
ABSTRACT
Gut bacteria regulate brain pathology of Alzheimer’s disease (AD) patients and animal models; however, the underlying mechanism remains unclear. In this study, 3-month-old APP-transgenic female mice with and without knock-out of Il-17a gene were treated with antibiotics-supplemented or normal drinking water for 2 months. The antibiotic treatment eradicated almost all intestinal bacteria, which led to a reduction in Il-17a-expressing CD4-positive T lymphocytes in the spleen and gut, and to a decrease in bacterial DNA in brain tissue. Depletion of gut bacteria inhibited inflammatory activation in both brain tissue and microglia, lowered cerebral Aβ levels, and promoted transcription of Arc gene in the brain of APP-transgenic mice, all of which effects were abolished by deficiency of Il-17a. As possible mechanisms regulating Aβ pathology, depletion of gut bacteria inhibited β-secretase activity and increased the expression of Abcb1 and Lrp1 in the brain or at the blood-brain barrier, which were also reversed by the absence of Il-17a. Interestingly, a crossbreeding experiment between APP-transgenic mice and Il-17a knockout mice further showed that deficiency of Il-17a had already increased Abcb1 and Lrp1 expression at the blood-brain barrier. Thus, depletion of gut bacteria attenuates inflammatory activation and amyloid pathology in APP-transgenic mice via Il-17a-involved signaling pathways. Our study contributes to a better understanding of the gut-brain axis in AD pathophysiology and highlights the therapeutic potential of Il-17a inhibition or specific depletion of gut bacteria that stimulate the development of Il-17a-expressing T cells.
Introduction
The microbial composition in the gut undergoes alterations in both Alzheimer’s disease (AD) patients and mouse models.Citation1-4 A prospective study of cognitively healthy individuals revealed that a decrease in butyrate-producing bacterial species (e.g., Roseburia inulinivorans and R. faecis) or an increase in pro-inflammatory bacteria (e.g., Veillonella dispar and V. atypica) correlates with subjective cognitive decline over a follow-up period of 2 to 4 yearsCitation5). Transplantation of gut bacteria from AD patients to gut bacteria-depleted rats results in impaired neurogenesis and cognitive function.Citation6 Gut bacteria-free Alzheimer’s precursor protein (APP)-transgenic or AppNL-G-F knock-in AD mice also exhibit reduced amyloid β peptide (Aβ) deposition and microgliosis in the brain.Citation7-11 Consequently, gut bacteria play an important role in AD pathogenesis, though the mechanism of how gut bacteria impact brain pathology in AD remains to be investigated.
However, the specific profile of intestinal bacteria associated with AD has not yet been defined. There is often a decrease in bacterial abundance in Firmicutes phylum, an increase in Proteobacteria phylum, and both a decrease and an increase in the proportion of Bacteroidetes bacteria.Citation1,Citation4,Citation12,Citation13 At the family and genus levels, the variability of results regarding AD-specific bacterial changes is even greater across different studies. For example, a consistent decrease in bacteria of the genus Butyricicoccus or Coprococcus or an increase in the genera Escherichia/Shigella (three genera are not changed in the same study) in AD patients is only found in two out of twenty independent studies.Citation4 Butyricicoccus and Coprococcus produce butyrate, which prevents excessive inflammation in the gut,Citation14,Citation15 while Escherichia/Shigella releases toxins and promotes inflammatory activation in the human body.Citation14 The variability in research findings also exists in AD animals.Citation4 It is a challenge to investigate the precise role of different bacterial taxa in AD pathogenesis. Germ-free or broad-spectrum antibiotic-treated mice are still often used to study the molecular mechanisms by which gut bacteria influence brain pathology in AD.
An important mechanism mediating the brain-gut axis is that gut bacteria produce short-chain free fatty acids (SCFAs, i.e., acetate, butyrate and propionate) that promote the maturation of microglia, innate immune responses and energy metabolism in a homeostatic state.Citation16,Citation17 Administration of SCFAs to germ-free or antibiotic-treated AD mice restores microglial proliferation and inflammatory activation; however, the effects on Aβ phagocytosis and Aβ accumulation in the brain are inconsistent between different studies.Citation16,Citation18,Citation19 Whether SCFAs act directly on microglia also remains unclear. We found that SCFA receptors, G protein-coupled receptor (Gpr) 41 and Gpr43 are absent in murine microgliaCitation20; however, deficiency of Gpr41 and Gpr43 likely inhibits microglial maturation under physiological conditionsCitation17 and increases microglial density and Aβ deposits in the brain of APP-transgenic mice.Citation21 Deficiency of the butyrate receptor Gpr109a, which is highly expressed in microglia,Citation20 has limited effects on microglial activation.Citation21 Since the SCFAs produced by intestinal bacteria activate Gpr41, Gpr43 and Gpr109a, promote anti-inflammatory properties and regulate the development of interleukin (Il)-17a or Il-10 producing T cells in the gut,Citation21–23 we hypothesized that gut bacteria alter microglial activation and brain pathology through circulating T lymphocytes.
