
ABSTRACT
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers worldwide. While a close correlation between chronic Helicobacter pylori infection and CRC has been reported, the role of the virome has been overlooked. Here, we infected Apc-mutant mouse models and C57BL/6 mice with H. pylori and conducted a comprehensive metagenomics analysis of H. pylori-induced changes in lower gastrointestinal tract bacterial and viral communities. We observed an expansion of temperate phages in H. pylori infected Apc+/1638N mice at the early stage of carcinogenesis. Some of the temperate phages were predicted to infect bacteria associated with CRC, including Enterococcus faecalis. We also observed a high prevalence of virulent genes, such as flgJ, cwlJ, and sleB, encoded by temperate phages. In addition, we identified phages associated with pre-onset and onset of H. pylori-promoted carcinogenesis. Through co-occurrence network analysis, we found strong associations between the viral and bacterial communities in infected mice before the onset of carcinogenesis. These findings suggest that the expansion of temperate phages, possibly caused by prophage induction triggered by H. pylori infection, may have contributed to the development of CRC in mice by interacting with the bacterial community.
Introduction
Colorectal cancer (CRC) is one of the most prevalent malignancies in the world, with low survival rates.Citation1 Multiple factors including genetics, lifestyle, and an altered microbiome are known to play a role in CRC, and recent studies have suggested a potential link between Helicobacter pylori infection and the development of the disease.Citation2 More than half of the world’s population carries H. pylori in the upper gastrointestinal tract, and colonization is often asymptomatic.Citation3 Chronic infection with this bacterium leads to gastric inflammation and may even induce gastric cancer.Citation4 In addition, increasing epidemiological studies have shown a close correlation between H. pylori infection and CRC.Citation2,Citation5
It has been shown that H. pylori infection contributes to tumorigenesis by deregulating gastrointestinal immunity and activating oncogenic signaling pathways in the gastrointestinal tract.Citation6–8 In addition, the infection significantly alters not only the gastric microbiotaCitation2,Citation9,Citation10 but also distant, intestinal microbial communities.Citation11 We recently reported a unique H. pylori-driven immune alteration signature characterized by a reduction in regulatory T-cells and proinflammatory T-cells, as well as H. pylori induced pro-carcinogenic STAT3 signaling, loss of goblet cells and increase of mucus Altogether, it was shown that H. pylori infection contributes to the development of CRC by disrupting gut homeostasis.Citation12
While changes in the gut bacterial community are associated with CRC, the role of gut viruses remains largely unexplored. Bacteriophages, or phages, represent the vast majority (97.7%) of the gut viruses in healthy western adults,Citation13 and their constant interactions with their bacterial hosts shape gut microbial communities and maintain gut homeostasis.Citation14 In addition to regulating the bacterial communities by specifically eliminating their bacterial hosts, phages influence the gut ecosystem by directly interacting with the immune cells and modulating the host immune response.Citation15 Recent studies have observed significant changes in the fecal virome, especially the phage population, in CRC patients.Citation16–19 In addition, the expansion of phages in a mouse model led to intestinal inflammation and colitis by activating IFN-γ through a TLR9-dependent pathway, which exacerbated colitis in the model.Citation20 Further, phages transfer the auxiliary metabolic genes (AMGs), which are prevalent in gut phage genomes,Citation21–23 to and between bacterial genomes through horizontal gene transfer (HGT).Citation24 These genes are largely involved in bacterial cellular processes and extracellular virulence,Citation25,Citation26 thus play a significant role in human health and diseases by altering bacterial metabolism.Citation15
Here we aim to study the role of gut viruses using a mouse model of H. pylori-promoted CRC.Citation12 We used shotgun viral metagenomic sequencing to characterize the viral community composition of the cecum, stool and intestinal samples from i) Apc+/1638N and ii) C57BL/6 mice, with and without H. pylori infection, as well as at different time points (before the onset of colonic tumors and afterward). We identified phage-encoded AMGs related to carcinogenesis. Finally, we studied the correlations between the changes in the viral communities and the bacterial communities with CRC development.
Results
H. pylori infection alters the gut viral community and expands temperate phages in Apc+/1638N mice
To investigate the impact of the virome and bacteriome on the development of extra gastric cancer promoted by H. pylori infection, tumor-prone Apc+/1638N and wild-type C57BL/6 mice were infected with H. pylori for 24 weeks (). We observed a significant increase in tumor number in the small intestine of H. pylori-infected Apc+/1638N mice at 12 weeks (Supplementary Figure S1a-c),Citation12 which further increased at 24 weeks post-infection (pi) as shown in previous study.Citation12 Colonic tumors were only observed 24 weeks pi in Apc+/1638N mice.Citation12 These data indicated that the carcinogenesis process was initiated earlier in H. pylori-infected mice (at 12 weeks pi), and thus defined this period as the pre-onset stage of CRC.
Figure 1. Altered gut virome in Apc+/1638N mice with H. pylori PMSS1 infection. (a) Animal setting in the study. (b) Principal coordinate analysis (PCoA) of Bray-Curtis distance of virome. Each dot represents pooled samples from three to five mice. Treatment groups are indicated respectively. The 95% confidence intervals of the control and H. pylori PMSS1 groups are shaded accordingly. (c) Distribution of shared and specific vOTUs: 4,301, 5,539 detected vOTUs in Apc+/1638N and C57BL/6 mice were assigned into two categories as shared vOTU, specific vOTU (to PMSS1 infection and to control). The proportion of numbers and the relative abundance of specific vOTUs in each sample. The scales on the y-axis are optimized to present the data. One dot/bar represents samples from three to five mice.
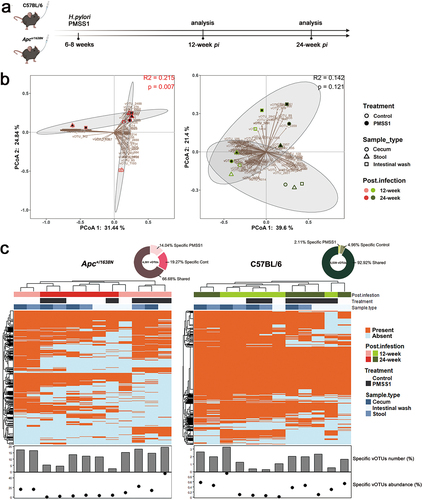
We characterized the bacterial and viral communities of the cecum, stool and intestine from both control and H. pylori-infected mice sacrificed 12- and 24 weeks pi. We identified a total of 3,649 unique amplicon sequence variants (ASVs) for the bacterial community. However, we did not observe a significant alteration in the bacterial composition, diversity and richness caused by H. pylori infection (Supplementary Figure S2).
