STAT1 activation.
Since MNoV_S99 is a natural strain commonly found in animal facilities that is able to establish persistent infections, we used it to further analyze whether the response to viral infections may rely on NOD2-associated signaling. Using the macrophage cell-line Raw264.7, we confirmed that MNoV_S99 is able to induce phosphorylation of STAT1 (Tyrosine 701) at different times at Moi 1 (Fig. S2). pSTAT1 was detectable as early as 15 min post-infection and its expression further increased at 30 min and 1 h, while only a slight increase in the STAT1 level was detectable at later time-points. In addition to the STAT1 activation through the Janus kinases JAK1 and Tyk2, we also examined the status of NFκB activation. For this purpose, pIκBα levels were quantified from infected cell lysates. Despite basal low-level activation in mock-infected cells, pIκBα intensity increased slightly at 30 min and 1 h post-infection even if it failed to reach significance. When checking AKT or the MAPK-ERK signaling axes in the same time frame, MNoV_S99 failed to induce any activation of these signaling pathways (Fig. S2). Next, we analyzed the response of BMDM derived from WT or Nod2-/- mice that were infected with increasing doses of MNoV_S99 for 30 or 60 min (). STAT1 activation increased over the course of the acute infection in cells derived from WT mice. In contrast, the levels of pSTAT1 were barely detectable in Nod2-/- cells at Moi 1. Similarly, with prolonged times of infection (24 h, Moi 0.1) we measured significantly lowered pStat1 levels in MNoV_S99-infected Nod2-/- cells in comparison with WT cells (). However, with a higher dose (Moi 5) this lowered phosphorylation of STAT1 in response to MNoV_S99 infection progressively vanished (), suggesting that NOD2-independent alternative mechanisms of action operate when the viral load is too important.
To examine whether the mitochondrial antiviral signaling (MAVS) adaptor protein is required for sensing MNoV_S99, BMDM derived from Mavs-deficient mice where generated. Interestingly, the higher pStat1 levels measured in WT cells (Moi 0.1, 1 h) were significantly decreased in BMDM from Mavs−/− cells ().
Altogether, these data show a mild but significant activation of STAT1 following MNoV_S99 infection in our in vitro cellular models. These signaling responses can be at least partially abrogated in the absence of MAVS and NOD2.
Lower levels of inflammation in Nod2−/− mice benefit noroviral propagation
MNoV_CR6 infection in DSS-induced colitis was previously shown to promote inflammation in a functional Atg16l1-dependent manner.Citation21 Using this viral strain that promotes STAT1 phosphorylation in similar degree than MNoV_S99 (), we checked how loss of Nod2 influenced resolution of inflammation in response to 3% DSS treatment in mice. As shown with the MNoV_S99 strain, loss of NOD2 improved the muscle thickening of the colon wall in mice infected with MNoV_CR6 (). However, a CR6 strain-specific effect can be observed in these experiments since significantly increased inflammation was only measured in Atg16l1 hypomorph mice but not in WT mice as opposed to MNoV_S99.
Figure 4. NOD2-dependent noroviral load. a)- WT, Nod2-/- and Atg16l1HM mice (n = 5 each) were treated with 3% DSS and infected with MNoV_CR6 (3×107 PFU). Inflammation was measured as percentage of increase in muscle thickness at the anal-rectal junction in colonic section images plotted after baseline subtraction relatively to the values obtained from WT mice. b)- the viral load measured in stools from WT, Nod2-/- and Atg16l1HM mice at days 3, 5, and 7 post-infection with MNoV_CR6 (3×107 PFU) (n = 8). c)- the viral load measured from indicated tissue samples from MNoV_CR6 infected (3×107 PFU) WT, Nod2-/- and Atg16l1HM mice that were treated for 1 week with D55 3% (n = 5). d)- MNoV_S99 genome quantification from WT and Nod2−/− BMDM cells infected with MNoV_S99 (Moi 0.1 for 24 h) (n = 3). All the statistical differences were measured with Student’s t-test.