Germ-free mice generate fewer Il-17a-producing CD4(+) T help (Th17) lymphocytes but more CD4(+)CD25(+)Foxp3(+) regulatory T (Treg) cells in the gut and spinal cord, which is associated with resistance to experimental autoimmune encephalomyelitis (EAE) in mice.Citation24 Depletion of intestinal bacteria by antibiotics reduces the accumulation of Il-17a-producing γδ T cells in the leptomeninges and ameliorates brain injury in a stroke mouse model.Citation25 In APP-transgenic mice, reductions in cerebral Aβ deposition and microglia after gut antibiotic treatments correlate with increased levels of Foxp3+ Treg cells in blood and brain.Citation10 However, transient depletion of Treg cells was shown to regulate microglia or/and infiltrated macrophages with differential effects on Aβ clearance and cognitive protection in two studies.Citation26,Citation27 Our recent study showed that deficiency of p38α-MAPK in peripheral myeloid cells decreases Th17 cells, which possibly increases microglial activation and Aβ clearance in AD mice.Citation28 Therefore, we asked whether Il-17a-expressing cells mediate the effects of gut bacteria on AD pathology.
Intestinal bacteria may also release structural components into the blood and bacteria themselves may even spread in the brain, both of which can directly activate microglia. Blood concentrations of lipopolysaccharide (LPS) together with inflammatory cytokines, e.g., IL-1β and tumor necrosis factor (TNF)-α, are increased in AD patients compared to non-AD individuals with and without cognitive impairment.Citation29 Components of Porphyromonas gingivalis, a bacterium often existing in chronic periodontitis, were found in the brains of AD patients.Citation30 We have observed that bacterial receptors CD14 and Toll-like receptor (TLR)-2 are receptors for aggregated Aβ,Citation31–33 implying that Aβ and bacterial components share receptors on microglia in the brain. MyD88 is an adaptor protein that is down-stream of most TLRs.Citation34 Our previous studies showed that deficiency of CD14, TLR2, TLR4, or MyD88 inhibits inflammatory activation of microglia, reduces cerebral Aβ and improves cognitive function in APP-transgenic mice.Citation20,Citation32,Citation33,Citation35,Citation36 Antibiotic therapy for AD patients has attracted interest.Citation37 A large cohort study suggests that sporadic use of antibiotics in older adults may decrease the risk of dementia.Citation38 Long-term antibiotic exposure reduces Th17 cells in the gut.Citation39 The question arises as to whether inhibition of Il-17a signaling mediates the efficacy of potential antibiotic therapy in AD.
AD pathology is more likely to lead to clinical dementia in women than in men. It is also believed that the prevalence or incidence of AD is higher in women than in men. The difference is not only due to the longer life expectancy of women, but also to biological or sociocultural factors that differ between women and men.Citation40 In this study, we treated female APP-transgenic mice with and without an antibiotic cocktail in the drinking water and observed that depletion of intestinal bacteria reduced inflammatory activation and Aβ load in the brain. These effects could be abolished by knockout of Il-17a gene.
Materials and methods
Animal models and cross-breeding
APP/PS1-transgenic mice (APPtg) over-expressing human mutated APP (KM670/671NL) and PS1 (L166P) under Thy-1 promotersCitation41 were gifts from M. Jucker, Hertie Institute for Clinical Brain Research, Tübingen, Germany. Il-17a knockout (Il17a−/−) mice were kindly provided by Y. Iwakura, Tokyo University of Science, Japan.Citation42 Il-17a-deficient AD mice were created by cross-breeding APP/PS1-transgenic mice and Il17a−/− mice to obtain APPtg/Il17a−/− genotype in our previous study.Citation28 APP/PS1-transgenic mice were also cross-bred with Il-17a-eGFP reporter mice (Il17aGFP/GFP; kindly provided by R. Flavell, Yale University, USA) to get APPtg/Il17aGFP/wt of genotype, in which eGFP is expressed under the control of endogenous Il-17a gene promoter.Citation43 All mice used in this project were on C57BL/6 genetic background.
Depletion of intestinal bacteria with antibiotics in drinking water
Sisters of Il-17a-deficient or wildtype APP-transgenic mice from each litter (≥2 mice per genotype) were randomly separated. Il-17a-deficient and wild-type female mice were cohoused and treated with and without vancomycin (500 mg/L), ampicillin (1 g/L), neomycin sulfate (1 g/L), streptomycin (1 g/L), and metronidazole (1 g/L) (all antibiotics were purchased from Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) in drinking water from 3 months of age for 2 months to remove gut bacteria according to a published protocol.Citation44 As controls, 3 or 22-month-old C57BL/6J female mice were treated in the same way. The water with antibiotics was changed every 7 days. In order to examine the off-target effects of oral antibiotics on neuroinflammation, we injected 5-month-old APP-transgenic mice via the peritoneal cavity daily for 7 days with 1.5 mg/kg/day vancomycin, 3 mg/kg/day ampicillin, neomycin and streptomycin according to the published protocolCitation45 and 30 mg/kg/day metronidazole, as it has a high oral bioavailability.Citation46 Animal experiments were conducted in accordance with national rules and ARRIVE guidelines, and authorized by Landesamt für Verbraucherschutz, Saarland, Germany (registration numbers: 56/2015, 46/2017 and 34/2019) and Nanchang University, China.
Tissue collection and isolation of blood vessels
Animals were euthanized at 5 months of age by inhalation of overdose isoflurane or by i.p. injection of ketamine (100 mg/kg)/xylazine (10 mg/kg). After intracardial perfusion with ice-cold PBS, the brain was removed and divided along the anterior-posterior axis. The left hemisphere was immediately fixed in 4% paraformaldehyde (Sigma-Aldrich) in PBS and embedded in paraffin for immunohistochemistry. The olfactory bulb was first removed from the right hemisphere, and the cortex and hippocampus were carefully separated from the brainstem, thalamus and striatum under microscope. A roughly 0.5-mm thick sagittal section of tissue was cut from the medial side of the tissue and homogenized in TRIzol (Thermo Fisher Scientific, Darmstadt, Germany) for RNA and DNA isolation. The remainder of the right hemisphere was snap-frozen in liquid nitrogen and stored at −80°C until biochemical analysis. The appendix together with a 0.5-cm-long segment of the neighboring colon was also collected, snap-frozen and stored at −80°C for isolation of intestinal bacteria.