In addition, we identified 5,687 unique viral operational taxonomic units (vOTUs) from these samples. We observed lower viral diversity and richness in Apc+/1638N compared to C57BL/6 mice regardless of H. pylori infection (Supplementary Figure S3a), emphasizing the importance of host genetics in shaping the viral communities. H. pylori infection reduced the virome diversity and richness in samples from C57BL/6 mice throughout the study, while it reduced the virome richness in Apc+/1638N mice only at 12 weeks pi (Supplementary Figure S3a). Yet, the changes in the virome composition were more pronounced in Apc+/1638N compared to C57BL/6 mice ().
We further characterized changes in the virome associated with H. pylori-promoted intestinal cancer. More specifically, we analyzed the presence or absence of different vOTUs in samples from Apc+/1638N and C57BL/6 mice with and without H. pylori infection. We identified many vOTUs which were only present in infected mice: 14.04% of 4,301 in infected Apc+/1638N mice and 2.11% of 5,539 in infected C57BL/6 mice (). Therefore, H. pylori infection altered the virome more strongly in Apc+/1638N mice than in C57BL/6 mice (). In addition, vOTUs specific to H. pylori infection accounted for a larger proportion of viral communities in infected Apc+/1638N mice at 12-week pi (14.20% in cecum, 22.31% in stool, 49.16% in intestinal wash) than that at 24-week pi (0.37% in cecum, 1.48% in stool, 4.06% in intestinal wash) (). This suggests more significant changes in viral communities before the onset (12-week pi) than after the onset of H. pylori-promoted CRC (24-week pi).
The majority of viruses found in the samples were phages that belonged to three classes: Caudoviricetes, Malgrandaviricetes, and Faserviricetes, which made up approximately 97.88% ± 3.60% on average (Supplementary Figure S4b). This aligns with previous research indicating that phages are the most prevalent viruses in the human gut.Citation13 We also predicted the replication cycle of these viruses or vOTUs using BACPHLIP, Deephage and Replidec and identified 4,128 vOTUs (72.59%) as temperate phages (). Interestingly, the relative abundance of temperate phages (79.78% in cecum, 73.79% in stool and 92.49% in intestinal wash) was high in H. pylori-infected Apc+/1638N mice at 12-week pi, while virulent phages were dominant at 24-week pi (88.19% in cecum, 98.33% in stool and 99.36% in intestinal wash) (). This suggests a higher probability for phage-mediated HGT in H. pylori-infected Apc+/1638N mice at 12-week pi relative to at 24-week pi.
Figure 2. Expanded temperate vOTUs, and specific vOTUs in infected Apc+/1638N mice carrying crucial AMGs in the pre-onset of H. pylori-promoted CRC. (a) Temperate vOTUs determination by three tools: BACPHLIP,Citation27 DeephageCitation28 and Replidec.Citation29 a vOTU is assigned to be temperate when agreed by at least two tools. (b) The ratio of temperate specific (red, the temperate vOTUs presented in either control or infected mice), temperate shared (black, the temperate vOTUs presented in both control and infected mice), virulent specific (light gray, the virulent vOTUs presented in either control or infected mice) and virulent shared vOTUs (dark gray, the virulent vOTUs presented in both control and infected mice) in gut virome of Apc+/1638N and C57BL/6 mice. (c) Heatmap shows the AMGs, overrepresented among significantly abundant temperate shared, temperate specific, virulent shared and virulent specific vOTUs in infected mice at 12-week pi. Functions of these AMGs were annotated using DRAM-v and UniProtKB.
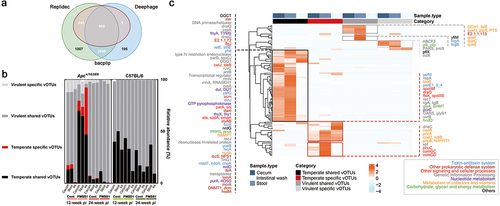
Prevalence of temperate phages and auxiliary metabolic genes precedes the onset of H. pylori-promoted CRC
We next predicted the host range of significantly abundant vOTUs (Sig.vOTUs: please see the material and method section for more details about viral groupings; Supplementary Figure S4 and S5, Supplementary Table S1 and S2) in H. pylori-infected Apc+/1638N mice to investigate the potential role of temperate vOTUs in tumorigenesis. We found several vOTUs that belong to the Caudoviricetes class that infect bacteria known to either prevent or promote colorectal cancer (CRC). The formers include Lactobacillus gallinarum,Citation30 Lactobacillus kefiranofaciens,Citation31 Lactobacillus acidophilus,Citation32 and Alistipes shahii.Citation33 On the other hand, some vOTUs infect bacteria associated with promoting CRC, such as Alistipes onderdonkii,Citation34 Alistipes finegoldii,Citation35 Flavonifractor plautii,Citation36 and Enterococcus faecalis, which has been linked to invasive phenotypes of colon cancer cells.Citation37 Interestingly, the vOTUs infecting E. faecalis were highly abundant in samples from infected mice at 12-week pi (Supplementary Figure S4a-b, and S5) while vOTUs infecting Alistipes spp. increased in abundance after 24 weeks (Supplementary Figure S5a-c). These findings suggest that there is a complex phage-bacterial infection network associated with H. pylori-promoted CRC, where temperate phages play a key role in the progression of CRC (Supplementary Figure S4b and S5b).
