
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Dysregulation of both the gut barrier and microbiota (dysbiosis) promotes susceptibility to and severity of Inflammatory Bowel Diseases (IBD). Leaky gut and dysbiosis often coexist; however, potential interdependence and molecular regulation are not well understood. Robust expression of claudin-3 (CLDN3) characterizes the gut epithelium, and studies have demonstrated a positive association between CLDN3 expression and gut barrier maturity and integrity, including in response to probiotics. However, the exact status and causal role of CLDN3 in IBD and regulation of gut dysbiosis remain unknown. Analysis of mouse and human IBD cohorts helped examine CLDN3 expression in IBD. The causal role was determined by modeling CLDN3 loss of expression during experimental colitis. 16S sequencing and in silico analysis helped examine gut microbiota diversity between Cldn3KO and WT mice and potential host metabolic responses. Fecal microbiota transplant (FMT) studies were performed to assess the role of gut dysbiosis in the increased susceptibility of Cldn3KO mice to colitis. A significant decrease in CLDN3 expression characterized IBD and CLDN3 loss of expression promoted colitis. 16S sequencing analysis suggested gut microbiota changes in Cldn3KO mice that were capable of modulating fatty acid metabolism and oxidative stress response. FMT from naïve Cldn3KO mice promoted colitis susceptibility in recipient germ-free mice (GFM) compared with GFM-receiving microbiota from WT mice. Our data demonstrate a critical role of CLDN3 in maintaining normal gut microbiota and inflammatory responses, which can be harnessed to develop novel therapeutic opportunities for patients with IBD.
Introduction
Gastrointestinal homeostasis depends on the establishment and preservation of gut compartmentalization, in which a single layer of epithelial cells separates the luminal microbiome from the rest of the body. This gastrointestinal epithelial lining forms a permeable barrier that is highly selective and continuously prevents the translocation of luminal antigens to other locations within the body. Tight junctions (TJ), which are intercellular junctions located at the apical end of the epithelial cell layer, are considered critical for regulating the paracellular movement of fluid and solutes and separating the luminal antigens and proinflammatory contents from the body.Citation1,Citation2 Altered TJ structure and epithelial permeability have been observed in inflammatory bowel disease (IBD).Citation3 Accordingly, impaired gut barrier function is positively associated with gut inflammation, and defects in intestinal epithelial barrier function characterize IBD.Citation3–5 However, recent studies have suggested that regulation of tight junction structure and function is rather complex.Citation3,Citation6,Citation7 Claudin proteins are integral to both structure and function of TJs; however, they also perform non-canonical functions, as genetic manipulation of claudin proteins in the gut epithelium has resulted in contrasting outcomes.Citation8–13 Thus, for therapeutic advantages, it is important to clearly understand the role of specific claudin proteins in gut homeostasis and in promoting IBD.
Claudin-3 (CLDN3) is one of the most highly expressed claudin proteins in the gut and shows a gradient with the highest expression at the crypt top among differentiated colonocytes and the lowest expression at the crypt bottom among poorly differentiated colonocytes.Citation13 In addition, CLDN3 serves as a receptor for Clostridium perfringens enterotoxin (CPE) and has been investigated extensively for this role.Citation14,Citation15 Further studies using IFN-γ, TNF-α, and IL-1β have shown that proinflammatory cytokines downregulate CLDN3 expression in the gut epithelium.Citation16–18 A seminal study using a murine model of postnatal gut maturation and probiotic use further showed that CLDN3 expression increases with gut maturation and barrier integrity.Citation19 Using Cldn3KO mice, we have reported that the loss of CLDN3 expression results in a hyperpermeable gut.Citation13 A leaky gut promotes IBD.Citation3,Citation4 However, the causal role of CLDN3 in IBD remains unclear.
The gut microbiome plays an important role in maintaining gut homeostasis and in regulating several essential functions, including host metabolism, synthesis of essential vitamins, and protection against pathogens.Citation20,Citation21 Dysbiosis is defined as a compositional and functional alteration in the microbiome of individuals with pathological conditions compared to healthy subjects.Citation20 A recent report further suggested that gut dysbiosis is not simply a result of inflammation; instead, the dysbiotic microbiome is functionally defective and contributes to inflammation.Citation22 Genome-wide association studies (GWAS) further identified links between inflammation and genes involved in microbial signaling, underlining the role of gut microbes in inflammatory gastrointestinal pathogenesis.Citation23–25 The clinical benefit of antibiotics or fecal stream diversion also supports the pathological role of bacteria in these disease states.Citation26,Citation27 Accumulating evidence suggests that intestinal inflammation alters the microbial community structure.Citation28–30 However, the role of gut barrier dysregulation in gut dysbiosis is poorly understood.
In the current study, we show that CLDN3 expression is downregulated to its nadir in IBD patients and murine models of colitis, and the loss of CLDN3 expression promotes the development of colitis and disease severity. We further show that the gut microbiome in Cldn3KO mice plays a major role in sensitivity to colitis, emphasizing the therapeutic significance of CLDN3 in promoting intestinal barrier maturity/integrity as well as maintaining normal gut microbiota.
Results
CLDN3 expression is markedly downregulated in IBD patients and murine models of colitis
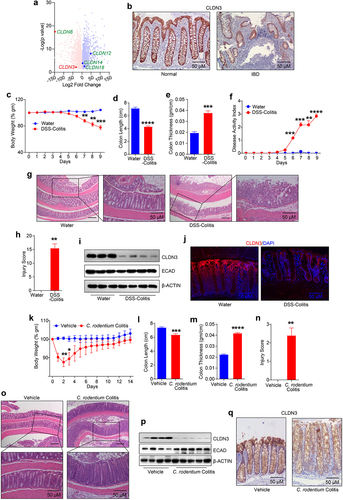
To determine the clinical relevance of CLDN3 expression in IBD, we first performed an in-silico analysis of CLDN3 mRNA expression in published IBD patient cohorts (GEO database; see Methods). The results demonstrated a significant downregulation of CLDN3 transcripts in IBD patients compared to that in healthy individuals (). Immunohistochemical analysis of biopsy specimens from patients with IBD showed a similar downregulation of CLDN3 protein expression (). We next examined status of CLDN3 expression in a murine model of colitis. The mice were subjected to DSS (Dextran Sodium Sulfate) and C. rodentium colitis. For DSS-induced colitis, mice administered dextran sulfate sodium (DSS; 2.5% (w/v)) in drinking water were evaluated for colitis-related parameters, including body weight loss, colon length, colon thickness, disease activity index (DAI) and histological grading (by a pathologist) to confirm colitis (). For C. rodentium colitis, mice received oral gavage of 5 × 108 cfu/100 µl C. rodentium (). Immunoblot analysis using total tissue lysates from the distal colon showed a sharp decrease in CLDN3 expression in DSS-treated mice compared to that in control mice. E-cadherin (ECAD) expression was not significantly affected in these samples (). We found a similar downregulation of CLDN3 expression in the colons of mice subjected C. rodentium colitis (). Overall, our data established that CLDN3 expression was sharply downregulated in IBD and murine models of colitis.
Figure 1. Robust decrease in CLDN3 expression characterize IBD patients and mice subjected to colitis.
CLDN3 loss-of-expression promotes colitis severity
In light of the above data, we sought to determine whether loss of CLDN3 expression in IBD is simply a bystander effect or has a causal role in IBD pathophysiology. We modeled CLDN3 loss in IBD patients using Cldn3KO mice and subjected these mice to DSS in their drinking water. Because of the susceptibility of these mice to higher doses of DSS (data not shown), we used 1% DSS (w/v) over an extended period of 11 days. Compared to WT littermate mice, we found that body weight loss in Cldn3KO mice started after day eight and was significantly lower on day-10 and −11 compared to WT-mice (). The colon length and thickness in DSS-treated Cldn3KO mice further suggested significant colonic atrophy and edema compared to those in WT mice (). DAI and histopathological analysis of the tissue sections of the colon, based on examination of epithelial degeneration, disappearance of crypts, and inflammatory cell infiltration, further revealed that DSS-treated Cldn3KO mice had severe mucosal damage compared to WT mice (). Considering that IBD is a chronic inflammatory condition, we further examined whether Cldn3 deficient mice were also susceptible to chronic colitis. For this purpose, we subjected Cldn3KO and WT mice to three cycles of DSS administration (5 days/cycle), followed by regular drinking water (16 days/cycle). As shown in , significant altrations in colon length and thickness were observed in DSS-treated Cldn3KO mice compared to WT mice. The cumulative injury score and histopathology analysis further supported colitis severity in Cldn3KO mice compared to WT mice () .
Figure 2. Cldn3KO mice are susceptible to colitis.