To isolate brain blood vessels, the cortex and hippocampus from 5-month-old APP-transgenic mice were carefully dissected and brain vessel fragments were isolated as we did previously.Citation20 Briefly, brain tissues were homogenized in HEPES-contained Hanks’ balanced salt solution (HBSS) and centrifuged at 4,400 g in HEPES-HBSS buffer supplemented with dextran from Leuconostoc spp. (molecular weight ~ 70,000; Sigma-Aldrich) to delete myelin. The vessel pellet was re-suspended in HEPES-HBSS buffer supplemented with 1% bovine serum albumin (Sigma-Aldrich) and filtered with 20 μm-mesh. The blood vessel fragments were collected on the top of filter and stored at −80°C for biochemical analysis.
Intestinal bacterial collection and 16S rRNA sequencing
Bacterial DNA was extracted from intestinal bacteria (100 mg) in the frozen cecum and colon with QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany). The amount of bacteria was evaluated by quantifying 16S rRNA with SYBR Green-based real-time PCR using the universal bacterial r16S gene primers (16S-V2-101F: 5-AGYGGCGIACGGGTGAGTAA-3, and 16S-V2-361 R: 5-CYIACTGCTGCCTCCCGTAG-3) as it was conducted in a published study.Citation25 The V3 -V4 region of the 16S rRNA-encoding gene was then amplified with the barcode fusion primers (338F: 5-ACTCCTACGGGAGGCAGCAG-3, and 806 R: 5-GGACTACHVGGGTWTCTAAT-3). After purification, PCR products were used for constructing libraries and sequenced on the Illumina MiSeq platform at Majorbio Co. Ltd. (Shanghai, China). The raw data was processed on Qiime2 (https://qiime2.org/.), reducing sequencing and PCR errors, and denoising to get the operational taxonomic unit (OTU) consensus sequences, which were mapped to the 16S Mothur-Silva SEED r119 database (http://www.mothur.org/). Alpha diversity including Sobs, Shannon, Ace, Chao and Simpson indexes were used for the analysis of bacterial richness and diversity in a single mouse. Principal coordinate analysis (PCoA) and analysis of similarity (ANOSIM) were used for β-diversity analysis to compare bacterial compositions on genus level between APP-transgenic mice with and without antibiotic treatments. The difference of bacterial compositions on genus level between these two groups were also compared with Wilcoxon rank sum test. All the analysis was performed using cloud-based tools with default analysis parameters (https://cloud.majorbio.com/page/tools.html).
Quantification of bacterial DNA in the brain tissue
DNA was extracted from the brain tissue using TRIzol (Thermo Fisher Scientific) according to the protocol provided by the company. To assess the presence and extent of bacterial dissemination into the brain, real-time PCR was conducted using universal bacterial 16S rRNA gene primers (16S-V2-101F and 16S-V2-361 R), and primers targeted mouse Gapdh gene (sense, 5’-ACAACTTTGGCATTGTGGAA-3’ and antisense, 5’-GATGCAGGGATGATGTTCTG-3’) as an internal control.
Positive selection of CD11b-positive and CD4-positive cells from the brain and spleen, respectively
The brain tissue (hippocampus and cortex) and spleen of 5-month-old APP-transgenic mice with and without treatments with antibiotics were prepared for single-cell suspensions using Neural Tissue Dissociation Kit (papain-based) and Spleen Dissociation Kit (mouse), respectively (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). After blocking with 50 µg/ml CD16/CD32 antibody (clone 2.4G2; BioXCell, Lebanon, USA), CD11b-positive brain cells were selected from the brain with microbeads-conjugated CD11b antibody (clone M1/70.15.11.5; Miltenyi Biotec) and CD4-positive spleen cells from the spleen with Dynabeads magnetic beads-conjugated CD4 antibody (clone L3T4; Thermo Fisher Scientific). Lysis buffer was immediately added to selected cells and total RNA was isolated using RNeasy Plus Mini Kit (Qiagen).
Microglial Aβ phagocytosis assay
Five-month-old APP-transgenic mice received an intraperitoneal injection of 10 mg/kg methoxy-XO4 (Bio-Techne GmbH, Wiesbaden-Nordenstadt, Germany) (2 mg/ml in a 1:1 mixture of DMSO and 0.9% NaCl [pH 12] (v/v)) after treatment with and without antibiotics according to a published protocol.Citation47 Methoxy-XO4 binds to β-sheet secondary structure of Aβ aggregates. Three hours later, a single cell suspension from the hippocampus and cortex was prepared using Neural Tissue Dissociation Kit (papain-based) (Miltenyi Biotec GmbH). After blocking with CD16/CD32 antibody (clone 2.4G2; BioXCell) and subsequent staining with PE-Cy5-conjugated CD11b antibody (clone M1/70; Thermo Fisher Scientific), fibrillar Aβ-containing CD11b-positive brain cells were detected by BD FACSVerse™ flow cytometry (BD Biosciences; Heidelberg, Germany).