To further understand the functional role of temperate vOTUs at 12-week pi and their contribution to CRC, we compared the abundance of AMGs in Sig. vOTUs of the samples from 12- () and 24-week pi (Supplementary Figure S6). We observed a higher abundance of AMGs in temperate vOTUs isolated from infected Apc+/1638N mice at 12-week pi () compared to 24-week pi, suggesting a more significant contribution of temperate phages to early carcinogenesis. In addition, the abundant AMGs encoded by Sig. vOTUs from the infected Apc+/1638N mice collected at 12-week pi were more diverse than those at 24-week pi (94 vs. 45 AMGs) ( and Supplementary Figure S6). In addition, 65.96% of the 94 AMGs were found abundant in temperate vOTUs from the infected Apc+/1638N mice at 12-week pi (). Then, we annotated these AMGs using DRAM-vCitation38 and UniProtKB (Supplementary Table S3 and S4), they mainly encode genetic information processing, signaling and cellular processes, metabolism, etc. (). Several AMGs were only detected in temperate vOTUs (Supplementary Table S3). These include toxin-antitoxin systems, which are linked to bacterial cell growth and death.Citation39 In addition, peptidoglycan hydrolase (flgJ) and N-acetylmuramoyl-L-alanine amidase (cwlJ, sleB) were also detected, which are associated with bacterial cell growth and mobility by affecting the survivability of their bacterial hosts.Citation40,Citation41 Furthermore, the presence of specific transporters, including trimeric autotransporter adhesin (ata, sadA, emaA), which plays a critical role in the virulence of bacteria,Citation42–44 was found to be more prominent in temperate vOTUs than in virulent vOTUs. These genes can be potentially transferred through phage-mediated HGT to bacteria, expand their virulence, and contribute to CRC development or progression.
Phage-bacteria co-occurrence network supports the strong phage-bacteria linkages prior to the onset of H. pylori-promoted CRC
To explore the effect of phage-bacteria and bacteria-bacteria interactions at different stages of CRC, we constructed multiple association networks for samples from H. pylori-infected Apc+/1638N mice and controls at 12- and 24-week pi ( and Supplementary Figure S7). We observed dissimilarities in the topologies of bacteria-bacteria and phage-bacteria networks between 12- and 24-week pi, suggesting different interaction patterns in pre-onset and onset of CRC.
Figure 3. Network of significant abundant vOTUs and bacterial community is centered at Ruminiclostridium, Ruminococcaceae and Helicobacter during the pre-onset of H. pylori-promoted CRC. (a) Network of 494 curial vOTUs related to the pre-onset of H. pylori-promoted CRC and bacterial communities and the degree centrality measures in control and H. pylori-infected Apc+/1638N mice at 12-week pi. (b) Network of 92 Sig. vOTUs related to the onset of H. pylori-promoted CRC and bacterial communities and the degree centrality measures in control and H. pylori-infected Apc+/1638N mice at 24-week pi. The node sizes reflect their degree of connection, those with a degree value above the empirical 95% quantile and are identified as hub taxa in the network of phage and bacteria in mice at 12- and 24-week pi (in navy). Hub taxa in both networks of phage and bacteria, and bacterial taxonomy are highlighted in red. Percentage of positive edges of each node is shown in the name of the Y-axis. The percentage of edge linked with Sig. vOTUs from four vOTU categories for bacteria is illustrated.
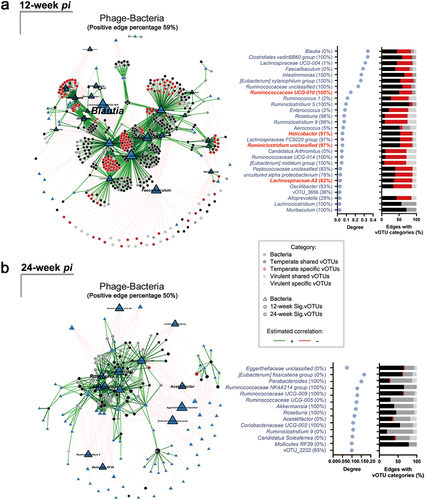
Multiple interaction networks with potential roles in CRC development were observed. For example, in H. pylori-infected samples from 12-week pi, we found correlations between Ruminiclostridium unclassified, Ruminococcaceae UCG-004, Ruminococcaceae UCG-010, and Helicobacter, as the hub taxa in the community (Supplementary Figure S7b). This is especially interesting since Ruminiclostridium and Ruminococcaceae have been previously linked to the development of CRC.Citation45,Citation46
Moreover, we found a correlation between vOTUs and specific bacterial taxa in the network. The bacterial taxa that were hubs in both phage-bacteria and bacteria-bacteria networks ( and Supplementary Figure S7b) surprisingly revealed the association between specific temperate vOTUs and hub taxa Ruminiclostridium unclassified (34.38%), Ruminococcaceae UCG-010 (36.47%), Helicobacter (34.38%) ( and Supplementary Table S5). In addition, we found that the specific temperate vOTUs were associated with a relatively higher number of hub taxa when considering their smaller proportion. These accounted for 28.54% of the significantly abundant vOTUs but contributed to 36.76% of the total phage-bacteria network (Supplementary Table S5).
However, we did not find any shared hub bacteria ( and Supplementary Figure S7d) in the samples collected at 24-week pi. Helicobacter was not the hub taxa in the bacterial community in infected mice (Supplementary Figure S7d). Only four hub bacteria in the phage-bacteria network (Ruminococcaceae UCG-004, Ruminococcaceae UCG-005, Ruminococcaceae UCG NK4A214 and Acetatifactor) showed a high eigenvector centrality within the bacterial community in infected mice (Supplementary Figure S7d). Furthermore, our data revealed that the phage community at 12-week pi exhibited strong associations with bacterial taxa that have previously been linked to CRC, such as a decrease in Blautia,Citation47 and Faecalibaculum,Citation48 as well as an increase in IntestinimonasCitation49 (). However, we did not observe significant differences in the relative abundance of the phages infecting these taxa among different treatments (Supplementary Figure S8). Our results indicate that there are strong phage-bacteria interactions during the pre-onset of H. pylori-promoted CRC.
Discussion
Colorectal cancer is the third most common cancer worldwide.Citation1 Several risk factors, including H. pylori infection, are known to play a role in the development of the disease.Citation12,Citation50 Although most H. pylori infections stay asymptomatic, chronic infections lead to gastric inflammation and cancer.Citation4 Phages play a significant role in maintaining gut homeostasis and are hypothesized to induce oncogenesis by altering bacterial community composition and physiology.Citation15,Citation17,Citation51 In addition, alterations in the fecal virome have been linked to CRC,Citation16,Citation19,Citation51 yet more mechanistic studies are needed to explore whether they play a causative role. Here, we studied gut viral community composition of H. pylori-promoted colorectal cancer in tumor-prone Apc+/1638N compared with wild-type C57BL/6 mice, and found significant changes from Apc+/1638N mice in 12 weeks pi which reflects the early stage of CRC.