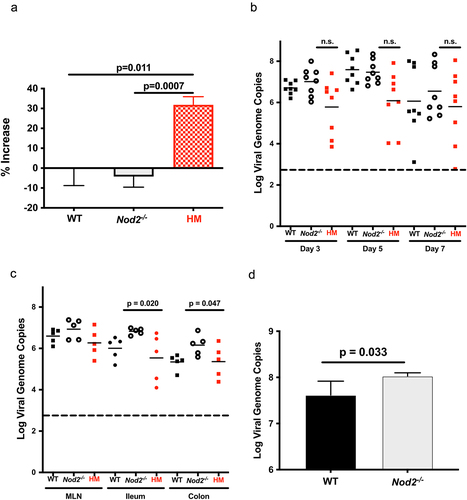
Next, we examined the impact of the pro-inflammatory signaling activation on viral propagation in these mice. In the absence of DSS-induced inflammation, no significant differences could be found when comparing the viral load in the stool of WT, Atg16l1HM or Nod2-/- mice (). By contrast, the measured viral load was significantly increased in ileum and colon of DSS-treated Nod2-/- mice in comparison with tissues of Atg16l1HM mice (). We have also analyzed the viral replication in BMDM derived from WT or Nod2-/- mice infected in vitro with MNoV_S99 (Moi 0.1 for 24 h). In this case, a slight but significant increase in the number of viral genomes was measured from Nod2-/- derived cultures (). Hence, the lower levels of inflammation found in Nod2-/- mice seem to favor a higher propagation of these two persistent strains.
MNoV_S99 infection promotes bacterial sensing by NOD2
To further explore the mechanisms behind the excessive inflammatory response associated with noroviral infections, we examined whether noroviral infection may enhance Nod2 expression levels using RT-qPCR. As what was previously observed with MNoV-1,Citation15 the transcription level of NOD2 was significantly enhanced in response to MNoV_S99 infection in either Raw264.7 cells or BMDM derived from WT mice ().
Figure 5. NOD2-dependent pro-inflammatory signaling associated with MNoV_S99 infection and bacterial MDP. Quantification of Nod2 mRNA levels measured by RT-qPCR in a)- in Raw264.7 cells infected with MNoV_S99 (Moi 0.1 or 1, for 24 h) and in b)- in BMDM from WT mice (Moi 0.1, for 24 h) normalized to ActB and relative to mock-infected cells (n = 3). c)- Representative western blot showing STAT1 and IκBα signaling pathways modulation in cell lysates from monocytes that were either infected with MNoV_S99 (Moi 5, 2 h), treated with MDP (10 ng/mL, 2 h), or a combination of both. d)- quantification and comparison of pSTAT1 and pIκbα signals between MNoV_S99 infected alone cells or in combination with MDP (10 ng/mL, 2 h), (n = 3), with the band intensity being normalized to β-ACTIN and relative to mock-treated cells. e) TNFα production by BMDC subjected to MNoV_S99’s ssRNA (10 µg/mL) alone, or supplemented with either MDP (1 µg/mL) or MDP-DD (1 µg/mL) overnight. f) TNFα production by BMDM cells infected with MNoV_S99 (Moi 0.1 or 1) for 6 h, prior to being treated with MDP (10 µg/mL) overnight. g) Net production of MNoV_S99 in Raw264.7 cells infected with MNoV_S99 (Moi 1 for 2 h before MDP treatment (100 ng/mL) for 24 h). Statistical differences were analyzed with Student’s t test.