Flow cytometric detection of Il17a-eGFP reporter in intestinal cells
A published protocolCitation48 was used to prepare single cell suspensions from both lamina propria and Peyer’s patches of the small intestine of 5-month-old APPtg/Il17aGFP/wt mice with and without two months of antibiotic treatment. After staining with APC-conjugated rat anti-CD4 monoclonal antibody (clone: GK1.5; Thermo Fisher Scientific), eGFP-expressing CD4-positive cells were detected by BD FACSCanto™ II flow cytometry (BD Biosciences).
Histological analysis
Serial 50-μm-thick sagittal sections were cut from the paraffin-embedded hemisphere. Four neighboring sections with 300 µm of interval were deparaffinized, labeled with rabbit anti-ionized calcium-binding adapter molecule (Iba)-1 antibody (Wako Chemicals, Neuss, Germany) and VectaStain Elite ABC-HRP kit (Cat.-No.: PK-6100, Vector Laboratories, Burlingame, USA), and visualized with diaminobenzidine (Sigma-Aldrich). Iba-1-positive microglia/brain macrophages were counted with Optical Fractionator in the hippocampus and cortex on a Zeiss AxioImager.Z2 microscope (Carl Zeiss Microscopy GmbH, Göttingen, Germany) equipped with a Stereo Investigator system (MBF Bioscience, Williston), as we did previously.Citation49
To evaluate the cerebral Aβ level, after deparaffinization, 4 serial brain sections from each animal were stained with rabbit anti-human Aβ antibody (clone D12B2; Cell Signaling Technology Europe, Frankfurt am Main, Germany) and Cy3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Europe Ltd. Cambridge, UK), or with methoxy-XO4 (Bio-Techne GmbH). After mounting, the whole section including hippocampus and cortex was imaged with Microlucida (MBF Bioscience) and merged. The positive staining and brain region analyzed were measured for the area with Image J tool “Analyse Particles” (https://imagej.nih.gov/ij/). The threshold for all compared samples was manually set and kept constantly. The percentage of Aβ coverage in the brain was calculated.
To determine the density of CD68-positive microglia, serial brain sections were stained with rabbit anti-CD68 antibody (clone E3O7V; Cell Signaling Technology Europe) and Cy3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Europe Ltd.). Since the single CD68-positive cell could not be clearly recognized (see ) and reliably counted, the percentage of CD68 coverage in the brain was calculated as for Aβ.
Analysis of microglial morphology
For the analysis of microglial morphology, our established protocol and Fiji Image J were used.Citation28 Paraffin-embedded 50-µm sagittal brain sections were used as described above. After fluorescent co-staining with Iba-1 and Aβ antibodies, total 10 Aβ plaques/mouse were randomly selected from the cortex dorsal to hippocampus and imaged under 40× objective with Z-stack scanning with 1 µm of interval. The serial images were Z-projected with maximal intensity, 8-bit grayscale transformed, Unsharp-Mask filter and despeckle-treated, and binarized to obtain a black and white image. The cells with complete nucleus and branches and without overlapping with neighboring cells were chosen for analysis. The single-pixel background noise was eliminated and the gaps along processes were filled under the view of the original image of the cell. The processed image was skeletonized and analyzed with the plugin Analyze Skeleton (2D/3D) (http://imagej.net/AnalyzeSkeleton) for the total number of primary branches, length of all branches, and the number of branch endpoints of each microglia. The whole analysis was done blinded to genotypes.
Western blot analysis
Frozen brain tissues and blood vessel isolates were homogenized in RIPA buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 0.1% SDS, 0.5% sodiumdeoxy-cholate, 1% NP-40, and 5 mM EDTA) supplemented with protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany) on ice. The proteins were separated by 10% or 12% Tris-glycine SDS/PAGE. Before loading on PAGE gel, vessel preparations were sonicated. Western blots were performed using rabbit monoclonal antibody against Abcb1 (clone E1Y7S) and rabbit polyclonal antibody against Lrp1 (Cat.-No.: 64099) (both antibodies were bought from Cell Signaling Technology), as well as rabbit polyclonal antibody against claudin-5 (Cat.-No.: GTX49370; GeneTex, Hsinchu, China). The detected proteins were visualized via the Plus-ECL method (PerkinElmer, Waltham, USA). To quantify proteins of interest, rabbit monoclonal antibody against β-actin (clone 13E5) or rabbit polyclonal antibody against α-tubulin (Cat.-No.: 2144) (both antibodies were from Cell Signaling Technology) was used as a protein loading control. Densitometric analysis of band densities was performed with Image-Pro Plus software version 6.0.0.260 (Media Cybernetics, Rockville, MD, USA). For each sample, the protein level was calculated as a ratio of target protein/loading control per sample.
Brain homogenates and ELISA assays of Aβ and Il-1β and Tnf-α
The frozen brain hemispheres were homogenized and extracted serially in Tris-buffered saline (TBS), TBS plus 1% Triton X-100 (TBS-T), guanidine buffer (5 M guanidine HCl/50 mM Tris, pH 8.0) as described in our previous study.Citation49 Aβ concentrations in three separate fractions of brain homogenates were determined by Invitrogen™ Amyloid β 42 and 40 Human ELISA kits (Cat.-No.: KHB3441 and KHB3481, respectively; both from Thermo Fisher Scientific). Results were normalized on the basis of the sample’s protein concentration.
We measured concentrations of Il-1β and Tnf-α in TBS-soluble brain homogenates with ELISA kits (Cat.-No.: DY401 and DY410, respectively, from R&D systems, Minneapolis, USA). The results were also adjusted by the protein concentration in the same sample.