Specifically, we observed differences in gut viral communities in samples from H. pylori-infected mice compared to non-infected controls. We found a decrease in diversity of the viral communities through the study period in C57BL/6 mice, and a reduction in richness of these communities after 12 weeks of H. pylori infection in Apc+/1638N mice. In addition, the abundance of vOTUs specific to H. pylori was higher at 12 weeks compared to 24 weeks post-infection. The significance of these changes was greater at 12 weeks post-infection than at 24 weeks, which suggests a stronger link between the alteration of these communities and tumorigenesis at earlier stage of CRC development.
The majority of the identified viruses were phages, belonging to Caudoviricetes, Malgrandaviricetes, and Faserviricetes classes. Interestingly, most of the expanded phages were temperate phages that can replicate either lysogenically as prophages or lytically by producing progeny. This is also consistent with previous studies that have reported an increase in temperate phages in patients with inflammatory bowel disease (IBD).Citation52 In addition, an association between temperate phages and CRC have been previously reported.Citation19 Lysogens or bacteria with integrated prophages in their genomes have been observed in high abundance in the murine gut,Citation53 and it has been suggested that inflammatory events may initiate prophage induction in these bacteria by activating oxidative stress and triggering an SOS response in bacteria.Citation54
Previous studies have shown that H. pylori infection causes immune and epithelial alterations in the intestinal tract by reducing regulatory T-cells and proinflammatory T-cells, inducing pro-carcinogenic STAT3 signaling and a loss of goblet cells.Citation12,Citation55 While these changes promote colorectal carcinogenesis in mice, they can also cause prophage induction, which could explain the expansion of temperate phages upon H. pylori infection in our study. This is specifically important as prophage induction is suggested to exacerbate disease severity in dysbiosis-associated diseases such as IBD, through immune responses triggered by bacterial lysis and released phage progeny.Citation52,Citation56 Similarly, the induction of prophages triggered by H. pylori infection could have contributed to CRC development in mice. More specifically, cell lysis due to prophage induction could release pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), which could trigger the release of proinflammatory cytokines, aggravating intestinal inflammation and dysbiosis.Citation57 In addition, cell death by prophage induction in commensal bacteria reduces their protective effects against pathobionts.Citation58 The induced phages could also trigger immune responses, including the production of IL-12, IL-6, IL-10, and IFN-γ-induced protein 10,Citation59 which have been associated with CRC.Citation60 Our host range predictions show that the expanded phages can target a wide diversity of bacterial taxa with distinct functions in gut health, including species with an anti-tumor effect like lactic acid-producer Lactobacillus and those associated with tumorigenesis such as E. faecalis, suggesting a general induction pattern in gut bacteria by H. pylori infection regardless of their taxa. The induction of prophages can significantly alter bacterial community composition in the gut, resulting in dysbiosis and horizontal gene transfer among bacteria with an unpredicted impact on gut health. Temperate phages, in general, encoded more AMGs than virulent phages, which is consistent with their potential role as contributors to bacterial fitness in the gut.Citation61,Citation62 In addition, our data indicated a higher abundance and diversity of AMGs in mice 12 weeks after infection with H. pylori, thus at the early stage of CRC development. The high prevalence of virulent genes such as flgJ and cwlJ, sleB in these phages and the possibility of temperate phages infecting taxa such as E. faecalis which is associated with CRC development further strengthens the hypothesis that they are involved in colorectal carcinogenesis. However, the underlying mechanisms remain to be explored.
We also found a positive correlation between H. pylori infection and an increased linkages between phages and Ruminiclostridium and Ruminococcaceae, two cancer-related bacterial genera.Citation45,Citation46 Interestingly, these changes were observed only 12 weeks after H. pylori infection, suggesting that they are mainly involved in the early stage of tumorigenesis while following a “driver passenger model”- the driver (H. pylori) causes changes in the tumor microenvironment enabling the expansion of passengers (cancer-causing taxa) that promote disease development.Citation63 These changes were only observed in tumor-prone Apc+/1638N and not wild-type mice, suggesting the importance of genetics in host-microbe interactions and colorectal carcinogenesis.
Our study has shed light on the effect of H. pylori-specific alterations in gut homeostasis and its knock-on effect on the gut virome and provides evidence of the role of temperate phages in the early stage of CRC development. We believe that the interactions between phages associated with CRC, the gut bacterial community, and host immunity has potentially impacted the disease progression, both directly and indirectly. Specifically, these phages may have changed cancer cell behavior by altering the gut bacterial community, interacting with immune cells, or transcytosing into colonic epithelial cells.Citation64 It is also possible that tumor cells have altered the composition of these viruses by secreting tumor-associated factors and interacting with the immune system and gut bacteria. However, to fully understand the role of phages in CRC and how they affect its development, it is essential to perform functional analyses of these viruses in the human gut.
Given the high prevalence of both lysogens and H. pylori in the gut, these findings could have potential implications for controlling H. pylori-promoted CRC. However, going forward, it will be necessary to further validate our results by assessing the effect of these phages on CRC development independent from H. pylori infection. It is also essential to investigate whether these findings are specific to H. pylori infection or if other gut pathogens can also stimulate the expansion of temperate phages, and how this would impact gut homeostasis.
Materials and methods
Study design and sample collection
Apc+/1638N mice were provided by Prof. Klaus-Peter Janssen (Klinikum rechts der Isar, Munich) and bred under specific pathogen-free conditions in the animal facility at the Technical University of Munich. C57BL/6 mice were purchased from Envigo RMS GmbH and acclimatized to the animal facility for 1–2 weeks as controls for Apc+/1638N mice. All animal experiments were conducted in compliance with European guidelines for the care and use of laboratory animals and were approved by the Bavarian Government (Regierung von Oberbayern, Az.55.2-1-54-2532-161-2017).
Six-eight weeks old Apc+/1638N and C57BL/6 mice were orally gavaged twice within 72 hours with 2 × 108 H. pylori PMSS1 (DSMZ No.105294). The cecum, fecal, and the intestinal wash samples were collected from mice at 12 and 24 weeks after infection for processing and downstream analysis (). To obtain the intestinal wash samples, we gently flushed the contents of the small intestine using 5 ml syringes filled with cold phosphate-buffered saline (PBS).