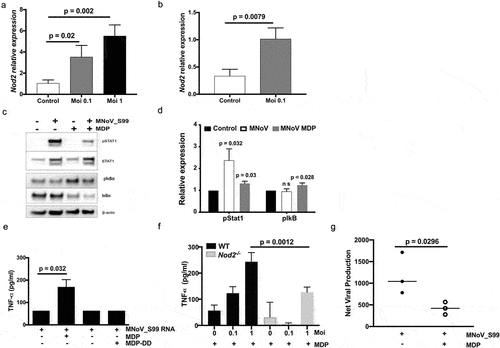
This increased Nod2 expression following norovirus infection could explain at least partially the viral-induced pathogenesis that we observed in our colitis models. Indeed, the greater expression of NOD2 should spike up when suddenly intestinal mononuclear phagocytes interact with intestinal commensal bacteria following DSS-induced epithelial abrasion. We next evaluated whether this heightened expression of NOD2 in response to MNoV_S99 infection may enhance bacterial sensing. While MNoV_S99 infection did induce Stat1 (but not IκBα) activation in monocytes isolated from WT mice (, MNoV_S99+, MDP- condition), MDP alone triggered potent IκBα activation without activating Stat1 (, MNoV_S99-, MDP+ condition). Interestingly, MNoV_S99 infection followed by the secondary MDP treatment was associated with significant phosphorylation of both Stat1 and IκBα (, MNoV_S99+, MDP+ vs mock treated cells). In the same line of investigation, we noticed a significantly augmented pro-inflammatory cytokine TNFα response to MDP in dendritic cells that were first primed with noroviral RNA (). Next, the secretion of TNFα was measured in the supernatants from WT and Nod2-/- BMDM cultures infected with MNoV_S99 followed by the MDP treatment. TNFα levels were increased with increasing doses of virus in WT cells, while they remained undetectable (Moi 0.1) or much lower (Moi 1) in Nod2-/- cells (). Equally of importance, viral titers measured in supernatants showed significantly decreased yields when infected cells were treated with MDP ().
Overall, these data show that the increased Nod2 transcription levels following noroviral infections may at least partially promote an inflammatory response to bacterial MDP, leading ultimately to a limitation in the viral propagation. Hence, lowered inflammation in Nod2−/- mice is associated with increased dissemination of MNoV_S99 and MNoV_CR6 strains, suggesting that bacterial sensing through NOD2 may regulate their persistence.
Discussion
Intestinal homeostasis is tightly regulated by the interplay between the microoganisms found in the gut microbiota, intestinal epithelial cells, and mononuclear phagocytes. The breach in gut flora homeostasis termed dysbiosis is becoming central to the onset of chronic inflammatory bowel diseases, such as Crohn’s disease.Citation28,Citation29 With the development of molecular biology techniques, alterations in the virome relevant to disease pathogenesis are thoroughly under investigation.Citation2 As such, the role played by noroviruses was studied in several gastrointestinal inflammatory models. An endemically circulating mouse norovirus strain was shown to promote intestinal barrier dysfunctions and inflammatory lesions caused by loss of tolerance to commensal bacteria in Il10-/- mice.Citation30 The secondary challenge with a pathogenic E. coli strain following MNoV-1’s infection can even cause lethal lesions due to the induction of an unresolved proinflammatory cytokine secretion in a Nod1/2 dependent mannerCitation19. The hypothesis of an environmental-driver in genetically susceptible hosts that could be at the origin of Crohn's-like disease was also explored with the MNoV_CR6 strain in the Atg16l1HM mice presenting defective autophagy in a DSS colitis model.Citation21
NOD2 is very well known for its pathogen-associated molecular pattern recognition activity against bacteria,Citation17 and recently, it appears it can also participate in the detection of certain viruses.Citation18 Using pro-inflammatory signaling activation downstream of NOD2 as a read-out, we have analyzed if NOD2 can also participate in MNoV detection and elimination. All our in vitro experiments with monocytic lineage derived primary cells (e.g. BMDM, BMDC, BM) and cell lines (e.g. Raw264.7) infected with different MNoV strains showed activation of the STAT1 signaling pathway implicated in the pro-inflammatory IFN and TNF responses. By contrast, these responses were abrogated in Nod2-/- and Mavs-/- mice derived from BMDM.
Among enteric viruses commonly found in the microbiota, noroviruses have been proposed to be good candidates in the hypothesis of an environmental-driver in genetically susceptible hosts at the origin of Crohn’s disease onset. MNoV_CR6 strain was shown to induce colitis in Atg16l1HM mice presenting defective autophagy in a DSS colitis model.Citation21
Several PRRs are implicated in viral sensing. MDA-5 and TLR3 have been shown to participate in the detection of either MNoV_1 or CW3.Citation15 These are both strains which do not persist in vivo, contrary to the S99 (Berlin) Citation25 and CR6 that were used in our study.