Quantitative PCR for analysis of gene transcripts
Total RNA was isolated from mouse brains with TRIzol or from selected CD11b or CD4-positive cells with RNeasy Plus Mini Kit (Qiagen) and reverse-transcribed. Gene transcripts were quantified with established protocolsCitation49,Citation50 and TaqMan gene expression assays of mouse Tnf-α, Il-1β, Chemokine (C – C motif) ligand 2 (Ccl-2), Il-10, Chitinase-like 3 (Chi3l3), Mannose receptor C type 1 (Mrc1), Apolipoprotein e (Apoe), Triggering receptor expressed on myeloid cells 2 (Trem2), Purinergic receptor P2Y, G-protein coupled 12 (P2ry12), C-X3-C motif chemokine receptor 1 (Cx3cr1), Lipoprotein lipase (Lpl), C-type lectin domain family 7, member a (Clec7a), Integrin alpha X (Itgax), Il-17a, Interferon γ (Ifn-γ), Il-4, Neprilysin and Insulin-degrading enzyme (Ide), Activity-regulated cytoskeleton-associated protein (Arc), Glutamate ionotropic receptor NMDA type subunit 1 (Grin1), Brain derived neurotrophic factor (Bdnf), and Gapdh (Thermo Fisher Scientific).
Statistical analysis
Data were presented as mean ± SEM and displayed using a scatter plot with a bar overlay in the figure, with the scatter plot representing individual data points. Means for two groups of cases were compared with two independent-samples Students t-test. The comparison of cerebral DNA levels of bacterial 16S rRNA gene between Il-17a-deficient and wild-type APP-transgenic mice with and without antibiotic treatment were conducted by Mann-Whitney-U-test, because the variables were apparently non-normally distributed. For multiple comparisons, we used one-way or two-way ANOVA followed by Bonferroni, Tukey, or Dunnett T3 post hoc test (dependent on the result of Levene’s test to determine the equality of variances). All statistical analyses were performed with GraphPad Prism 8 version 8.0.2. for Windows (GraphPad Software, San Diego, USA) or SPSS software for Windows (Version 26.0, IBM, Armonk, USA). Other statistical methods in microbiome analysis offered by external companies have been described above. Statistical significance was set at p < 0.05.
Results
Oral treatment with an antibiotic cocktail depletes almost all bacteria in the gut of APP-transgenic mice
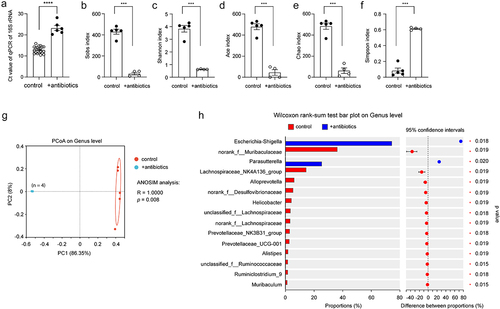
To investigate the relationship between gut and brain, we treated 3-month-old APP-transgenic female littermate mice with and without an antibiotic cocktail in drinking water for 2 months. By quantifying gut bacterial 16S rRNA gene with real-time PCR, we found that antibiotic treatment depleted almost all bacteria in the gut, as the number of remaining bacteria was only 0.08% of that of normal drinking water control mice (; ΔCt value: 10.34 in real-time PCR; t test, p < 0.0001), which was consistent with previous studies that have demonstrated the depletion of gut bacteria by anaerobic and aerobic culture of gut contents.Citation44,Citation51 Not surprisingly, the richness and diversity of the remaining bacteria in the gut of antibiotics-treated AD mice were dramatically reduced, as indicated by decreased Sobs, Ace, Chao, and Shannon indices and increased Simpson index in the α-diversity analysis (; t test, p < 0.05). Similarly, the β-diversity-based PCoA analysis clearly showed the difference in intestinal bacterial architecture between APP-transgenic mice with and without antibiotic treatment (; ANOSIM, R = 1.0000, p = 0.008). We further observed that the remaining antibiotic-resistant bacteria belonged almost exclusively to the genera Escherichia-Shigella and Parasutterella (; Wilcoxon rank-sum test, p < 0.05).
Figure 1. Antibiotic treatment successfully depletes the bacteria in the gut of APP-transgenic mice.