VLP isolation and metagenomic sequencing
Three to five samples from each group were pooled to get sufficient viral DNA for sequencing. Samples were vortexed vigorously at 4°C overnight, then centrifuged at 4,000 × g for 30 minutes to separate VLPs from bacteria and biological matters. The supernatant containing VLPs was filtered through 0.22 μm filters (Merck Millipore, SLGPR33RS) to remove the remaining unwanted materials. The filtrate was further concentrated to 5 mL by 10 kDa Amicon® Ultra Centrifugal Filters (Merck Millipore, UFC900324) and then purified using CsCl density gradient centrifugation at 24,000 rpm, for 4 h at 4°C. The following concentrations of CsCl were used to create the gradient: 1.2, 1.4, 1.5, and 1.65 g/cm3. Densities ranging from 1.39 to 1.51 g/cm3 were collected and concentrated to achieve 100 μL of highly concentrated VLPs. The samples were subjected to DNA extraction as described by Ma et al.Citation25 The final concentration and quality of viral DNA were determined using the Qubit dsDNA HS kit (Invitrogen, Q32854) and sequenced using the Illumina Novaseq 6000 platform by Novogene.
Metagenome assembly and analysis
Viromic sequencing data were analyzed using the ViroProfiler pipeline.Citation65 Briefly, raw reads were filtered with fastp (v0.23.1)Citation66 to remove adaptors and regions with low quality. Reads showing nucleotide identity to the mammalian host were removed by mapping clean reads to a masked host reference genomeCitation67 using bbmap.sh.Citation68 The host reference genome was downloaded from the NCBI genome database and viral sequences were downloaded from viral RefSeq and neighbor nucleotide records in the NCBI nucleotide database, before being shredded using shred.sh script.Citation68 The shredded virus sequences were then mapped to the host reference genome using bbmap.sh script. Mapped regions were masked using bbmask.sh scriptCitation68 to create a virus-free host genome database. Clean reads were assembled into contigs using metaSPAdes (v3.15.4).Citation69 Contigs (>3kb) generated from different samples were combined into a contig library (cclib). Duplicated contigs were removed using seqkitCitation70 and dedupe.sh.Citation68
CheckV (v0.8.1)Citation71 was used to extract viral regions from contigs that contain proviruses. To further reduce the redundancy of the contig library, we clustered contigs following the “rapid genome clustering based on pairwise ANI” protocol in CheckV, which clustered contigs that share more than 95% identity and 80% coverage. The longest contigs from each cluster were selected to create the non-redundant contig library (nrclib). VirSorter2 (v2.2.3)Citation72 and VIBRANTCitation73 were used for the identification of viral sequences in nrclib. MMseqs2 taxonomy moduleCitation74 and vConTACT2 (v0.11.1)Citation75 were used for taxonomy annotation of viral operational taxonomic units (vOTUs). ORFs were predicted using Prodigal (v2.6.3)Citation76 for each vOTUs.
Relative abundance data were obtained by mapping clean reads to nrclib using Bowtie2,Citation77 and coverage and sequencing depth of each vOTU were calculated using CoverM (v0.6.1) (https://github.com/wwood/CoverM). Transcripts per million (TPM) was used to normalize the abundance table. BACPHLIP (v0.9.6),Citation27 Deephage (v1.0)Citation28 and in-house software, ReplidecCitation29 were used to predict the replication cycle of each vOTU. DRAM-vCitation38 was used to identify and annotate the AMGs in vOTUs and manually curated using the UniProtKB database. Hosts of vOTUs were predicted using iPHoP.Citation78
16S rRNA gene amplicon identification
The 16S rRNA data was obtained from Ralser’s study.Citation12 Overlapping paired-end reads were processed with the DADA2 pipelineCitation79 using the open-source software QIIME 2 v.2020.112.2 (https://qiime2.org).Citation80 Unique amplicon sequence variants (ASVs) were assigned a taxonomy and aligned to the SILVA reference database (v138).Citation81
Statistical analysis
The diversity() function in the vegan package was used to calculate alpha (Shannon and Chao1 index) and beta diversities and the adonis() function to run PERMANOVA (permutational multivariate analysis of variance). The significantly abundant vOTUs (Sig. vOTUs) in samples from Apc+/1638N infected by H. pylori PMSS1 were identified using the analysis of the composition of microbiomes with bias correction (ANCOM-BC) in R (v1.0.2).Citation82 The cutoff of the log (fold change) and adjusted p-value (Benjamini – Hochberg) were set at 2 and 0.05. Co-occurrence networks inference on clr-normalized abundances was performed using the Spearman’s correlation in corrplot (v0.84) and NetCoMi package (v1.0.3)Citation83 and significant edges were selected using cor.mtest(). Community structures were estimated using greedy optimization of modularity. The cutoff of the correlation and the p-value in the network of intra-bacteriome was set at 0.3 and 0.05, while the network of phage-bacteria was at 0.8 and 0.001, respectively. The ggplot2 package and GraphPad Prism 9.0 were used for plotting the graphs.
Contributions
S.L., L.D. and M.G. conceived and designed the study. A.R., and R.ML. conducted animal experiments. J.R. assembled and sorted the raw reads of viral metagenomic data. X.P. established and ran the in-house Replidec for the data. S.L. analyzed the data and drafted the manuscript. S.L., M.K.M., J.X. and L. D. contributed to data interpretation. M.K.M. and L.D. revised the manuscript. L.D., M.G., and S.L. acquired the funding. All authors read and reviewed the articles.
Supplemental Material
Download Zip (11.8 MB)Acknowledgments
The authors thank Sophie E. Smith for proofreading the manuscript, the reviewers and the editor for their constructive comments, as well as members of the Deng Lab for technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Sequence data have been deposited with links to BioProject accession number PRJNA808836 (16S) and PRJNA938071 (viral metagenomes) in the NCBI BioProject database.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2257291
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Morgan E, Arnold M, Gini A, Lorenzoni V, Cabasag CJ, Laversanne M, Vignat J, Ferlay J, Murphy N, Bray F. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023;72(2):338–14. doi:10.1136/gutjnl-2022-327736.
- Butt J, Epplein M, Leong JM. Helicobacter pylori and colorectal cancer—A bacterium going abroad? PLoS Pathog. 2019;15(8):e1007861. doi:10.1371/journal.ppat.1007861.
- Salih BA. Helicobacter pylori infection in developing countries: the burden for how long? Saudi J Gastroenterol. 2009;15(3):201–207. doi:10.4103/1319-3767.54743.
- Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–739. doi:10.1128/CMR.00011-10.
- Kim TJ, Kim ER, Chang DK, Kim Y-H, Baek S-Y, Kim K, Hong SN. Helicobacter pylori infection is an independent risk factor of early and advanced colorectal neoplasm. Helicobacter. 2017;22(3):e12377. doi:10.1111/hel.12377.
- Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002;99(22):14428–14433. doi:10.1073/pnas.222375399.
- Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe. 2014;15(3):306–316. doi:10.1016/j.chom.2014.02.008.
- Dooyema SDR, Noto JM, Wroblewski LE, Piazuelo MB, Krishna U, Suarez G, Romero-Gallo J, Delgado AG, Peek RM. Helicobacter pylori actively suppresses innate immune nucleic acid receptors. Gut Microbes. 2022;14(1):2105102. doi:10.1080/19490976.2022.2105102.
- Guo Y, Zhang Y, Gerhard M, Gao J-J, Mejias-Luque R, Zhang L, Vieth M, Ma J-L, Bajbouj M, Suchanek S, et al. Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut. 2020;69(9):1598–1607. doi:10.1136/gutjnl-2019-319696.
- Bakhti SZ, Latifi-Navid S. Interplay and cooperation of Helicobacter pylori and gut microbiota in gastric carcinogenesis. BMC Microbiol. 2021;21(1):258. doi:10.1186/s12866-021-02315-x.
- Kienesberger S, Cox LM, Livanos A, Zhang X-S, Chung J, Perez-Perez GI, Gorkiewicz G, Zechner EL, Blaser MJ. Gastric Helicobacter pylori infection affects local and distant microbial populations and host responses. Cell Rep. 2016;14(6):1395–1407. doi:10.1016/j.celrep.2016.01.017.
- Ralser A, Dietl A, Jarosch S, Engelsberger V, Wanisch A, Janssen KP, Middelhoff M, Vieth M, Quante M, Haller D, et al. Helicobacter pylori promotes colorectal carcinogenesis by deregulating intestinal immunity and inducing a mucus-degrading microbiota signature. Gut. 2023;72(7):1258–1270. doi:10.1136/gutjnl-2022-328075.
- Gregory AC, Zablocki O, Zayed AA, Howell A, Bolduc B, Sullivan MB. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe. 2020;28(5):724–740.e8. doi:10.1016/j.chom.2020.08.003.
- Mirzaei MK, Deng L. New technologies for developing phage-based tools to manipulate the human microbiome. Trends Microbiol. 2022;30(2):131–142. doi:10.1016/j.tim.2021.04.007.
- Tiamani K, Luo S, Schulz S, Xue J, Costa R, Khan Mirzaei M, Deng L. The role of virome in the gastrointestinal tract and beyond. FEMS Microbiol Rev. 2022;46(6):fuac027. doi:10.1093/femsre/fuac027.
- Nakatsu G, Zhou H, Wu WKK, Wong SH, Coker OO, Dai Z, Li X, Szeto C-H, Sugimura N, Lam T-T, et al. Alterations in enteric virome are associated with colorectal cancer and survival outcomes. Gastroenterology. 2018;155(2):529–541.e5. doi:10.1053/j.gastro.2018.04.018.
- Handley SA, Devkota S. Going viral: a novel role for bacteriophage in colorectal cancer. mBio. 2019;10(1):e02626–18. doi:10.1128/mBio.02626-18.
- Marongiu L, Landry JJM, Rausch T, Abba ML, Delecluse S, Delecluse H-J, Allgayer H. Metagenomic analysis of primary colorectal carcinomas and their metastases identifies potential microbial risk factors. Mol Oncol. 2021;15(12):3363–3384. doi:10.1002/1878-0261.13070.
- Hannigan GD, Duhaime MB, Ruffin MT, Koumpouras CC, Schloss PD, Miller JF. Diagnostic potential and interactive dynamics of the colorectal cancer virome. mBio. 2018;9(6):e02248–18. doi:10.1128/mBio.02248-18.
- Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, Ajami NJ, Wong MC, Ghazaryan A, Valentine JF, et al. Expansion of bacteriophages is linked to aggravated intestinal inflammation and colitis. Cell Host & Microbe. 2019;25(2):285–299.e8. doi:10.1016/j.chom.2019.01.008.
- Mathieu A, Dion M, Deng L, Tremblay D, Moncaut E, Shah SA, Stokholm J, Krogfelt KA, Schjørring S, Bisgaard H, et al. Virulent coliphages in 1-year-old children fecal samples are fewer, but more infectious than temperate coliphages. Nat Commun. 2020;11(1):378. doi:10.1038/s41467-019-14042-z.
- de Jonge PA, Wortelboer K, Scheithauer TPM, van den Born B-J, Zwinderman AH, Nobrega FL, Dutilh BE, Nieuwdorp M, Herrema H. Gut virome profiling identifies a widespread bacteriophage family associated with metabolic syndrome. Nat Commun. 2022;13(1):3594. doi:10.1038/s41467-022-31390-5.
- Nayfach S, Páez-Espino D, Call L, Low SJ, Sberro H, Ivanova NN, Proal AD, Fischbach MA, Bhatt AS, Hugenholtz P, et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat Microbiol. 2021;6(7):960–970. doi:10.1038/s41564-021-00928-6.
- Borodovich T, Shkoporov AN, Ross RP, Hill C. Phage-mediated horizontal gene transfer and its implications for the human gut microbiome. Gastroenterol Rep (Oxf). 2022;10:goac012. doi:10.1093/gastro/goac012.
- Ma T, Ru J, Xue J, Schulz S, Mirzaei MK, Janssen K-P, Quante M, Deng L. Differences in gut virome related to Barrett Esophagus and esophageal adenocarcinoma. Microorganisms. 2021;9(8):1701. doi:10.3390/microorganisms9081701.
- Mangalea MR, Paez-Espino D, Kieft K, Chatterjee A, Chriswell ME, Seifert JA, Feser ML, Demoruelle MK, Sakatos A, Anantharaman K, et al. Individuals at risk for rheumatoid arthritis harbor differential intestinal bacteriophage communities with distinct metabolic potential. Cell Host & Microbe. 2021;29(5):726–739.e5. doi:10.1016/j.chom.2021.03.020.