Our in vivo data show significantly exacerbated inflammation when applying either 3% or 5% DSS colitis protocol to WT or Atg16l1HM mice. The excessive MNoV_S99 or MNoV_CR6 associated inflammatory response was abrogated in Nod2-/- mice or in BMDM derived from these mice.
Hence, NOD2-dependent signaling adds a layer of regulation to the viral sensing and propagation. While in our norovirus + DSS model knocking-down NOD2 prevents gut inflammation, it has the opposite effect with influenza A virus (IAV) infection in the lung of Nod2-/- or Ripk2-/- mice.Citation31 Indeed, the NOD2-RIPK2 signaling ax has been shown to downmodulate the NLRP3-dependent mitophagy activation and the associated inflammatory response in the lung tissue of infected mice.
Norovirus propagation or prolonged persistence is tightly regulated by the interplay between several signaling pathways. MNoV_1 replication was shown to be inhibited in macrophages following LPS mediated NF-κB activation that in turn triggers IFN-β-dependent JAK-STAT antiviral activity.Citation32 Interestingly, in our cellular models, in addition to the phosphorylation of STAT1 that we observed when cells were infected with MNoV_S99, the heightened Nod2 expression levels preceded a greater ability of cells to respond to bacterial MDP. Consequently, the subsequent treatment with MDP induced an increased NfκB signaling pathway via IκBα activation. The sequential activation of these two pro-inflammatory signaling pathways resulted in the inhibition of the MNoV_S99 propagation on the one hand and the augmented production of the pro-inflammatory cytokine TNFα in response to MDP on the other hand. Hence, the aggravated inflammatory pathology we observed in our DSS colitis model can be explained by this NOD2-dependent signaling loop in MNoV_S99 or MNoV_CR6 infected mice.
In conclusion, our study shows a NOD2-dependent bacterial sensing that prevents dissemination of the persistent MNoV_S99 and MNoV_CR6 strains.
Methods
Unless otherwise indicated, all media and reagents were purchased from Thermo Fischer Scientific.
Colitis mouse model and viral infection
All animal experiments were approved by the local ethical committee n° CEEA - 2016030717519,903. Age and gender-related Nod2−/− and wild-type C57BL/6J mice (CDTA, Orléans, France) were housed four to six per cage, under a strict pathogen-free environment. Mice were gavaged with 5x107 TCID50/mL of MNoV_S99 diluted in 200µL PBS or mock treated with PBS only. 7 d post-infection, drinking water was substituted with a 3–5% dextran sodium sulfate (DSS) (35,000–40,000 MW; TdB Consultancy) solution replaced every 2 d for a total duration of 7 d. 3% DSS colitis experiments with MNoV_CR6 were conducted as previously described in.Citation21
Cells and virus
Mouse leukemic monocyte macrophage cell line RAW264.7 were purchased from ECCAC (Sigma-Aldrich) and maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% (v/v) fetal bovine serum, 1% penicillin/streptomycin, and 1% sodium pyruvate.
Bone marrow-derived macrophages (BMDMs) were isolated from femurs of wild-type, Nod2−/−, or Mavs-/- mice of C57BL/6J background.Citation33 Using a 26 G ½” needle, bone marrow cells were flushed out of the bones with Iscove’s modified Dulbecco’s medium (IMDM), supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% non-essential amino acid, 1% sodium pyruvate, 1% glutamine, and 20% (v/v) conditioned-media from L929 cells. Red blood cells were lysed using a 160 mM NH4CL and 170 mM Tris solution for 5 min at RT. 3 to 6 × 106 viable cells were plated in non-cell-culture-treated petri dishes and grown for 5–7 d in the above mentioned fully supplemented IMDM medium.
Bone marrow-derived dendritic cells were isolated similarly as BMDM cells. Then, 2 × 106 viable cells were plated in non-cell-culture-treated petri dishes and grown for 7 d in RPMI 1640, supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% L-glutamine, 1% HEPES, and 20% (v/v) conditioned-media from J558 cell line producing murine granulocyte monocyte colony stimulating factor (GM-CSF).
Monocytes were isolated from bone marrow cells by using the mouse Monocyte Isolation Kit (Miltenyi Biotec) and a QuadroMACS separator.