Depletion of gut bacteria reduces Il-17a-expressing CD4-positive lymphocytes in APP-transgenic mice
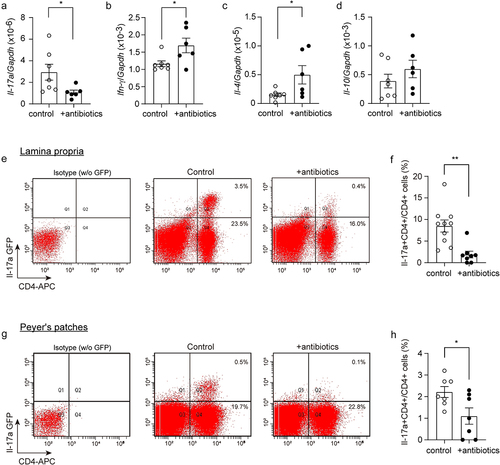
Our recent study showed that Il-17a-expressing CD4-positive lymphocytes increase in the gut and spleen of APP-transgenic mice.Citation28 We therefore selected CD4-positive cells with a magnetic beads-conjugated antibody from the spleen of antibiotics-treated APP-transgenic mice and quantified the transcripts of characteristic genes for Th1, Th2, Th17 and Treg lymphocytes. As shown in , depletion of gut bacteria significantly decreased the transcription of Il- 17a, but increased the transcription of Ifn-γ and Il-4 (t test, p < 0.05). We further treated 3-month-old APP-transgenic mice expressing Il-17a-eGFP reporterCitation43 with and without antibiotics, and observed that depletion of gut bacteria for 2 months significantly reduced eGFP-expressing CD4+ lymphocytes in both lamina propria and Peyer’s patches of the gut compared to control AD mice with normal drinking water (; t test, p < 0.05). Therefore, depletion of intestinal bacteria leads to a significant reduction in Il-17a-expressing CD4-positive T lymphocytes in AD mice. We also found some Il-17a-eGFP-expressing CD4-negative cells in both lamina propria and Peyer’s patches (see upper-left quadrant in ); however, these cells could not form a clearly distinguishable population in both the antibiotics-treated and non-treated AD mice, which made it difficult to reliably quantify cells. Thus, our results do not exclude the possibility that depletion of gut bacteria also regulates Il-17a expression in other immune cells, such as lymphoid-tissue inducer-like cells, natural killer cells, and Paneth cells.Citation52 In our APP-transgenic animal models, expression of GFP was not detected in γδ T lymphocytes in the gut.Citation28
Figure 2. Depletion of gut bacteria reduces Il-17a-expressing CD4-positive lymphocytes.
In additional experiments, we performed an immunohistochemical analysis of GFP in the brain of APP-transgenic mice and of EAE mice that were established in our previous study,Citation53 both of which expressed Il-17a-eGFP. We could not detect GFP-expressing cells in the brain parenchyma of AD mice, but clearly saw GFP-positive cells surrounding blood vessels in the EAE model (Supplementary Fig. S1, A). Similarly, we did not detect Il-17a gene transcripts in the brain tissue from APP-transgenic mice within 40 cycles of real-time PCR (data not shown). These results suggest that gut bacterial depletion alters brain pathology by regulating Il-17a-expressing cells outside the brain.
Depletion of gut bacteria reduces bacterial DNA in the brain of APP-transgenic mice
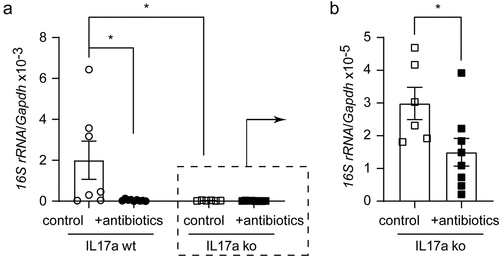
Intestinal bacteria may release structural components into the blood that circulate to the brain, or the bacteria spread directly to the brain.Citation30,Citation54 We then asked whether depletion of gut bacteria alters the potential presence of bacterial components in the brain. Since we observed that depletion of gut bacteria reduced the number of Il-17a-expressing lymphocytes, we asked whether Il-17a affected the amount of bacterial substance in the brain. We treated Il-17a-deficient and wild-type APP-transgenic female mice with antibiotics as described above. Both groups of mice after antibiotic treatment showed enlargement of the ceca and dark-colored cecal contents as shown in published studiesCitation17 (data not shown). Interestingly, antibiotic treatment significantly decreased the DNA level of the bacterial 16S rRNA gene in the brains of both Il-17a-deficient and wild-type APP-transgenic mice (; Mann-Whitney-U-Test, p < 0.05). The DNA level of 16S rRNA gene was significantly lower in Il-17a-deficient than in Il-17a-wild-type APP-transgenic mice without antibiotic treatment (; 16S rRNA/Gapdh: 2.99 ± 0.49 × 10−5 vs. 2.00 ± 0.93 × 10−3; Mann-Whitney-U-Test, U = 4, p = 0.015). After antibiotic treatment, the DNA level of the 16S rRNA gene did not differ between Il-17a-deficient and wild-type AD mice (16S rRNA/Gapdh: 1.49 ± 0.42 × 10−5 [Il-17a-deficient] vs. 4.32 ± 1.32 × 10−5 [Il-17a-wild-type]); Mann-Whitney-U-Test, U = 19, p = 0.102). Thus, Il-17a deficiency potentially blocked the translocation of bacterial components from the intestine to the brain.
Figure 3. Depletion of gut bacteria reduces bacterial DNA in the brains of both Il-17a-deficient and wild-type APP-transgenic mice.