- Hockenberry AJ, Wilke CO. BACPHLIP: predicting bacteriophage lifestyle from conserved protein domains. PeerJ. 2021;9:e11396. doi:10.7717/peerj.11396.
- Wu S, Fang Z, Tan J, Li M, Wang C, Guo Q, Xu C, Jiang X, Zhu H. DeePhage: distinguishing virulent and temperate phage-derived sequences in metavirome data with a deep learning approach. GigaScience. 2021;10(9):giab056. doi:10.1093/gigascience/giab056.
- Peng X, Ru J, Mirzaei MK, Deng L. Replidec - use naive Bayes classifier to identify virus lifecycle from metagenomics data. bioRxiv. 2022;2022–07.
- Sugimura N, Li Q, Chu ESH, Lau HCH, Fong W, Liu W, Liang C, Nakatsu G, Su ACY, Coker OO, et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut. 2021;71(10):2011–2021. doi:10.1136/gutjnl-2020-323951.
- Zhao J, Wang Y, Wang J, Lv M, Zhou C, Jia L, Geng W. Lactobacillus kefiranofaciens ZW18 from kefir enhances the anti-tumor effect of anti-programmed cell death 1 (PD-1) immunotherapy by modulating the gut microbiota. Food Funct. 2022;13(19):10023–10033. doi:10.1039/D2FO01747D.
- Kahouli I, Malhotra M, Westfall S, Alaoui-Jamali MA, Prakash S. Design and validation of an orally administrated active L. fermentum-L. acidophilus probiotic formulation using colorectal cancer ApcMin/+ mouse model. Appl Microbiol Biotechnol. 2017;101(5):1999–2019. doi:10.1007/s00253-016-7885-x.
- Zitvogel L, Daillère R, Roberti MP, Routy B, Kroemer G. Anticancer effects of the microbiome and its products. Nat Rev Microbiol. 2017;15(8):465–478. doi:10.1038/nrmicro.2017.44.
- Hasan R, Bose S, Roy R, Paul D, Rawat S, Nilwe P, Chauhan NK, Choudhury S. Tumor tissue-specific bacterial biomarker panel for colorectal cancer: bacteroides massiliensis, Alistipes species, Alistipes onderdonkii, bifidobacterium pseudocatenulatum, Corynebacterium appendicis. Arch Microbiol. 2022;204(6):348. doi:10.1007/s00203-022-02954-2.
- Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ, Hackl H, Pfister A, Schilling J, Moser PL, et al. Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host Microbe. 2016;19(4):455–469. doi:10.1016/j.chom.2016.03.007.
- Gupta A, Dhakan DB, Maji A, Saxena R, Vishnu Prasoodanan PK, Mahajan S, Pulikkan J, Kurian J, Gomez AM, Scaria J, et al. Association of flavonifractor plautii, a flavonoid-degrading bacterium, with the gut microbiome of colorectal cancer patients in India. mSystems. 2019;4(6):e00438–19. doi:10.1128/mSystems.00438-19.
- Williamson AJ, Jacobson R, van Praagh JB, Gaines S, Koo HY, Lee B, Chan W-C, Weichselbaum R, Alverdy JC, Zaborina O, et al. Enterococcus faecalis promotes a migratory and invasive phenotype in colon cancer cells. Neoplasia. 2022;27:100787. doi:10.1016/j.neo.2022.100787.
- Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, Liu P, Narrowe AB, Rodríguez-Ramos J, Bolduc B, et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020;48(16):8883–8900. doi:10.1093/nar/gkaa621.
- Singh G, Yadav M, Ghosh C, Rathore JS. Bacterial toxin-antitoxin modules: classification, functions, and association with persistence. Current Res Microbial Sci. 2021;2:100047. doi:10.1016/j.crmicr.2021.100047.
- Coloma-Rivero RF, Gómez L, Alvarez F, Saitz W, Del Canto F, Céspedes S, Vidal R, Oñate AA. The role of the flagellar protein FlgJ in the virulence of Brucella abortus. Front Cell Infect Microbiol. 2020;10:178. doi:10.3389/fcimb.2020.00178.
- Amon JD, Yadav AK, Ramirez-Guadiana FH, Meeske AJ, Cava F, Rudner DZ, Henkin TM. SwsB and SafA are required for CwlJ-Dependent spore germination in Bacillus subtilis. J Bacteriol. 2020;202(6):e00668–19. doi:10.1128/JB.00668-19.
- Mintz KP. Identification of an extracellular matrix protein adhesin, EmaA, which mediates the adhesion of actinobacillus actinomycetemcomitans to collagen. Microbiol. 2004;150(8):2677–2688. doi:10.1099/mic.0.27110-0.
- Raghunathan D, Wells TJ, Morris FC, Shaw RK, Bobat S, Peters SE, Paterson GK, Jensen KT, Leyton DL, Blair JMA, et al. SadA, a trimeric autotransporter from Salmonella enterica Serovar Typhimurium, can promote biofilm formation and provides limited protection against infection. Infect Immun. 2011;79(11):4342–4352. doi:10.1128/IAI.05592-11.
- Weidensdorfer M, Ishikawa M, Hori K, Linke D, Djahanschiri B, Iruegas R, Ebersberger I, Riedel-Christ S, Enders G, Leukert L, et al. The Acinetobacter trimeric autotransporter adhesin Ata controls key virulence traits of Acinetobacter baumannii. Virulence. 2019;10(1):68–81. doi:10.1080/21505594.2018.1558693.
- Wu Y, Jiao N, Zhu R, Zhang Y, Wu D, Wang A-J, Fang S, Tao L, Li Y, Cheng S, et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat Commun. 2021;12(1):3063. doi:10.1038/s41467-021-23265-y.
- Du X, Li Q, Tang Z, Yan L, Zhang L, Zheng Q, Zeng X, Chen G, Yue H, Li J, et al. Alterations of the gut microbiome and fecal metabolome in colorectal cancer: implication of intestinal metabolism for tumorigenesis. Front Physiol. 2022;13:854545. doi:10.3389/fphys.2022.854545.
- Liu X, Mao B, Gu J, Wu J, Cui S, Wang G, Zhao J, Zhang H, Chen W. Blautia—a new functional genus with potential probiotic properties? Gut Microbes. 2021;13(1):1875796. doi:10.1080/19490976.2021.1875796.