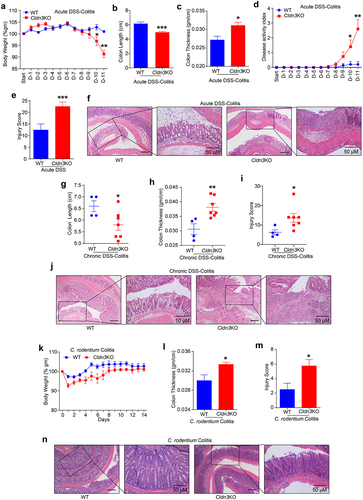
However, IBD is a complex disease, and none of the murine models represents the true pathobiology of human diseases. Therefore, we further examined if Cldn3 deficient mice were also susceptible to C. rodentium colitis, which is commonly used to model IBD.Citation10 Similar to DSS-colitis, when subjected to C. rodentium colitis (for 14 days), significantly altered body weight (% change) and colonic thickness were observed in Cldn3KO mice compared to WT mice (). To examine whether altered colonic thickness correlated with disease pathobiology, H&E-stained colon sections 14 days post-infection with C. rodentium were analyzed. Based on the pathologist scoring of epithelial injury, acute inflammation, chronic inflammation, and depth of inflammation, Cldn3KO mice showed significantly higher pathological scores than their WT littermates (). Overall, these data affirmed the causal role of CLDN3 in maintaining normal gut homeostasis, and that the loss of CLDN3 promotes colitis.
CLDN3 loss-of-expression modulates mucosal inflammatory cytokines
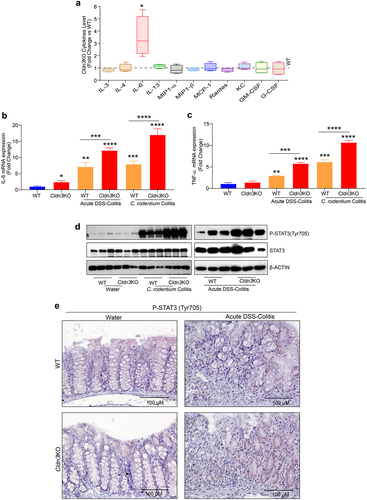
We previously reported that the gut in Cldn3KO mice is leakier than that in WT mice and shows spontaneous immune cell infiltration.Citation13 Evidence suggests that resident infiltrating immune cells in the gut secrete cytokines and chemokines.Citation31 Thus, we hypothesized that inflammatory cytokine expression would be dysregulated in Cldn3KO mice. Interestingly, a detailed immune analysis using total colon lysates (distal colon) and Multiplex Cytokine ELISA revealed a significant deregulation of basal mucosal immune homeostasis in Cldn3KO mice compared to naïve mice. We found that IL-6 levels were significantly upregulated in Cldn3KO mice compared to those in WT controls (). There were no changes in IL-3, IL4, MIP-1α/β, IL-13, MCP-1, Rantes, KC, GM-CSF, or G-CSF levels in Cldn3KO mice compared to WT mice (). In summary, Cldn3KO mice exhibited a specific increase in the expression of IL-6 in their colon.
Figure 3. Deletion of Cldn3 expression exhibits deregulated inflammatory signaling.
CLDN3 expression suppresses inflammatory cytokine production and inflammatory signaling pathways during colitis
Considering that IL-6 expression was upregulated in naïve Cldn3KO mice, we further examined whether the production of TNF-α, a proinflammatory cytokine targeted in treating IBD, is also altered in Cldn3KO mice under conditions of colitis. We measured cytokine levels in colonic tissue harvested from the mice subjected to DSS and C. rodentium colitis. Interestingly, upon colitis induction by DSS or C. rodentium infection, the loss of CLDN3 expression significantly exacerbated the expression of TNF-α and IL-6 (). Considering the persistent increase in IL-6 expression and the fact that STAT3 activation is associated with IBD, we further evaluated the status of STAT3 activation in colon tissues of experimental mice.Citation32,Citation33 We examined STAT3 activation by western blot analysis using an antibody specific for STAT3 phosphorylated at Tyr705. As shown in , we found a significant upregulation of STAT3Y705 expression in Cldn3KO mice compared to WT mice when subjected to colitis. Immunohistochemical analysis to determine the source of activated STAT3 suggested increased activation of STAT3 in both epithelial and immune cells in Cldn3KO mice compared to WT mice (). In summary, our data suggest that CLDN3 loss of expression promotes activation of key proinflammatory cytokines and signaling pathways during colitis.
Cldn3 loss-of-expression promotes susceptibility to colitis in manners dependent on the gut microbiota
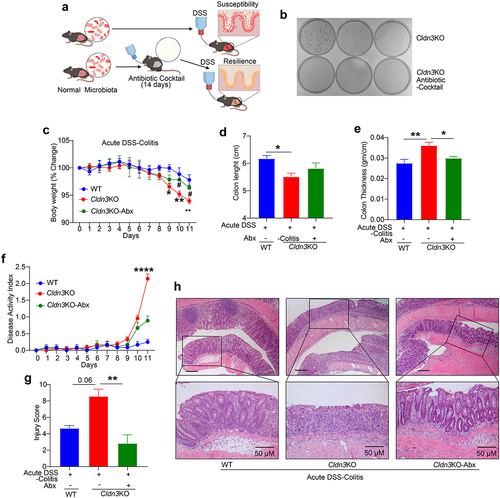
The gut microbiome has emerged as a principal factor in the development and function of the immune system, and published research has linked changes in the gut microbiota to IBD risk and disease severity.Citation20 Probiotic treatment promotes CLDN3 expression to promote gut barrier integrity.Citation19 Therefore, we examined whether gut microbiome in Cldn3KO mice was responsible for their susceptibility to colitis. Cldn3KO mice were treated with a broad-spectrum antibiotic (Abx) cocktail in drinking water for 14 days before the induction of experimental colitis to create pseudo-germ-free mice (). As shown in , Abx treatment resulted in robust depletion of the gut microbiota in treated mice. Based on the body weight change and behavior including drinking and eating habits, Abx treatment had no adverse effects on the health of Cldn3KO mice. When subjected to DSS-induced colitis, these pseudo-germ-free Cldn3KO mice showed striking improvements in body weight loss, colon atrophy or shortening, and edema compared to Cldn3KO mice that were subjected to DSS-induced colitis without Abx treatment (). DAI and the blinded histological colitis injury scores in Abx-treated Cldn3KO mice were also significantly lower than Cldn3KO mice without Abx (). The histological evaluation and injury scoring based on H&E analysis further showed that the Abx-treated Cldn3KO mice had more mucus-filled crypts and reduced inflammatory cell infiltrates compared to Cldn3KO mice not treated with Abx (). These findings suggest that susceptibility to DSS colitis in Cldn3KO mice is dependent on the gut microbiota.
Figure 4. Pseudo-germ-free (microbial depletion) Cldn3KO mice show reduced severity of DSS-colitis.
Cldn3KO mice harbor a more diverse microbial community than WT-mice
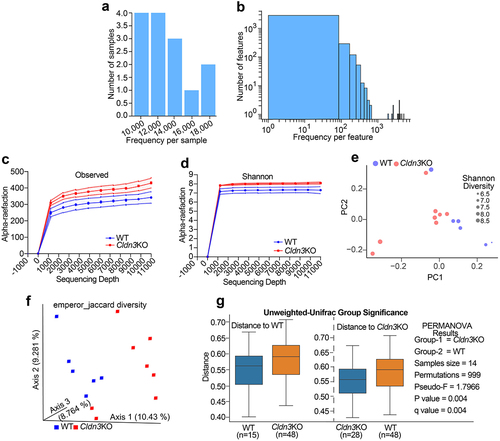
Based on the above data, the gut microbiome of Cldn3KO mice was analyzed to understand how they differ from that of WT mice and the potential mechanisms by which they may affect the host. We collected stool samples from Cldn3KO (8 mice) and WT (6 mice) littermate mice and performed 16S rRNA gene sequencing. The resultant data were subjected to a bioinformatics analysis pipeline, which was reflected by the amplicon sequencing variant (ASV) and the frequency distribution in each sample. ) show the distribution of the ASVs frequency in each sample and the observed frequency per feature, respectively. Interestingly, the Observed and Shannon rarefaction curves showed less diversity in WT littermates than in Cldn3KO mice (). Principal coordinate analysis based on Shannon diversity suggested no significant differences between groups (). Jaccard and unweighted-UniFrac β-diversity analyses were performed to compare the gut microbiota community structures across the groups. As shown in , we found significant dissimilarities between the microbiomes of Cldn3KO and WT samples using Jaccard and unweighted-UniFrac β-diversity analyses. Taken together, these data suggest a distinct microbiome in Cldn3KO mice compared to that in WT littermates.
Figure 5. Loss of endogenous Cldn3 expression alters gut microbial composition.