Depletion of intestinal bacteria inhibits proinflammatory activation in the brain of APP-transgenic mice, but not in Il-17a-deficient AD mice
Depletion of intestinal bacteria has been shown to suppress inflammation in the brain of APP-transgenic mice.Citation8,Citation11 We did observe that intestinal antibiotic treatment for 2 months significantly reduced the number of Iba1-positive microglia in both the hippocampus and cortex of female APP-transgenic mice (Hippocampus: from 17.13 ± 1.29 × 103 cells/mm3 to 11.85 ± 0.73 × 103 cells/mm3, and Cortex: from 24.31 ± 0.84 × 103 cells/mm3 to 17.46 ± 2.55 × 103 cells/mm3; ; t test, p < 0.05). Interestingly, in Il-17a-deficient APP-transgenic mice, depletion of gut bacteria did not alter the number of Iba-1-positive cells in the hippocampus (; 16.21 ± 0.57 × 103 cells/mm3 vs. 15.85 ± 0.83 × 103 cells/mm3; t test, p > 0.05). We measured transcripts of proinflammatory genes (Tnf-α, Il-1β, and Ccl-2) and anti-inflammatory genes (Il-10, Chi3l3, and Mrc1) in brains of APP-transgenic mice. As shown in , depletion of intestinal bacteria down-regulated the transcription of Il-1β and Ccl-2, but up-regulated Il-10 transcription in Il-17a-wildtype APP-transgenic mice (t test, p < 0.05). Intestinal bacterial depletion did not change the transcription of Tnf-α, Chi3l3 and Mrc-1 in APP-transgenic mice ( t test, p > 0.05). In Il-17a-deficient AD mice, depletion of intestinal bacteria did not modulate the transcription of Tnf-α, Il-1β, Ccl-2, Il-10, and Mrc-1 (; t test, p > 0.05), except decreasing the transcription of Chi3l3 (; t test, p < 0.05). The results on antibiotics-induced transcriptional regulations of Tnf-α and Il-1β genes were confirmed by ELISA measurements of their protein levels, which showed that depletion of gut bacteria significantly decreased the Il-1β protein concentration (; t test, p < 0.05) and tended to reduce the amount of Tnf-α protein (; t test, p = 0.075) in brains of Il-17a-wildtype but not Il-17a-deficient mice.
Figure 4. Depletion of gut bacteria reduces inflammatory activation in the brain of Il-17a-wildtype, but not Il17a-deficient APP-transgenic mice.
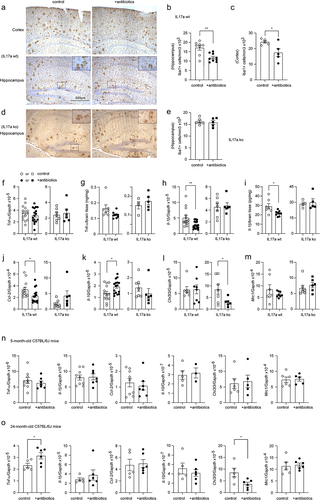
In order to investigate whether the intestinal bacterial depletion-induced inflammatory inhibition was specific for mice with AD pathology, we treated 3 and 22-month-old female C57BL/6J mice with and without antibiotics for 2 months. Depletion of intestinal bacteria did not change transcription of all genes tested in 5-month-old C57BL/6J mice (Tnf-α, Il-1β, Ccl-2, Il-10, Chi3l3, and Mrc1; ; t test, p > 0.05). In 24-month-old C57BL/6J mice, depletion of intestinal bacteria increased Tnf-α transcription and decreased Chi3l3 transcription (; t test, p < 0.05). These experiments also suggested that it was intestinal bacterial depletion instead of antibiotics themselves modifying the inflammatory activation in the brain of APP-transgenic mice.
In order to examine the off-target effects of oral treatment of antibiotics, we injected 5-month-old APP-transgenic mice daily for 7 days with the antibiotic cocktail (1.5 mg/kg/day vancomycin, 3 mg/kg/day ampicillin, neomycin and streptomycin) according to the published protocol.Citation45 We injected metronidazole at the maximal dose of 30 mg/kg/day, because it has a high oral bioavailability.Citation46 Intraperitoneal injection of the antibiotic cocktail did not change the transcription of proinflammatory genes, Tnf-α, Il-1β, and Ccl-2 (Supplementary Fig. S2, A – C; t test, p > 0.05); however, significantly decreased the transcription of anti-inflammatory genes, Il-10, and Mrc1 (Supplementary Fig. S2, D and F; t test, p < 0.05), and tended to inhibit Chi3l3 gene transcription (Supplementary Fig. S2, E; t test, p = 0.055). The pattern of transcriptional modification in the brain by intraperitoneal injection of antibiotic cocktail is obviously different from that in oral antibiotics-treated mice; therefore, the effects of oral antibiotic treatment were due to the depletion of bacteria in the gut and not the antibiotics themselves.
Depletion of intestinal bacteria inhibits microglial activation in the brain of APP-transgenic mice, which is abolished by knockout of Il-17a gene
To understand the mechanism, through which intestinal antibiotic treatment modified neuroinflammation in AD mice, we selected CD11b+ brain cells from 5-month-old female APP-transgenic mice with and without 2-month treatments of antibiotics and detected transcripts of disease-associated microglia (DAM) signature genes.Citation55 Depletion of intestinal bacteria significantly reduced transcription of Il-1β and Ccl-2 genes (; t test, p < 0.05), and tended to decrease the transcription of Tnf-α, Il-10 and Itgax genes (; t test, 0.05 < p < 0.10) in cells derived from Il-17a-wildtype APP-transgenic mice, but not in cells from Il-17a-deficient AD mice. Depletion of intestinal bacteria even increased transcription of Il-10 gene in Il-17a-deficient APP-transgenic mice ( t test, p < 0.05). Gut bacterial depletion down-regulated transcription of Apoe gene in both Il17a-deficient and wild-type APP-transgenic mice (; t test, p < 0.05). Antibiotic treatment did not significantly change the transcription of other genes (Trem2, Cx3cr1, P2ry12, Clec7a, and Lpl) tested in both Il-17a-deficient and wild-type mice (; t test, p > 0.05).
Figure 5. Depletion of gut bacteria inhibits inflammatory activation in microglia in the brain of Il-17a-wildtype, but not Il17a-deficient APP-transgenic mice.