The MNoV_S99 strain (GenBank accession no. DQ911368) was provided by Prof. P. Maris from ANSES Fougères Laboratory (France) and was propagated in RAW 264.7 cells. MNoV_CR6 concentrated stocks were prepared as described in.Citation34 Briefly, supernatant from 293T cells transfected with a plasmid containing the viral genome was applied to RAW264.7 cells to amplify virus production, and virions were concentrated by ultracentrifugation and resuspension in endotoxin-free PBS. Concentration of stocks was determined by plaque assay.
For in vitro infection experiments, 3–5×105 viable cells were seeded in six well plates. The next day, just before viral infection, cells were counted from one well to determine precisely the multiplicity of infection (Moi). In the remaining wells medium was removed and replaced with variable quantities of MNoV_S99 from a common viral stock in a final volume of 1 mL fresh medium per well. Mock treated cells were incubated in parallel with matching quantities of culture media from non-infected cells that were generated at the same time as viral stocks.
At various time-points after infection, supernatant was removed and cells were washed once with PBS, before lysis in RIPA buffer for WB analysis or RLT buffer for total RNA extraction (RNAeasy kit, Qiagen) for RT-qPCR experiments.
Immunohistochemistry, ELISA, western blot, RT-qPCR, and titration
For immunohistochemistry analysis, formalin-fixed colon samples were embedded in paraffin. 5 µm sections were stained with H&E. Slides were imaged with an AxioPlan 2 (Zeiss) microscope and blindly scored for inflammation severity by two investigators as previously described.Citation35 Briefly, tissue lesions were scored from 0 to 3, so was the infiltration of inflammatory cells in the lamina propria. A combined score was calculated, ranging from 0 (no changes) to 6 (wide-spread cellular infiltrations and extensive tissue damage).
The muscularis propria thickness was measured from light microscopy images of H&E stained colon sections using Image J (NCBI). Five measurements from different locations were performed for each animal and compared with colon sections from mock treated animals.
For ELISA analysis, briefly 1 cm colon samples were recovered from mice, luminal content was flushed with PBS, samples were cut opened and resuspended overnight in 250 µl DMEM supplemented with 1% penicillin/streptomycin in 24 well plate at 37°C. The following day, supernatants were collected and stored at −80°C before cytokine measurement. TNFα and Interleukin-6 protein levels were measured specifically using pre-coated 96 well ELISA plates (R&D System) following manufacturers’ indications.
For TNFα ELISA from BMDC, 5 × 105 cells were plated in 24 well plates and treated with MNoV_S99’s ssRNA (extracted with RNeasy Qiagen kit) with Lipofectamine 2000Citation18 ± MDP or MDP-DD (Invivogen) overnight and supernatants were measured for TNFα levels.
For TNFα ELISA from BMDM, 2 × 105 cells were plated in 96 well plates infected with MNoV_S99 (Moi 0.1 or 1) for 6 h prior to MDP treatment (10µg/mL) overnight, and supernatants were measured for TNFα levels.
For western blot analysis, total proteins from cell lysates were quantified by BCA protein assay kit (Pierce), equalized amounts of proteins (10–20 µg) were resolved by 4–15% gradient SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked in 0.05% PBS-Tween, 5% skimmed milk solution, before incubation subsequently with primary and secondary antibodies diluted (see ) in a blocking solution. Following secondary antibodies incubations, signals of interest were detected using a chemiluminescence reader (ImageQuantTM Las 4000, GE Healthcare Life Sciences), images were processed, and band ROIs were quantified with Image J software (NCBI). The mean band intensities ± SDs of protein of interest were normalized to β-ACTIN. Relative values to control mock-infected conditions were compared between different experiments.
Table 1. Antibodies used for western blotting.