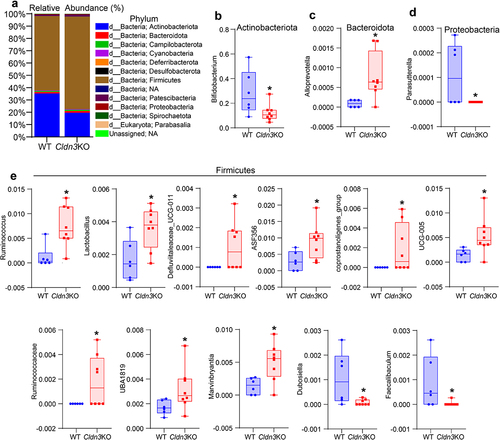
Relative abundance of bacterial communities in Cldn3KO mice differs significantly from that in WT mice
Given that Cldn3KO mice exhibit an altered gut microbiota compared to WT animals, we further explored the taxonomic composition of these microbiomes, which showed that Firmicutes (Bacillota) and Actinobacteria (Actinomycetota) were the predominant phyla in all samples from Cldn3KO and WT mice (). However, the proportions of taxa from these phyla differed between the two groups. Interestingly, the Cldn3KO group, compared to the WT group, exhibited increased levels of the phylum Firmicutes (75.08% vs. 64.41% WT); however, Actinobacteria was reduced from 35.30% (Cldn3KO) to 19.65% (vs. WT; ). Overall, this observation indicates an altered Firmicutes/Actinobacteria ratio in these mice. We further found ten genera from Bacteroidetes (Bacteriodota) (1) and Firmicutes (9), which were significantly increased in Cldn3KO mice compared to WT mice. In contrast, four genera from Firmicutes (2), Proteobacteria (pseudomonadota) (1), and Actinobacteria (1) were lower in Cldn3KO mice than in WT (). Overall, the above data validated a significant alteration in gut microbiota community profiles in Cldn3KO mice compared with WT controls.
Figure 6. Relative abundance of bacterial communities in Cldn3KO differ from WT.
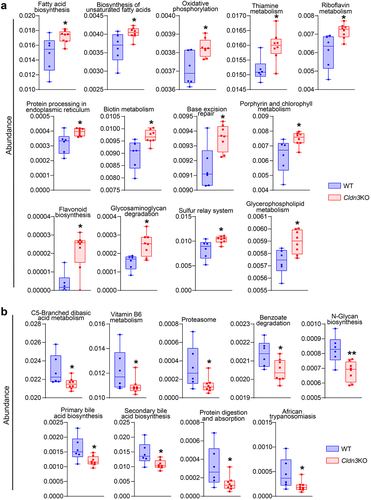
Predicted bacterial functions in Cldn3KO mice implicated significant metabolic changes similar to IBD patients
Recent studies have highlighted the significance of microbial metabolites in the causal interplay between microbiome and host homeostasis.Citation34 Therefore, to determine how gut dysbiosis in Cldn3KO mice may affect their susceptibility to colitis, we determined bacterial gene functions via 16S rRNA gene-based microbial compositions using the R-packages algorithm and the KEGG annotation database. presents the significantly upregulated functional pathways associated with changes in the gut microbiota in Cldn3KO mice, including fatty acid metabolism and oxidative phosphorylation, which are known to be altered in IBD patients and promote intestinal inflammation.Citation35 The downregulated pathways included the N-glycan, primary, and secondary bile acid biosynthesis pathways compared with WT mice. The metabolism of vitamin B6 and C5-Branched dibasic acid was also downregulated in the microbiomes of Cldn3KO mice (). Collectively, these data suggest that predicted alterations in gut microbial functions due to Cldn3 deficiency could potentially lead to profound changes in bacterial metabolic profiles, which can further affect host metabolism in a manner similar to the metabolic changes in IBD patients.
Figure 7. Gut dysbiosis in Cldn3KO mice leads to altered host metabolism.
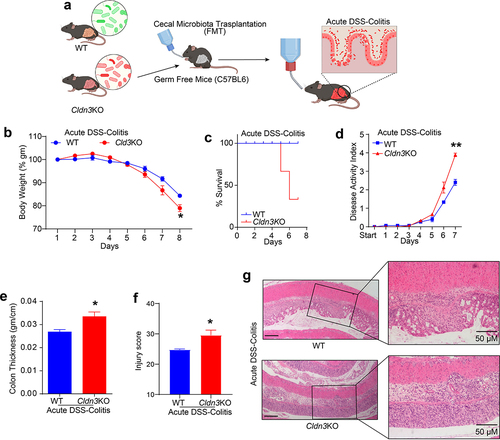
Fecal microbiota transplant (FMT) from Cldn3KO mice to germ-free mice (GFM) promotes colitis
Considering that Cldn3KO mice are susceptible to colitis and display gut dysbiosis, we hypothesized that the altered microbiome in these mice promotes susceptibility to colitis. To test this hypothesis, we transplanted fecal microbiota from Cldn3KO and WT mice into germ-free recipient mice (). After two weeks, no clinical signs of diarrhea or colonic inflammation were noted in the recipient mice. However, when subjected to DSS-induced colitis after the two-week inoculation period, colitis-associated parameters such as body weight loss, overall survival, and disease activity index suggested that recipient mice colonized with the microbiota from Cldn3KO mice had severe colitis compared to those colonized with the microbiota from WT mice (). We also observed a significant increase in colon edema measured by the weight/length ratio in DSS-treated recipient mice harboring a Cldn3KO versus WT microbiota (). Blinded histopathological analysis further supported the severity of inflammation in DSS-treated recipient mice with Cldn3KO versus WT mice microbiota (). Taken together, our data suggest that the gut microbiota from WT and Cldn3KO mice have differential effects on colitis.
Figure 8. Fecal microbiota transplantation (FMT) from Cldn3KO mice to germ-free C57BL/6 mice induces susceptibility to DSS colitis.
Epithelial–microbial interaction drives inflammatory signaling in Cldn3KO mice
To further elucidate the inflammation-promoting potential of dysbiosis in Cldn3KO mice, confluent monolayers of Caco-2 cells were exposed to the fecal microbiota content from both Cldn3KO and WT mice (). As shown in , phosphorylation of STAT3 (y705) and NF-kB (s536), a measure of proinflammatory signaling, was substantially upregulated in Caco-2 cells exposed to fecal microbiota from Cldn3KO mice compared to WT mice. Further analysis for the source of such changes revealed marked upregulation of IL-6 and TNF- expressions, which were higher in cells exposed to the fecal microbial content from Cldn3KO mice compared to WT mice (). We further examined whether observed upregulation of IL-6 and TNF-α were compartmentalized by determining their release in the apical versus basal compartments of the transwell chambers where Caco-2 cells were cultured. As shown in , expression of both TNF-α and IL-6 was higher in the apical compartment than the basal compartment. In light of these findings, we further examined whether exposure of the Caco-2 cells to the gut microbial contents from the WT versus Cldn3KO mice affected the barrier integrity. Trans-epithelial resistance (TEER) was measured in control and treated cells over 48 hours. The results using Chopstick Electrode Set (EVOM) showed a significant decrease in TEER in cells treated with the gut microbial content from WT-mice which was further downregulated significantly in cells treated with Cldn3KO mice fecal microbiota (). Our data suggest a key role of CLDN3 in gut barrier integrity. Therefore, we wondered if CLDN3 expression was changed in response to the above treatment. Immunoblotting using the total cell lysates showed that CLDN3 expression was indeed downregulated in cells treated with WT-mice fecal microbial content which appeared exacerbated in cells exposed to the Cldn3KO mice fecal
microbial content (). In same lysates, expression of claudin-1, yet other tight junction protein, was not changed. Our additional analysis to examine possible changes in cell proliferation (Cyclin-D1) and death (Cleaved Caspase-3) further showed an increase in the expression of Cleaved Caspase-3 in cells treated with WT-mice microbial contents which was further increased in cells treated with Cldn3KO mice microbial contents. Expression of Cyclin-D1 remained largely unaltered (). Overall, our data suggested that fecal microbiota induced proinflammatory signaling, barrier dysfunction and cell death in IECs is potentiated by the fecal microbiota content in Cldn3KO mice, similar to colitis in vivo.
Figure 9. Cldn3KO microbiota potentiates inflammatory signaling in intestinal epithelial cells.