- Zagato E, Pozzi C, Bertocchi A, Schioppa T, Saccheri F, Guglietta S, Fosso B, Melocchi L, Nizzoli G, Troisi J, et al. Endogenous murine microbiota member faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nature Microbiology. 2020;5(3):511–524. doi:10.1038/s41564-019-0649-5.
- Osman MA, Neoh H, Ab Mutalib N-S, Chin S-F, Mazlan L, Raja Ali RA, Zakaria AD, Ngiu CS, Ang MY, Jamal R. Parvimonas micra, peptostreptococcus stomatis, Fusobacterium nucleatum and Akkermansia muciniphila as a four-bacteria biomarker panel of colorectal cancer. Sci Rep. 2021;11(1):2925. doi:10.1038/s41598-021-82465-0.
- Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16(12):713–732. doi:10.1038/s41575-019-0189-8.
- Li Y, Zhang F, Zheng H, Kalasabail S, Hicks C, Fung KY, Preaudet A, Putoczki T, Beretov J, Millar EKA, et al. Fecal DNA virome is associated with the development of colorectal neoplasia in a murine model of colorectal cancer. Pathogens. 2022;11(4):457. doi:10.3390/pathogens11040457.
- Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’Regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP, et al. Whole-virome analysis Sheds light on viral Dark Matter in inflammatory Bowel disease. Cell Host & Microbe. 2019;26(6):764–778.e5. doi:10.1016/j.chom.2019.10.009.
- Kim M-S, Bae J-W. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 2018;12(4):1127–1141. doi:10.1038/s41396-018-0061-9.
- Henrot C, Petit M-A. Signals triggering prophage induction in the gut microbiota. Mol Microbiol. 2022;118(5):494–502. doi:10.1111/mmi.14983.
- Algood HMS. T Cell cytokines impact epithelial Cell responses during Helicobacter pylori infection. J Immunol. 2020;204(6):1421–1428. doi:10.4049/jimmunol.1901307.
- Duerkop BA, Kleiner M, Paez-Espino D, Zhu W, Bushnell B, Hassell B, Winter SE, Kyrpides NC, Hooper LV. Murine colitis reveals a disease-associated bacteriophage community. Nat Microbiol. 2018;3(9):1023–1031. doi:10.1038/s41564-018-0210-y.
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune System. Int Rev Immunol. 2011;30(1):16–34. doi:10.3109/08830185.2010.529976.
- Kamada N, Chen GY, Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14(7):685–690. doi:10.1038/ni.2608.
- Sartorius R, Trovato M, Manco R, D’Apice L, De Berardinis P. Exploiting viral sensing mediated by toll-like receptors to design innovative vaccines. NPJ Vaccines. 2021;6(1):1–15. doi:10.1038/s41541-021-00391-8.
- Braumüller H, Mauerer B, Andris J, Berlin C, Wieder T, Kesselring R. The cytokine network in colorectal cancer: implications for new treatment strategies. Cells. 2023;12(1):138. doi:10.3390/cells12010138.
- Boling L, Cuevas DA, Grasis JA, Kang HS, Knowles B, Levi K, Maughan H, McNair K, Rojas MI, Sanchez SE, et al. Dietary prophage inducers and antimicrobials: toward landscaping the human gut microbiome. Gut Microbes. 2020;11(4):721–734. doi:10.1080/19490976.2019.1701353.
- Wendling CC, Refardt D, Hall AR. Fitness benefits to bacteria of carrying prophages and prophage-encoded antibiotic-resistance genes peak in different environments. Evolution. 2021;75(2):515–528. doi:10.1111/evo.14153.
- Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver–passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10(8):575–582. doi:10.1038/nrmicro2819.
- Nguyen S, Baker K, Padman BS, Patwa R, Dunstan RA, Weston TA, Schlosser K, Bailey B, Lithgow T, Lazarou M, et al. Bacteriophage transcytosis provides a mechanism to cross epithelial Cell layers. mBio. 2017;8(6):e01874–17. doi:10.1128/mBio.01874-17.
- Ru J, Khan Mirzaei M, Xue J, Peng X, Deng L. ViroProfiler: a containerized bioinformatics pipeline for viral metagenomic data analysis. Gut Microbes. 2023;15(1):2192522. doi:10.1080/19490976.2023.2192522.
- Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. doi:10.1093/bioinformatics/bty560.
- Handley SA Virus+ sequence masked mouse reference genome (GRCm38) [internet]. 2020. https://zenodo.org/record/4116248
- Bushnell B. Bbmap short-read aligner, and other bioinformatics tools. 2015.
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSpades: a new versatile metagenomic assembler. Genome Res. 2017;27(5):824–834. doi:10.1101/gr.213959.116.
- Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One. 2016;11(10):e0163962. doi:10.1371/journal.pone.0163962.
- Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides NC. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol. 2021;39(5):578–585. doi:10.1038/s41587-020-00774-7.
- Guo J, Bolduc B, Zayed AA, Varsani A, Dominguez-Huerta G, Delmont TO, Pratama AA, Gazitúa MC, Vik D, Sullivan MB, et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. 2021;9(1):37. doi:10.1186/s40168-020-00990-y.
- Kieft K, Zhou Z, Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020;8(1):90. doi:10.1186/s40168-020-00867-0.
- Mirdita M, Steinegger M, Breitwieser F, Söding J, Levy Karin E, Kelso J. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics. 2021;37(18):3029–3031. doi:10.1093/bioinformatics/btab184.
- Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol. 2019;37(6):632–639. doi:10.1038/s41587-019-0100-8.
- Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics. 2012;28(17):2223–2230. doi:10.1093/bioinformatics/bts429.
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923.
- Roux S, Camargo AP, Coutinho FH, Dabdoub SM, Dutilh BE, Nayfach S, Tritt A. iPhop: an integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLoS Biol. 2023;21(4):e3002083. doi:10.1371/journal.pbio.3002083.
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi:10.1038/nmeth.3869.
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi:10.1038/s41587-019-0209-9.
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–6. doi:10.1093/nar/gks1219.
- Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11(1):3514. doi:10.1038/s41467-020-17041-7.
- Peschel S, Müller CL, von Mutius E, Boulesteix A-L, Depner M. NetCoMi: network construction and comparison for microbiome data in R. Brief Bioinform. 2021;22(4):bbaa290. doi:10.1093/bib/bbaa290.