For Nod2 quantification from MNoV_S99 infected BMDM or RAW264.7 cells, total RNA extraction was performed using the RNeasy Mini Kit (Qiagen). One µg of RNA was reversed transcribed in the presence of 2.5 mM of oligo-dT using the Reverse Transcription kit (Roche) following the manufacturer’s instructions. 75 ng of cDNAs were used for the qPCR analyses using Q5 High-Fidelity 2X Master Mix (New England BioLabs). Nod2 and Actb were specifically amplified using the Fwd_5’GGAAACAACATTGGCAGCAT3’ and Rev_5’TCTTGAGTCCTTCTGCGAGA3’ Fwd_5’TTCTTTGCAGCTCCTTCGTT3’ and Rev_5’ATGGAGGGGAATACAGCCC3’, respectively. Actb was used as an internal reference gene in order to normalize the transcript levels. The relative mRNA levels (2−ΔΔCt) were determined by comparing (a) the PCR cycle thresholds (Ct) for Nod2 and Actb (ΔCt) and (b) ΔCt values for treated and control groups (ΔΔCt).
MNoV_S99 load was determined by measuring the MNoV genome copy numbers using RT-qPCR analysis as previously describedCitation36. Briefly, total RNA was extracted from BMDM cell cultures using the RNeasy Mini Kit (Qiagen). 5 µL of RNA were used per reaction of RT-qPCR. Viral genome levels were determined using the Takyon™Dry No Rox One-Step RT Probe Mastermix (Eurogentec). Viral genomes were specifically amplified using the Fwd_5’GTGCGCAACACAGAGAAAACG3’ and Rev_5’CGGGCTGAGCTTCCTGC3’ primers that target the MNoV ORF1 region combined with the TaqMan probe 5’[6-FAM]- CTAGTGTCTCCTTTGGAGCACCTA-[BHQ1]3’ using the Stratagene Mx3005P (Agilent Technologies) with 1 RT cycle and 40 cDNA amplification cycles. The quantification of viral genome copies was obtained using a standard curve (ranging from 2 × 101 to 2 × 106 genome copies) with serial dilutions of the plasmid pMNoV1 corresponding to the full genome of MNoV_1 (Dr Christiane Wobus, University of Michigan, USA). Viral load for MNoV_CR6 strain was determined by measuring genome copy numbers using RT-qPCR analysis as previously described inCitation21.
The net production of MNoV_S99 was determined using the TCID50 method as described inCitation35. Briefly, Raw264.7 cells were infected at Moi 1 with MNoV_S99 for 2 h, before adding MDP (100 ng/mL) or not in the supernatant for 24 h. Titers (n = 3) were obtained from each supernatant. The net production is calculated by normalizing the amount of viruses produced with the amount of viruses used for the infection.
NOD2GFP immunohistochemistry imaging
NOD2GFP miceCitation37 in the C57BL/6 background were maintained at the Institut Pasteur Central Animal facility. We used heterozygous NOD2GFP mice since the presence of one functional copy of NOD2 ensures that physiological responses dependent on functional NOD2 are not impaired. Heterozygous NOD2GFP female mice (8–12 weeks of age) were sacrificed, and several centimeters of the distal ileum were removed, opened longitudinally, and washed in PBS. The tissue was spread flat over a 4% w/v LMP agarose (Merck) pad and fixed with 4% v/v paraformaldehyde (VWR). The ileal tissue was then embedded in a 4% LMP agarose block and cut into 200 µm-thick sections using a Microm HM 650 V Vibration microtome.
For staining, tissues were blocked and permeabilized overnight in a solution containing 0.4% Triton X-100 (Merck) and marked with an anti-GFP (rabbit polyclonal antibody A-11122, Invitrogen) primary antibody and secondary Alexa Fluor 488 goat anti-rabbit antibody (Invitrogen). Tissue sections were counter stained with DAPI (1 µg/mL, BD Biosciences) and Phalloidin-iFluor 647 (1:200 dilution, Abcam) and mounted using ProLong Gold Antifade reagent. Confocal acquisitions were performed using a Leica HyD SP5 confocal microscope. Image analysis was performed using Icy version 2.4.2.0.Citation38
Authors’ contribution
G.M. and M.T. performed the majority of the experimental procedures. T.G. contributed to the preclinical studies in mice. M.D., T.G., and O.B. performed RT-qPCR and ELISA analysis. M.D. contributed to histological analysis of colon sections. E.W. performed viral genome quantification using RT-qPCR. R.W. and I.G.B. performed a histological analysis of tissue sections from NOD2-β-Gal reporter mice. All authors contributed to the interpretation of the raw data and critically reviewed and/or modified the manuscript. G.M. and M.C. conceived, designed, and wrote the paper.