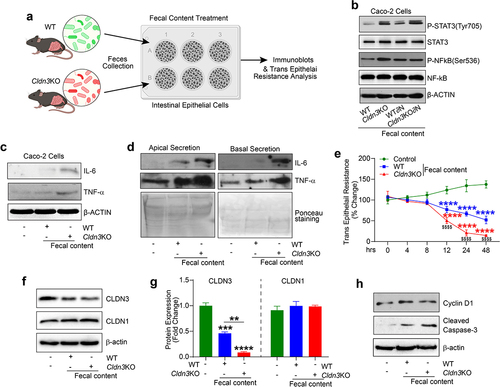
Signaling pathways and gene expression modulated by an altered gut microbiota show significant association with CLDN3 expression in IBD patients
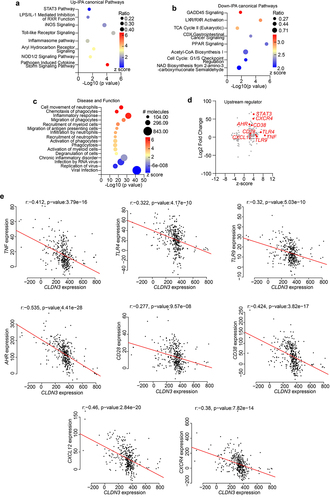
To determine the clinical significance of our findings, we performed an unbiased in silico meta-analysis of published high-throughput transcriptomic data from patients with IBD (see Methods). We applied the QIAGEN Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) tool to the DEGs generated using public databases and determined the up- and down-regulated DEGs. In total, 131 significant canonical pathways were identified, among which the upregulated pathways included the STAT3 signaling pathway, LPS/IL-1 Mediated Inhibition of RXR function, iNOS Signaling, Toll-like Receptor Signaling, Inflammasome pathway, aryl hydrocarbon receptor pathway, NOD1/2 signaling pathway, and pathogen-induced cytokine storm signaling pathway (). There were 41 significantly downregulated pathways, including LXR/RXR Activation, TCA Cycle II, CDX Gastrointestinal cancer signaling, PPAR Signaling, and Cell Cycle: G1/S Checkpoint Regulation (). IPA disease and function tool analysis further indicated that bacteria-associated diseases and functions were upregulated in patients with IBD (). The upstream regulator analysis tool is a novel function in IPA analysis that can identify potential upstream regulators, including transcription factors, transmembrane receptors, and any gene or small molecule that has been observed experimentally to affect gene expression, by analyzing the linkage to the DEGs through coordinated expression. STAT3, CD28, CD38, AHR, CXCR12, CXCR4, TNF, and TLR4/9 have also been identified as critical upstream regulators of these genes.Citation36 These genes are known to be involved in microbiome-associated signaling pathways, diseases, and functions ().Citation37 One of our key findings in IPA upstream regulator analysis was a robust upregulation in host genes that can be affected by the microbiome in the IBD patient dataset ().Citation37 In IBD, the expression of upstream regulatory genes is potentially associated with intestinal inflammation and disease severity.Citation36 Therefore, we examined the potential association between CLDN3 and these genes in IBD patients. Using the same patient cohort utilized in the analysis described in , Spearman’s correlation demonstrated a significant inverse correlation between CLDN3 and TNF, TLR4, TLR9, AHR, CD28, CD38, CXCR12, and CXCR4 expression in the patient cohort. Collectively, our analysis suggests that loss of CLDN3 modulates microbiome-associated signaling pathways in patients with IBD.
Figure 10. Microbiome associated signaling is altered in IBD and corelates with CLDN3 expression.
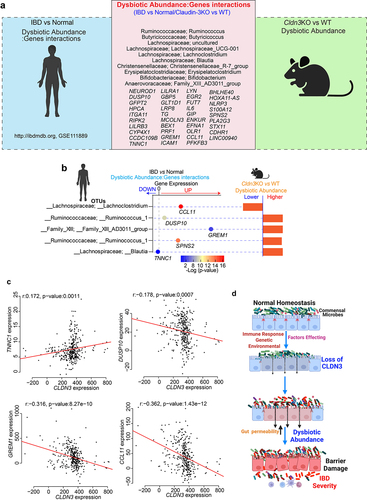
Gut microbe changes in IBD patients overlap with the microbiome changes in Cldn3KO mice
Having demonstrated that the loss of CLDN3 expression in the mouse gut promotes gut dysbiosis and that these mice have an increased susceptibility to colitis, we further examined the relevance of CLDN3 expression in IBD and associated microbiome changes. To this end, we took advantage of a previously published report in which the authors identified significant gene-taxon pairs in the ileum and rectum of IBD patients.Citation37 We compared the dysbiotic taxa from Cldn3KO naïve mice (vs. WT littermates) with taxons from a published study.Citation37 As shown in a Venn diagram (), we found 10 taxons in naïve Cldn3KO mice that aligned positively with dysbiotic taxa in patients with IBD. These dysbiotic taxa included Ruminococcus, Butyricicoccus, Lachnospiraceae_UCG-001, Erysipelatoclostridium, Lachnoclostridium, Blautia, Christensenellaceae_R-7_group, Bifidobacterium, and Family_XIII_AD3011_group (). Further gene/dysbiotic taxon interaction analyses revealed that 39 genes were positively or negatively related to dysbiotic OTUs. Based on the literature, we found genes such IL-6, RIPK2, PLA2G3, CCL11, ICAM1, and NLRP3, which are associated with specific bacterial species present in IBD patients, suggesting that these species are the most susceptible to the activity of these genes.Citation37 Further analysis revealed that at least four taxons of IBD patients overlapped with those of Cldn3KO mice. These taxons were further associated with critical host genes (). We further examined the potential association between CLDN3 and these five genes in IBD patients using Spearman’s correlation. Of these five host genes, four showed a significant association with CLDN3 expression. As shown in , TNNC1 was positively associated with CLDN3, DUSP10, GREM1, and CCL11 were negatively correlated with CLDN3. Overall, these results indicate that the loss of CLDN3 is associated with gut microbial dysbiosis, which promotes inflammation ().
Figure 11. Host-microbiome interactions in IBD patient cohort show overlap with Cldn3KO mice.
Discussion
Despite the knowledge that deregulation of gut barrier function and microbiome helps promote susceptibility to intestinal inflammatory diseases, including IBD, and/or disease severity, the causal interdependence between gut barrier dysfunction and dysbiosis remains poorly understood. While frequent co-existence of a leaky gut and microbiota dysbiosis has often been reported, it primarily relates to disease condition(s), and causal relationships are difficult to interpret. In the present study, we used a comprehensive approach that includes microbial profiling in naïve mice, murine modeling of IBD, and in silico analysis of the high-throughput data for causal integration between IBD patients and Cldn3KO mice. We demonstrate an essential role for CLDN3 in maintaining the normal microbiome. To the best of our knowledge, this is the first report to demonstrate that the loss of CLDN3 expression is sufficient to induce alterations in the gut microbiota that promote susceptibility to IBD as determined by the FMT studies. We previously reported that CLDN3 loss of expression induces gut permeability in mice.Citation13 Taken together, CLDN3 appears to be the link connecting the gut barrier and the microbiota. This postulation is supported by a published report that CLDN3 expression is positively associated with the maturity of the gut barrierCitation19. Moreover, beneficial bacteria (probiotics) promote gut barrier integrity by specifically promoting CLDN3 expression.Citation19 Other studies have shown similar positive association of CLDN3 expression with gut barrier integrity.Citation38 These data, when combined with the fact that CLDN3 expression is markedly suppressed in IBD patients and murine models of colitis, emphasize the significance of promoting CLDN3 in improving the prognosis of IBD.
Importantly, our data showing that CLDN3 loss-of-expression not only promotes gut permeability but also induces gut dysbiosis is significant, as regulation of the gut barrier structure/function is complex.Citation3,Citation6,Citation7 The claudin family of proteins is now accepted as the principal regulator of tight junctions, and the dysregulated expression of multiple claudin proteins has been reported in IBD.Citation39,Citation40 However, related studies, especially those using mice genetically manipulated for specific claudin proteins, have identified non-canonical roles for specific claudin proteins and are possible contributors to intestinal pathobiology. Overexpression of claudin-1 protein in the gut epithelium, to model claudin-1 expression in IBD, promotes colitis by primarily affecting IECs differentiation in Notch- and Wnt-dependent manners.Citation41 Similar murine modeling of claudin-2, the leaky claudin, in mice to determine a causal role in IBD, has presented diverse outcomes depending on the colitis model, which suggests complex roles of claudin-2 in intestinal homeostasis.Citation8,Citation10,Citation11,Citation42 While the status of claudin-4 in IBD is not clear, based on a recent preliminary report, it appears that claudin-4 upregulation may exacerbate colitis by regulating epithelial injury/repair.Citation43 Similarly, results in mice genetically manipulated for claudin-7, yet another high expression of claudin protein in the gut suggests a critical role for claudin-7 in regulating cell-extracellular matrix interactions and stem cell functions.Citation44 Of note, claudin-7 KO mice also demonstrate a dysbiotic gut microbiota, but only after being subjected to DSS-colitis.Citation44 Claudin-15, another claudin protein expressed in the intestine at high levels, is also reported to be downregulated in IBD.Citation45 However, simple genetic manipulation of claudin-15 leads to a mega-intestine phenotype, suggesting that this protein also manifests functions other than tight junctions in the gut.Citation46 Taken together, our data show that CLDN3 loss not only induces a leaky gut phenotype but also promotes gut dysbiosis and susceptibility to IBD, suggesting that its targeting (to promote) can not only correct the gut barrier integrity but also improve dysbiosis and colitis risk. Our previously published report that CLDN3 loss of expression promotes the risk of colitis-associated cancer (CAC) further supports this postulation.Citation13 However, CLDN3 loss also affects the normal homeostasis of other vital organs, including the liver and bile acid secretion.Citation47 Changes in bile acid levels are associated with gut inflammation and colitis.Citation37,Citation48–50 Thus, further studies are needed to delineate the role of the gut epithelium specific changes in CLDN3 expression upon gut dysbiosis and colitis and are part of our ongoing studies.
Although the precise etiology of IBD remains unclear, it is thought that the altered immune homeostasis characterizing IBD can result from crosstalk among genetic susceptibility, barrier dysregulation, and gut microbiota, potentially leading to gut dysbiosis, thus allowing the growth of pathogenic microorganisms.Citation25,Citation37,Citation51,Citation52 Accordingly, gut dysbiosis in IBD is characterized by a decrease in alpha diversity.Citation53 We observed a decrease in alpha diversity in Cldn3KO mice. In addition, Jaccard and unweighted-UniFrac β-diversity analyses showed dissimilarities between the gut microbiome of Cldn3KO mice and WT littermates.Citation37,Citation52 Specifically, the predominant changes were altered ratios of four major phyla, Firmicutes, Actinobacteria, Bacteroidetes, and Proteobacteria, in Cldn3KO mice compared to WT mice. Notably, Bifidobacterium increases with gut maturation and produces short-chain fatty acids (SCFA). Bifidobacterium administration significantly alters the abundance of Lactobacillus and contributes to the remission of intestinal inflammation and injury by constituting a favorable gut ecosystem, possibly modulating regulatory T cells and the SCFA environment.Citation54 Additionally, Bifidobacterium levels are reduced in the gut of IBD patients.Citation55 Most notably, similar to IBD patients, we observed a markedly low abundance of Bifidobacterium in Cldn3KO mice.Citation55,Citation56 Despite increased colitis susceptibility in Cldn3KO mice, our data demonstrated an unexpected increase in the beneficial phylum Firmicutes in Cldn3KO mice guts; however, the overall diversity of the bacterial communities in this phylum was altered. Specifically, we found a higher abundance of proinflammatory genera such as Ruminococcus and Marvinbryantia in Cldn3KO mice, which are known to be associated with gut inflammation and bowel dysfunction.Citation57–59 Recent studies using mouse modeling of colitis and associated colon cancer showed a similar reduced abundance of Firmicutes organisms, such as Dubosiella and Faecalibaculum as found in Cldn3KO mice, supporting a colitis-promoting environment in Cldn3KO mice.Citation60,Citation61 In this regard, a recent study elegantly showed that the autochthonous murine microbiota member Faecalibaculum rodentium and its human homolog Holdemanella biformis protect against intestinal tumor growth.Citation62 Overall, these data support the possible role of colitis-promoting gut microbiota in the higher risks of colitis in Cldn3KO mice. However, further investigations using these altered microbes are warranted to outline the role of intestinal CLDN3 expression in gut dysbiosis and IBD.
Importantly, dysbiosis promotes a shift in intestinal homeostasis toward a proinflammatory environment and thus increases the expression of proinflammatory mediators and response gene expression.Citation20,Citation63 In this context, our analysis of the published transcriptomic data from IBD patients in relation to gut microbiome revealed STAT3, iNOS, Toll-like receptor, inflammasome, and NOD1/2 signaling pathways as upregulated pathways. We also found increased expression of IL-6 and STAT3 signaling in Cldn3KO mice with colitis. In this regard, Butyricicoccus, associated with higher IL-6 levels in IBD patients, showed overlapping presence between IBD patients and Cldn3KO mice.Citation37 On the other hand, Bifidobacterium and Dubosiella which are low in Cldn3KO mice associate negatively with IL-6, STAT3, and NFkB expression.Citation64,Citation65 We speculate that these dysbiotic microbiota under condition of Cldn3 loss promote pro-inflammatory signaling. In support, exposure of polarized IECs to the microbial contents from WT mice induced expression of IL-6 and TNF-α which was exacerbated by gut microbial contents from Cldn3KO mice. Notably, NFkB can promote secretion of TNF-α, and IL-6; however, high IL-6 levels can also trigger the NFkB-dependent release of TNF-α, which can result in a feedback loop to aggravate inflammation.Citation66,Citation67 We have reported upregulation of gp130, the IL-6 receptor, in Cldn3KO mice and CLDN3 knockdown Caco-2 cells.Citation13 Taken together, our data presents two plausible scenarios: 1) microbiota-dependent activation of an autocrine IL-6/gp130/STAT3 signaling under low CLDN3 conditions; and 2) microbiota-mediated upregulation of IL-6 in IECs diffuses basally to activate the immune cells, triggering proinflammatory signaling pathways, including STAT3 activation and NFkB, which then aggravate gut inflammation by feedback mechanism. Published studies support compartmentalized and context-dependent activation of the IL-6/STAT3 signaling during inflammation.Citation68,Citation69 However, additional studies are needed to mechanically verify above postulations and part of our ongoing studies.
Next, we examined the potential association between CLDN3 and these genes in IBD patients. Notably, when we examined the same IBD patient cohort used in the above analysis, Spearman’s correlation demonstrated a significant inverse correlation between CLDN3 and TNF, TLR4, TLR9, AHR, CD28, CD38, CXCR12, and CXCR4 expression. Collectively, our analysis supported the postulation that the loss of CLDN3 can modulate microbiome-associated signaling pathways in IBD patients and thus promote disease severity.
However, to increase the risk of IBD and/or disease severity, it is critical that host metabolism is affected by genetic or environmental factors. In this context, gut microbiota plays a critical role in regulating host metabolism.Citation34 Accordingly, our analysis using the R-package algorithm for microbial abundance based on KEGG annotations revealed that the gut microbiome of Cldn3KO mice had the potential to significantly upregulate functional pathways, including fatty acid metabolism and oxidative phosphorylation. The downregulated pathways included the N-glycan, primary, and secondary bile acid biosynthesis pathways compared to WT mice. The metabolism of vitamin B6 and C5-Branched dibasic acid was also downregulated in Cldn3KO mice. In this regard, an interplay between the microbiome and systemic and fecal metabolic environments has recently been described.Citation34 Collectively, these data suggest that alterations in the gut microbiota due to Cldn3 deficiency lead to changes in the metabolic profiles of the mouse gut.
Overall, our data in the current study provide strong evidence for a critical role for CLDN3 protein in regulating gut barrier functions and microbiota composition. Further analysis using IBD patient cohorts and murine models of colitis provides additional evidence for the causal role of CLDN3 loss in promoting IBD. Considering that Cldn3KO mice present both phenotypes, leaky gut and microbial dysbiosis, it remains unanswered whether increased gut permeability precedes gut dysbiosis or vice versa. Irrespective, these data provide novel information regarding the regulation of gut leakiness and microbiota homeostasis in IBD and its future clinical relevance.
Materials and Methods
IBD patient samples
All work in this study was carried out under the Institutional Review Board approved for Clinical Research at the University of Nebraska Medical Center (UNMC), Omaha Nebraska, USA. All deidentified IBD patient sample slides were obtained from archived tissue cores and confirmed by a pathologist by histological examination.
Cldn3KO mouse
All mice used in this study had a C57BL/6J background. The generation of constitutive Cldn3KO mouse has been described.Citation13 All animal experiments were performed according to institutional guidelines. All mice were kept in individually ventilated cages in compliance with the Animal Welfare Act to avoid microbial cross-contamination with Cldn3KO and WT mice. Mice were housed under a 12-hr day and night cycle with free access to food and water. Cldn3KO mice were bred with their WT littermates. After genotyping, the mice were separated into two groups and maintained for at least four weeks to ensure that microbiota contamination was not an issue.
Models of experimental colitis
All mouse studies were approved by the UNMC Animal Care and Use Committee. All mice were provided a regular chow diet ad libitum. Matched Cldn3KO and WT littermate mice (8–10 weeks old male and female) were used. The experimental colitis models used in this study are described below:
Acute and chronic DSS colitis
Male and female mice from Cldn3KO and WT littermates were grouped randomly into respective groups, and colitis was induced by dextran sodium sulfate (DSS; 40 kDa; TdB Labs, Sweden) as described previously.Citation8
C. rodentium induced colitis
C. rodentium strain DBS100 (ATCC 51,459; USA) was grown by shaking overnight in Luria-Bertani broth at 37°C. Mice were infected by oral gavage using 0.1 ml of Luria broth containing 5 × 108 cfu of C. rodentium. To minimize the differences between infections, both Cldn3KO and WT littermate mice were infected with the same bacterial preparation. The mice were monitored for clinical symptoms and signs of colitis, and their body weight was recorded every day. On day 14 following infection, mice were euthanized, and colon tissues were prepared for histological, protein, and RNA analyses.
Evaluation of colonic thickness/edema
The colonic weight/length ratio is an indirect method to determine the colon thickness or edema in the murine models of experimental colitis, which correlates well with the histological score.Citation8 Briefly, the mouse colon was opened longitudinally, and the fat and connective tissues were removed, washing out the fecal materials. After that, Kimwipe was used to clean and soak the extra water in the colon. The colon length and weight were measured to determine the colon thickness.
Assessment of disease activity index
During the DSS-induced colitis studies, a disease activity index (DAI) score was assessed to evaluate the progression of colitis as described previously.Citation8 The DAI was a composite score determined by relative body weight loss, stool softness, and blood in mouse stool or rectum.