Supplemental Material
Download PNG Image (518.8 KB){kind=link}
Supplemental Material
Download PNG Image (124.5 KB){kind=link}
Supplemental Material
Download MS Word (15.9 KB)Acknowledgments
We thank the UTechS PBI for the NODGFP tissue imaging assistance. We thank the members of the Institut Pasteur Central Animal Facility for their assistance with animal studies. .We thank T. Durand and MP. Fourmaux for excellent technical assistance.
Disclosure statement
K.C. has received research support from Pfizer, Takeda, Pacific Biosciences, Genentech, and AbbVie; consulted for or received honoraria from Vedanta, Genentech, and AbbVie; and holds U.S. patent 10,722,600 and provisional patent 62/935,035 and 63/157,225.
Data availability statement
The data that support the findings of this study are available from the corresponding authors, GM and MC, upon request on HAL. https://hal.science/hal-04101382.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2249960
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Cadwell K, Pfeiffer J. Expanding the role of the virome: commensalism in the gut. J Virol. 2015;89(4):1951–15. doi:10.1128/JVI.02966-14.
- Norman JM, Handley S, Baldridge M, Droit L, Liu C, Keller B, Kambal A, Monaco C, Zhao G, Fleshner P, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160(3):447–460. doi: 10.1016/j.cell.2015.01.002.
- Pfeiffer JK, Virgin HW. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Sci. 2016;351(6270):aad5872. doi:10.1126/science.aad5872.
- Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng X-L, Qu L, et al. Replication of human noroviruses in stem cell-derived human enteroids. Sci. 2016;353(6306):1387–1393. doi: 10.1126/science.aaf5211.
- Estes MK, Ettayebi K, Tenge VR, Murakami K, Karandikar U, Lin S-C, Ayyar BV, Cortes-Penfield NW, Haga K, Neill FH, et al. Human norovirus cultivation in nontransformed stem cell- derived human intestinal enteroid cultures: Success and challenges. Viruses. 2019;11(7):E638. doi: 10.3390/v11070638.
- Ludwig-Begall LF, Mauroy A, Thiry E. Noroviruses—the state of the art, nearly fifty years after their initial discovery. Viruses. 2021;13(8):1541. doi:10.3390/v13081541.
- Newman KL, Leon JS. Norovirus immunology: Of mice and mechanisms. Eur J Immunol. 2015;45(10):2742–2757. doi:10.1002/eji.201545512.
- Wobus CE, Karst SM, Thackray LB, Chang K-O, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW, et al. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004;2(12):e432. doi: 10.1371/journal.pbio.0020432.
- Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, Kelley ST, Virgin HW. Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J Virol. 2007;81(19):10460–10473. doi:10.1128/JVI.00783-07.
- Nice TJ, Strong DW, McCune BT, Pohl CS, Virgin HW. A single- amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J Virol. 2013;87(1):327–334. doi:10.1128/JVI.01864-12.
- Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Sci. 2015;347(6219):269–273. doi:10.1126/science.1258100.
- Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. STAT1-dependent innate immunity to a Norwalk-like virus. Sci. 2003;299(5612):1575–1578. doi:10.1126/science.1077905.
- Hwang S, Maloney N, Bruinsma M, Goel G, Duan E, Zhang L, Shrestha B, Diamond M, Dani A, Sosnovtsev S, et al. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host & Microbe. 2012;11(4):397–409. doi: 10.1016/j.chom.2012.03.002.
- Thorne LG, Goodfellow IG. Norovirus gene expression and replication. J Gen Virol. 2014;95(2):278–291. doi:10.1099/vir.0.059634-0.
- McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin IV HW, Colonna M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008;4(7):e1000108. doi:10.1371/journal.ppat.1000108.
- Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, Zheng Y, Rongvaux A, Sun Q, Yang G, et al. Nlrp6 regulates intestinal antiviral innate immunity. Sci. 2015;350(6262):826–830. doi: 10.1126/science.aab3145.
- Chamaillard M, Girardin SE, Viala J, Philpott DJN. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol. 2003;5(9):581–592. doi:10.1046/j.1462-5822.2003.00304.x.
- Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–1080. doi:10.1038/ni.1782.
- Kim Y-G, Park J-H, Reimer T, Baker D, Kawai T, Kumar H, Akira S, Wobus C, Núñez G. Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host & Microbe. 2011;9(6):496–507. doi:10.1016/j.chom.2011.05.006.
- Mumphrey SM, Changotra H, Moore TN, Heimann-Nichols ER, Wobus CE, Reilly MJ, Moghadamfalahi M, Shukla D, Karst SM. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J Virol. 2007;81(7):3251–3263. doi:10.1128/JVI.02096-06.
- Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi:10.1016/j.cell.2010.05.009.
- Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim Y-G, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. doi: 10.1038/ni.1823.
- Hugot JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi:10.1038/35079107.
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. doi: 10.1038/ng1954.
- Niendorf S, Klemm U, Mas Marques A, Bock C-T, Höhne M, Boon AC. Infection with the persistent murine norovirus strain MNV-S99 suppresses IFN-Beta release and activation of Stat1 in vitro. PLoS One. 2016;11(6):e0156898. doi:10.1371/journal.pone.0156898.
- Hrdý J, Alard J, Couturier-Maillard A, Boulard O, Boutillier D, Delacre M, Lapadatescu C, Cesaro A, Blanc P, Pot B, et al. Lactobacillus reuteri 5454 and Bifidobacterium animalis ssp. lactis 5764 improve colitis while differentially impacting dendritic cells maturation and antimicrobial responses. Sci Rep. 2020;10(1):5345. doi: 10.1038/s41598-020-62161-1.
- Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL, McAllaster MR, Balce DR, Feehley T, Brestoff JR, et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Sci. 2018;360(6385):204–208. doi: 10.1126/science.aar3799.
- Hold GL, Smith M, Grange C, Watt ER, El-Omar EM, Mukhopadhya I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol. 2014;20(5):1192–1210. doi: 10.3748/wjg.v20.i5.1192.
- Kim DH, Cheon JH. Pathogenesis of inflammatory bowel disease and recent advances in biologic therapies. Immune Netw. 2017;17(1):25–40. doi:10.4110/in.2017.17.1.25.
- Basic M, Keubler LM, Buettner M, Achard M, Breves G, Schröder B, Smoczek A, Jörns A, Wedekind D, Zschemisch NH, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm Bowel Dis. 2014;20(3):431–443. doi: 10.1097/01.MIB.0000441346.86827.ed.
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14(5):480–488. doi: 10.1038/ni.2563.
- Yu P, Li Y, Wang Y, Peppelenbosch MP, Pan Q. Lipopolysaccharide restricts murine norovirus infection in macrophages mainly through NF-kB and JAK-STAT signaling pathway. Virology. 2020;546:109–121. doi:10.1016/j.virol.2020.04.010.
- Michallet M-C, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, Poeck H, Bscheider M, Hartmann G, König M, et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28(5):651–661. doi: 10.1016/j.immuni.2008.03.013.
- Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature. 2014;516(7529):94–98. doi:10.1038/nature13960.
- Hwang S, Alhatlani B, Arias A, Caddy SL, Christodoulou C, Bragazzi Cunha J, Emmott E, Gonzalez‐Hernandez M, Kolawole A, Lu J, et al. Murine norovirus: propagation, quantification, and genetic manipulation. Curr Protoc Microbiol. 2014;33(1):15K.2.1–61. doi: 10.1002/9780471729259.mc15k02s33.
- Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S, Neurath MF. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc. 2017;12(7):1295–1309. doi:10.1038/nprot.2017.044.
- Barreau F, Meinzer U, Chareyre F, Berrebi D, Niwa-Kawakita M, Dussaillant M, Foligne B, Ollendorff V, Heyman M, Bonacorsi S, et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS One. 2007;2(6):e523. doi: 10.1371/journal.pone.0000523.
- de Chaumont F, Dallongeville S, Chenouard N, Hervé N, Pop S, Provoost T, Meas-Yedid V, Pankajakshan P, Lecomte T, Le Montagner Y, et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods. 2012;9(7):690–696. doi: 10.1038/nmeth.2075.