Gut microbial composition analysis
Fecal contents were flash frozen, and bacterial DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen Valencia, CA, USA) with a bead-beating step as previously described. Amplicon sequencing and detailed composition of the fecal microbiota using 16S rRNA bacterial gene sequencing were performed using the Illumina software at the University of Nebraska Medical Center’s Genomic Core facility. QIIME2 (Quantitative Insights into Microbial Ecology version 2, 2020.2) software with the DADA2 algorithm was used to determine the taxonomic diversity profiles of the microbiota in Cldn3KO and WT littermates. Sequences were denoised and clustered into Amplicon Sequencing Variants (ASVs) using the DADA2 algorithm. Taxonomic classification was performed using the QIIME2 workflow with the SILVA 132 reference set. For the microbiota-related analysis, the two genotypes, Cldn3KO and WT, were compared using the Wilcoxon t-test. A statistical difference of at least P < .05 was used to select the significant features between the genotypes. Alpha and beta diversity, relative frequency, and box plots were plotted using the R package.
Fecal Microbial Transplantation (FMT)
To validate the role of gut dysbiosis in IBD following CLDN3 loss, FMT was performed using cecal microbial transplantation into germ-free mice. In brief, ceca with microbial content from Cldn3KO and WT littermates were snap-frozen. At the time of inoculum preparation, frozen ceca were introduced into an anaerobic chamber (Bactron600 Shel Lab, Cornelius, OR) to avoid oxygen exposure, and slurries were prepared by suspending cecal contents in pre-reduced sterile PBS with 10% glycerol (pH 7) at a dilution of 1:10. The cecal communities of both Cldn3KO and WT mice were transferred into germ-free C57BL/6J mice raised by the Nebraska Gnotobiotic Mouse Program at the University of Nebraska-Lincoln. After day 14 post-inoculation of cecal microbial content, acute DSS colitis was induced to study the effect of CLDN3 loss on microbial dysbiosis and colitis. At the end of the study, colon specimens were fixed in 10% formalin, embedded in paraffin, and subjected to H&E staining to evaluate histology and pathology. The degree of colitis pathology was assessed by pathologists at the University of Nebraska Medical Center, Omaha, in a blinded manner.
Generation of pseudo-germ-free mice
To deplete the gut microbiota, Cldn3KO mice were treated with a cocktail of broad-spectrum antibiotics (ampicillin 1 g/L, neomycin sulfate 1 g/L, metronidazole 1 g/L, vancomycin 0.5 g/L) in drinking water for 2 weeks as previously described.Citation70 The mice were monitored for adverse effects of the broad-spectrum antibiotic cocktail. After two weeks, fecal content was used to determine microbial depletion using standard bacterial inoculation on Luria broth agar culture plates. After confirming gut microbial depletion, the mice were subjected to DSS acute colitis.
In vitro stimulation of intestinal epithelial cells by dysbiotic fecal content
To investigate the potential inflammatory responses of intestinal epithelial cells to gut dysbiosis, Caco-2 cells were challenged with fecal content from Cldn3KO and WT littermates. Briefly, Cldn3KO and WT mice were kept in separate cages for at least three weeks, after which fecal pellets were collected under sterile conditions. Equal amounts of fecal content were dissolved in phosphate-buffered saline and sonicated for 10 seconds at 20% amplitude. Following sonication, samples were divided into two parts, native and heat-inactivated for 10 minutes at 95°C respectively. 20 µl of native and heat-inactivated dysbiotic fecal content were added apically to the monolayer of intestinal epithelial cells for stated times points then cells were collected for immunoblot analysis.
Measurements of trans-epithelial resistance
Cells were plated on transwell filters (0.4 μm). The confluent monolayer was treated apically with the fecal microbial content from both Cldn3KO and WT mice for 48 hrs. The trans-epithelial resistance was determined as described previously and is represented as Ω xcm2.Citation71
Immunoblot, immunofluorescence and immunohistochemistry analysis
Immunoblot, immunofluorescence, and immunohistochemistry analyses were performed as described previously.Citation71
RNA isolation for quantitative RT-PCR
Total RNA from mouse colon samples was extracted from 20 to 30 mg of tissue using the Quick-RNA Miniprep Kit (Zymo Rsearch, Irvine, CA) with DNase digestion. Total RNA (1ug) from each sample was reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). PCR was performed using a cytokine primer set (Supplementary Table S1). Each well-contained SYBR Green Master Mix (Bio-Rad), primer sets, and cDNA (30ng). Samples were loaded in duplicates in 96 well plates and run on a Bio-Rad CFX Connect (Bio-Rad, Hercules, CA, USA) real-time PCR machine. The Ct values were used to calculate the fold change. The normalization was performed using β-actin.
GEO data collection, pre-processing and analysis of IBD patients
Whole-transcriptome data from mucosal biopsies of IBD patients were downloaded from the Gene Expression Omnibus (GEO) website (www.ncbi.nlm.nih.gov/geo/) using the GEO query package in R programming language. Publicly available datasets were used for the analysis (GSE53306, GSE111889, GSE48634, GSE65114, GSE16879, GSE36807, GSE13367, GSE14580, GSE38713, GSE75214, GSE59071, GSE9452, and GSE48958).Citation37,Citation72–83 All the data were batch normalized and log 2 transformed using ‘sva’ package in R bioinformatics software.Citation84,Citation85
Statistical analysis
Statistical analyses were performed using data from at least triplicate experiments unless stated otherwise in the figure legends. Mean values with standard errors were used for independent Student’s t-tests or one-way ANOVA, and corrections for multiple comparisons were made using Tukey’s multiple comparison tests in Prism 9.5 (GraphPad Software Inc.). Statistical significance was set at p < .05.
Supplementary Table.docx
Download MS Word (13.2 KB)Acknowledgments
We thank Dr. Laurey Steinke, Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, for help with preparation of fecal DNA from mice for 16S sequencing. The University of Nebraska DNA Sequencing Core received partial support from the National Institute for General Medical Science (NIGMS) INBRE - P20GM103427-14 and COBRE - 1P30GM110768-01 grants as well as The Fred & Pamela Buffett Cancer Center Support Grant - P30CA036727.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data are available upon request. The data supporting the findings of this study are available from the corresponding author upon request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2282789
Additional information
Funding
References
- Mandel LJ, Bacallao R, Zampighi G. Uncoupling of the molecular ‘fence’ and paracellular ‘gate’ functions in epithelial tight junctions. Nature. 1993;361:552–26. doi:10.1038/361552a0.
- Ivanov AI. Structure and regulation of intestinal epithelial tight junctions: current concepts and unanswered questions. Adv Exp Med Biol. 2012;763:132–148.
- Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol. 2017;10(2):307–317. doi:10.1038/mi.2016.128.
- Miele E, Pascarella F, Quaglietta L, Giannetti E, Greco L, Troncone R, STAIANO A. Altered intestinal permeability is predictive of early relapse in children with steroid-responsive ulcerative colitis. Aliment Pharmacol Ther. 2007;25(8):933–939. doi:10.1111/j.1365-2036.2007.03291.x.
- Soderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C, Järnerot G, Sjödahl R. Different intestinal permeability patterns in relatives and spouses of patients with Crohn’s disease: an inherited defect in mucosal defence? Gut. 1999;44(1):96–100. doi:10.1136/gut.44.1.96.
- Krug SM, Fromm M. Special issue on “the tight junction and its proteins: more than just a barrier”. Int J Mol Sci. 2020;21(13):21. doi:10.3390/ijms21134612.
- Otani T, Furuse M. Tight junction structure and function revisited. Trends Cell Biol. 2020;30(12):1014. doi:10.1016/j.tcb.2020.10.001.
- Ahmad R, Chaturvedi R, Olivares-Villagomez D, Habib T, Asim M, Shivesh P, Polk DB, Wilson KT, Washington MK, Van Kaer L, et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014;7(6):1340–1353. doi:10.1038/mi.2014.21.
- Gowrikumar S, Ahmad R, Uppada SB, Washington MK, Shi C, Singh AB, Dhawan P. Upregulated claudin-1 expression promotes colitis-associated cancer by promoting β-catenin phosphorylation and activation in Notch/p-AKT-dependent manner. Oncogene. 2019;38(26):5321–5337. doi:10.1038/s41388-019-0795-5.
- Tsai PY, Zhang B, He WQ, Zha JM, Odenwald MA, Singh G, Tamura A, Shen L, Sailer A, Yeruva S, et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host & Microbe. 2017;21(6):671–681.e4. doi:10.1016/j.chom.2017.05.009.
- Nishida M, Yoshida M, Nishiumi S, Furuse M, Azuma T. Claudin-2 regulates colorectal inflammation via myosin light chain kinase-dependent signaling. Dig Dis Sci. 2013;58(6):1546–1559. doi:10.1007/s10620-012-2535-3.
- Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. 2015;64(10):1529–1538. doi:10.1136/gutjnl-2014-308419.
- Ahmad R, Kumar B, Chen Z, Chen X, Muller D, Lele SM, Washington MK, Batra SK, Dhawan P, Singh AB, et al. Loss of claudin-3 expression induces IL6/gp130/Stat3 signaling to promote colon cancer malignancy by hyperactivating Wnt/β-catenin signaling. Oncogene. 2017;36(47):6592–6604. doi:10.1038/onc.2017.259.
- Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, Tsukita S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J Cell Biol. 1999;147(1):195–204. doi:10.1083/jcb.147.1.195.
- Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000;476(3):258–261. doi:10.1016/S0014-5793(00)01744-0.
- Huang X, Oshima T, Tomita T, Fukui H, Miwa H. Butyrate alleviates cytokine-induced barrier dysfunction by modifying claudin-2 levels. Biology (Basel). 2021;10(3):10. doi:10.3390/biology10030205.
- Haines RJ, Beard RS Jr., Eitner RA, Chen L, Wu MH. TNFα/IFNγ mediated intestinal epithelial barrier dysfunction is attenuated by MicroRNA-93 downregulation of PTK6 in mouse colonic epithelial cells. PloS One. 2016;11(4):e0154351. doi:10.1371/journal.pone.0154351.
- Haines RJ, Beard RS Jr., Chen L, Eitnier RA, Wu MH. Interleukin-1β mediates β-catenin-driven downregulation of claudin-3 and barrier dysfunction in Caco2 cells. Dig Dis Sci. 2016;61(8):2252–2261. doi:10.1007/s10620-016-4145-y.
- Patel RM, Myers LS, Kurundkar AR, Maheshwari A, Nusrat A, Lin PW. Probiotic bacteria induce maturation of intestinal claudin 3 expression and barrier function. Am J Pathol. 2012;180(2):626–635. doi:10.1016/j.ajpath.2011.10.025.
- Lama Tamang R, Juritsch AF, Ahmad R, Salomon JD, Dhawan P, Ramer-Tait AE, Singh AB. The diet-microbiota axis: a key regulator of intestinal permeability in human health and disease. Tissue Barriers. 2023;11(2):2077069. doi:10.1080/21688370.2022.2077069.
- Kocot AM, Jarocka-Cyrta E, Drabinska N. Overview of the importance of biotics in gut barrier integrity. Int J Mol Sci. 2022;23(5):23. doi:10.3390/ijms23052896.
- Nagao-Kitamoto H, Shreiner AB, Gillilland MG 3rd, Kitamoto S, Ishii C, Hirayama A, Kuffa P, El-Zaatari M, Grasberger H, Seekatz AM, et al. Functional characterization of inflammatory bowel disease–associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol. 2016;2(4):468–481. doi:10.1016/j.jcmgh.2016.02.003.
- Hong SN, Park C, Park SJ, Lee CK, Ye BD, Kim YS, et al. Deep resequencing of 131 Crohn’s disease associated genes in pooled DNA confirmed three reported variants and identified eight novel variants. Gut. 2016;65:788–796. doi:10.1136/gutjnl-2014-308617.
- Doecke JD, Simms LA, Zhao ZZ, Huang N, Hanigan K, Krishnaprasad K, Roberts RL, Andrews JM, Mahy G, Bampton P, et al. Genetic susceptibility in IBD: overlap between ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2013;19(2):240–245. doi:10.1097/MIB.0b013e3182810041.
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. doi:10.1038/ng.3359.
- Kedia S, Virmani S, Vuyyuru SK, Kumar P, Kante B, Sahu P, Kaushal K, Farooqui M, Singh M, Verma M, et al. Faecal microbiota transplantation with anti-inflammatory diet (FMT-AID) followed by anti-inflammatory diet alone is effective in inducing and maintaining remission over 1 year in mild to moderate ulcerative colitis: a randomised controlled trial. Gut. 2022;71(12):2401–2413. doi:10.1136/gutjnl-2022-327811.
- Turner D, Bishai J, Reshef L, Abitbol G, Focht G, Marcus D, Ledder O, Lev-Tzion R, Orlanski-Meyer E, Yerushalmi B, et al. Antibiotic Cocktail for Pediatric Acute Severe Colitis and the Microbiome: The PRASCO Randomized Controlled Trial. Inflamm Bowel Dis. 2019;26(11):1733–1742. doi:10.1093/ibd/izz298.
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of enterobacteriaceae. Cell Host & Microbe. 2007;2(3):204. doi:10.1016/j.chom.2007.08.002.
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Sci. 2013;339(6120):708–711. doi:10.1126/science.1232467.
- Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14(7):685–690. doi:10.1038/ni.2608.
- Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front Immunol. 2018;9:1270. doi:10.3389/fimmu.2018.01270.
- Li Y, de Haar C, Chen M, Deuring J, Gerrits MM, Smits R, Xia B, Kuipers EJ, van der Woude CJ. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59(2):227–235. doi:10.1136/gut.2009.184176.
- Mudter J, Weigmann B, Bartsch B, Kiesslich R, Strand D, Galle PR, Lehr HA, Schmidt J, Neurath MF. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am J Gastroenterol. 2005;100(1):64–72. doi:10.1111/j.1572-0241.2005.40615.x.
- Visconti A, Le Roy CI, Rosa F, Rossi N, Martin TC, Mohney RP, Li W, de Rinaldis E, Bell JT, Venter JC, et al. Interplay between the human gut microbiome and host metabolism. Nat Commun. 2019;10(1):4505. doi:10.1038/s41467-019-12476-z.
- Li X, Wei X, Sun Y, Du J, Li X, Xun Z, Li YC. High-fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am J Physiol Gastrointest Liver Physiol. 2019;317(4):G453–G462. doi:10.1152/ajpgi.00103.2019.
- Park SC, Jeen YT. Genetic studies of inflammatory bowel disease-focusing on Asian patients. Cells. 2019;8(5):404. doi:10.3390/cells8050404.
- Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi:10.1038/s41586-019-1237-9.
- Ganapathy AS, Saha K, Wang A, Arumugam P, Dharmaprakash V, Yochum G, Koltun W, Nighot M, Perdew G, Thompson TA, et al. Alpha-tocopherylquinone differentially modulates claudins to enhance intestinal epithelial tight junction barrier via AhR and Nrf2 pathways. Cell Rep. 2023;42(7):112705. doi:10.1016/j.celrep.2023.112705.
- Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88(10):1110–1120. doi:10.1038/labinvest.2008.78.
- Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, Collins JE. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85(9):1139–1162. doi:10.1038/labinvest.3700316.
- Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK, Beauchamp RD, Singh AB, Dhawan P. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of notch-signalling. Gut. 2014;63(4):622–634. doi:10.1136/gutjnl-2012-304241.
- Raju P, Shashikanth N, Tsai PY, Pongkorpsakol P, Chanez-Paredes S, Steinhagen PR, Kuo W-T, Singh G, Tsukita S, Turner JR, et al. Inactivation of paracellular cation-selective claudin-2 channels attenuates immune-mediated experimental colitis in mice. J Clin Invest. 2020;130(10):5197–5208. doi:10.1172/JCI138697.
- Shashikanth N, Liu Y, Xing T, Turner J. NOVEL CONTRIBUTIONS of CLAUDIN-4 to EPITHELIAL BARRIER REGULATION and COLITIS. Inflamm Bowel Dis. 2022;28(Supplement_1):SS51–SS52. doi:10.1093/ibd/izac015.081.
- Xiao Y, Lian H, Zhong XS, Krishnachaitanya SS, Cong Y, Dashwood RH, Savidge TC, Powell DW, Liu X, Li Q, et al. Matrix metalloproteinase 7 contributes to intestinal barrier dysfunction by degrading tight junction protein claudin-7. Front Immunol. 2022;13:1020902. doi:10.3389/fimmu.2022.1020902.
- Chatterjee I, Zhang Y, Zhang J, Lu R, Xia Y, Sun J. Overexpression of vitamin D receptor in intestinal epithelia protects against colitis via upregulating tight junction protein claudin 15. J Crohn’s Colitis. 2021;15:1720–1736. doi:10.1093/ecco-jcc/jjab044.
- Tamura A, Kitano Y, Hata M, Katsuno T, Moriwaki K, Sasaki H, Hayashi H, Suzuki Y, Noda T, Furuse M, et al. Megaintestine in claudin-15–Deficient mice. Gastroenterology. 2008;134(2):523–534.e3. doi:10.1053/j.gastro.2007.11.040.
- Baier FA, Sanchez-Taltavull D, Yarahmadov T, Castella CG, Jebbawi F, Keogh A, Tombolini R, Odriozola A, Dias MC, Deutsch U, et al. Loss of claudin-3 impairs hepatic metabolism, biliary barrier function, and cell proliferation in the murine liver. Cell Mol Gastroenterol Hepatol. 2021;12(2):745–767. doi:10.1016/j.jcmgh.2021.04.003.
- Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat microbiol. 2019;4(2):293–305. doi:10.1038/s41564-018-0306-4.
- Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–259. doi:10.1194/jlr.R500013-JLR200.
- Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, Sim D, Jarr K, Spear ET, Singh G, et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host & Microbe. 2020;27(4):659–670.e5. doi:10.1016/j.chom.2020.01.021.
- Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387(10014):156–167. doi:10.1016/S0140-6736(15)00465-1.
- Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, Ananthakrishnan AN, Andrews E, Barron G, Lake K, et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat microbiol. 2018;3(3):337–346. doi:10.1038/s41564-017-0089-z.
- Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, Brostoff J, Parkhill J, Dougan G, Petrovska L, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11(1):7. doi:10.1186/1471-2180-11-7.
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. doi:10.1186/gb-2012-13-9-r79.
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song S, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host & Microbe. 2014;15(3):382–392. doi:10.1016/j.chom.2014.02.005.
- Duranti S, Gaiani F, Mancabelli L, Milani C, Grandi A, Bolchi A, Santoni A, Lugli GA, Ferrario C, Mangifesta M, et al. Elucidating the gut microbiome of ulcerative colitis: bifidobacteria as novel microbial biomarkers. FEMS Microbiol Ecol. 2016;92(12):fiw191. doi:10.1093/femsec/fiw191.
- Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019;116(26):12672–12677. doi:10.1073/pnas.1904099116.
- Wang Y, Qi W, Song G, Pang S, Peng Z, Li Y, Wang P. High-fructose diet increases inflammatory cytokines and alters gut microbiota composition in rats. Mediators Inflamm. 2020;2020:1–10. doi:10.1155/2020/6672636.
- Wang Y, Xie Q, Sun S, Huang B, Zhang Y, Xu Y, Zhang S, Xiang H. Probiotics-fermented Massa Medicata Fermentata ameliorates weaning stress in piglets related to improving intestinal homeostasis. Appl Microbiol Biotechnol. 2018;102(24):10713–10727. doi:10.1007/s00253-018-9438-y.
- Ni Y, Zhang Y, Zheng L, Rong N, Yang Y, Gong P, Yang Y, Siwu X, Zhang C, Zhu L, et al. Bifidobacterium and lactobacillus improve inflammatory bowel disease in zebrafish of different ages by regulating the intestinal mucosal barrier and microbiota. Life Sci. 2023;324:121699. doi:10.1016/j.lfs.2023.121699.
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Sci. 2013;341(6145):569–573. doi:10.1126/science.1241165.
- Zagato E, Pozzi C, Bertocchi A, Schioppa T, Saccheri F, Guglietta S, Fosso B, Melocchi L, Nizzoli G, Troisi J, et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat microbiol. 2020;5(3):511–524. doi:10.1038/s41564-019-0649-5.
- Otte ML, Lama Tamang R, Papapanagiotou J, Ahmad R, Dhawan P, Singh AB. Mucosal healing and inflammatory bowel disease: therapeutic implications and new targets. World J Gastroenterol. 2023;29(7):1157–1172. doi:10.3748/wjg.v29.i7.1157.
- Li D, Cui L, Gao Y, Li Y, Tan X, Xu H. Fecal microbiota transplantation improves intestinal inflammation in mice with ulcerative colitis by modulating intestinal flora composition and down-regulating NF-kB signaling pathway. Microb Pathog. 2022;173:105803. doi:10.1016/j.micpath.2022.105803.
- Singh S, Bhatia R, Khare P, Sharma S, Rajarammohan S, Bishnoi M, Bhadada SK, Sharma SS, Kaur J, Kondepudi KK, et al. Anti-inflammatory Bifidobacterium strains prevent dextran sodium sulfate induced colitis and associated gut microbial dysbiosis in mice. Sci Rep. 2020;10(1):18597. doi:10.1038/s41598-020-75702-5.
- Cabrera-Rivera GL, Madera-Sandoval RL, Leon-Pedroza JI, Ferat-Osorio E, Salazar-Rios E, Hernandez-Aceves JA, Guadarrama-Aranda U, López-Macías C, Wong-Baeza I, Arriaga-Pizano LA, et al. Increased TNF-α production in response to IL-6 in patients with systemic inflammation without infection. Clin Exp Immunol. 2022;209(2):225–235. doi:10.1093/cei/uxac055.
- Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 induces NF-κB activation in the intestinal epithelia. J Immunol. 2003;171(6):3194–3201. doi:10.4049/jimmunol.171.6.3194.
- Serrano C, Galan S, Rubio JF, Candelario-Martinez A, Montes-Gomez AE, Chanez-Paredes S, Cedillo-Barrón L, Schnoor M, Meraz-Ríos MA, Villegas-Sepúlveda N, et al. Compartmentalized response of IL-6/STAT3 signaling in the colonic mucosa mediates colitis development. J Immunol. 2019;202(4):1239–1249. doi:10.4049/jimmunol.1801060.
- Piedra-Quintero ZL, Serrano C, Villegas-Sepulveda N, Maravillas-Montero JL, Romero-Ramirez S, Shibayama M, Medina-Contreras O, Nava P, Santos-Argumedo L. Myosin 1F regulates M1-polarization by stimulating intercellular adhesion in macrophages. Front Immunol. 2018;9:3118. doi:10.3389/fimmu.2018.03118.
- Amorim N, McGovern E, Raposo A, Khatiwada S, Shen S, Koentgen S, Hold G, Behary J, El-Omar E, Zekry A, et al. Refining a Protocol for Faecal Microbiota Engraftment in Animal Models After Successful Antibiotic-Induced Gut Decontamination. Front Med. 2022;9:770017. doi:10.3389/fmed.2022.770017.
- Yadav SK, Ahmad R, Moshfegh CM, Sankarasubramanian J, Joshi V, Elkhatib SK, Chhonker YS, Murry DJ, Talmon GA, Guda C, et al. Repeated social defeat stress induces an inflammatory gut milieu by altering the mucosal barrier integrity and gut microbiota homeostasis. Biol Psychiatry Global Open Sci. 2023;3(4):824–836. doi:10.1016/j.bpsgos.2023.03.005.
- Zhao X, Fan J, Zhi F, Li A, Li C, Berger AE, Boorgula MP, Barkataki S, Courneya J-P, Chen Y, et al. Mobilization of epithelial mesenchymal transition genes distinguishes active from inactive lesional tissue in patients with ulcerative colitis. Hum Mol Genet. 2015;24(16):4615–4624. doi:10.1093/hmg/ddv192.
- Smith PJ, Levine AP, Dunne J, Guilhamon P, Turmaine M, Sewell GW, Oʼshea NR, Vega R, Paterson JC, Oukrif D, et al. Mucosal transcriptomics implicates under expression of BRINP3 in the pathogenesis of ulcerative colitis. Inflamm Bowel Dis. 2014;20(10):1802–1812. doi:10.1097/MIB.0000000000000169.
- Balfe ALG, O’Connell R. The GSE65114 dataset. The Gene Expression Omnibus 2015.
- Arijs I, De Hertogh G, Lemaire K, Quintens R, Van Lommel L, Van Steen K, Leemans P, Cleynen I, Van Assche G, Vermeire S, et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PloS One. 2009;4(11):e7984. doi:10.1371/journal.pone.0007984.
- Montero-Melendez T, Llor X, Garcia-Planella E, Perretti M, Suarez A, Calogero RA. Identification of novel predictor classifiers for inflammatory bowel disease by gene expression profiling. PloS One. 2013;8(10):e76235. doi:10.1371/journal.pone.0076235.
- Bjerrum JT, Hansen M, Olsen J, Nielsen OH. Genome-wide gene expression analysis of mucosal colonic biopsies and isolated colonocytes suggests a continuous inflammatory state in the lamina propria of patients with quiescent ulcerative colitis. Inflamm Bowel Dis. 2010;16(6):999–1007. doi:10.1002/ibd.21142.
- Arijs I, Li K, Toedter G, Quintens R, Van Lommel L, Van Steen K, Leemans P, De Hertogh G, Lemaire K, Ferrante M, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58(12):1612–1619. doi:10.1136/gut.2009.178665.
- Planell N, Lozano JJ, Mora-Buch R, Masamunt MC, Jimeno M, Ordas I, Esteller M, Ricart E, Piqué JM, Panés J, et al. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut. 2013;62(7):967–976. doi:10.1136/gutjnl-2012-303333.
- Vancamelbeke M, Vanuytsel T, Farre R, Verstockt S, Ferrante M, Van Assche G, Rutgeerts P, Schuit F, Vermeire S, Arijs I, et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23(10):1718–1729. doi:10.1097/MIB.0000000000001246.
- Vanhove W, Peeters PM, Staelens D, Schraenen A, Van der Goten J, Cleynen I, De Schepper S, Van Lommel L, Reynaert NL, Schuit F, et al. Strong upregulation of AIM2 and IFI16 Inflammasomes in the mucosa of patients with active inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(11):2673–2682. doi:10.1097/MIB.0000000000000535.
- Olsen J, Gerds TA, Seidelin JB, Csillag C, Bjerrum JT, Troelsen JT, Nielsen OH. Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm Bowel Dis. 2009;15(7):1032–1038. doi:10.1002/ibd.20879.
- Van der Goten J, Vanhove W, Lemaire K, Van Lommel L, Machiels K, Wollants WJ, De Preter V, De Hertogh G, Ferrante M, Van Assche G, et al. Integrated miRNA and mRNA expression profiling in inflamed colon of patients with ulcerative colitis. PloS One. 2014;9(12):e116117. doi:10.1371/journal.pone.0116117.
- Davis S, Meltzer PS. Geoquery: a bridge between the gene expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–1847. doi:10.1093/bioinformatics/btm254.
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. doi:10.1093/bioinformatics/bts034.