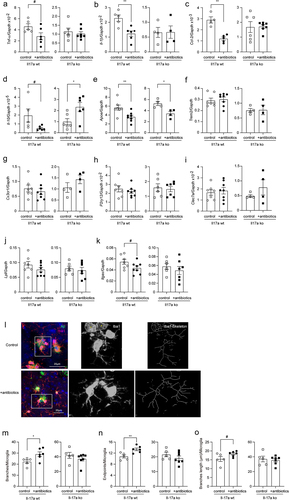
In further experiments, we labeled microglia with Iba-1 antibodies and analyzed the morphology of microglia in the vicinity of Aβ deposits as we had done previously.Citation28 Depletion of intestinal bacteria increased the total number and end points of branching microglial processes (; t test, p < 0.05), and tended to increase branch length (; t test, p = 0.078) in 5-month-old APP-transgenic mice, consistent with previous observations.Citation16,Citation19 However, all changes in microglial morphology caused by gut bacteria depletion were absent in Il-17a-deficient AD mice (; t test, p > 0.05).
As we observed bacterial DNA in the brain, we further investigated whether Il-17a deficiency alters the response of microglia to bacterial components. We found that 5-month-old Il-17a-deficient and wild-type APP-transgenic mice without antibiotic treatment did not differ in the transcription of Myd88, Cd14, Tlr1, Tlr2, Tlr4 and Tlr9 (Supplementary Fig. S1, B), suggesting that knockout of Il-17a gene does not change microglial reactivity to bacteria. Surprisingly, our additional experiments and a previous study showed that the lack of Il-17a did not reduce the transcription of inflammatory genes, Tnf-α, Il-1β, Ccl-2 and Il-10 in microglia (Supplementary Fig. S1, C) and even activate microglia.Citation28
Depletion of intestinal bacteria reduces cerebral Aβ in Il-17a-wildtype but not in Il-17a-deficient APP-transgenic mice
Extracellular Aβ plaques are a pathological hallmark of AD. Depletion of gut bacteria was reported to attenuate Aβ deposits in APP-transgenic mice.Citation7,Citation11 We treated 3-month-old female APP-transgenic mice with an antibiotic cocktail for 2 months. As shown in , 2-month treatments with antibiotics significantly reduced immunoreactive Aβ density from 6.02% ± 0.32% to 4.42% ± 0.64% in the cortex and from 5.67% ± 0.34% to 4.16% ± 0.58% in the hippocampus (t test, p < 0.05) of Il-17a-wildtype AD mice. In Il-17a-deficient APP-transgenic mice, treatment with antibiotics changed the density of Aβ in neither hippocampus nor cortex (; t test, p > 0.05). The brain section was also stained with methoxy-XO4 that typically binds to the β sheet structure of Aβ aggregates. Similarly, treatments with antibiotics significantly reduced Aβ deposits in the cortex (; t test, p < 0.05) and tended to decrease Aβ in the hippocampus (; t test, p = 0.060) of Il-17a-wildtype APP-transgenic mice, but did not change density of methoxy-XO4-stained Aβ deposits in Il-17a-deficient AD mice (; t test, p > 0.05).
Figure 6. Depletion of gut bacteria reduces Aβ in the brain of Il-17a-wildtype, but not Il17a-deficient APP-transgenic mice.
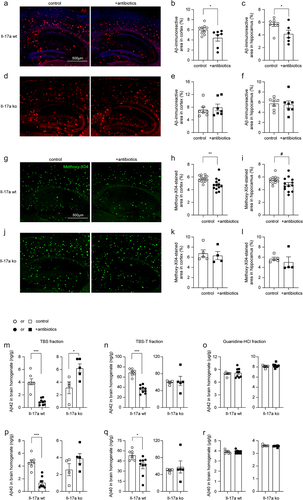
We went on measuring Aβ in the brain homogenate with ELISA. In Il-17a wild-type APP-transgenic mice, antibiotic treatment significantly decreased the levels of both Aβ42 and Aβ40 in TBS- and TBS-T fractions (; t test, p < 0.05), but not in guanidine-soluble fraction (; t test, p > 0.05). TBS-, TBS-T-, and guanidine-soluble brain homogenates are enriched with monomeric, oligomeric and high-molecular-weight aggregated Aβ, respectively.Citation33 In Il-17a-deficient APP-transgenic mice, treatment with antibiotics did not reduce Aβ42 and Aβ40 in all three fractions of brain homogenates (). On the contrary, antibiotic treatment even increased the concentration of Aβ42 in TBS-soluble fraction of brain homogenate (; t test, p < 0.05).
Depletion of intestinal bacteria does not increase microglial Aβ phagocytosis and extracellular Aβ degradation, but potentially reduces Aβ production
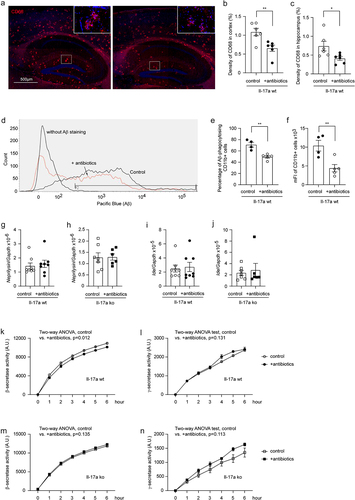
In following experiments, we asked how depletion of intestinal bacteria attenuated the amyloid pathology in AD mice. First, we stained brain tissue with an antibody targeting the lysosomal protein CD68, a suggested marker of phagocytosis. As shown in , depletion of gut bacteria significantly decreased the density of CD68 immunofluorescence (t test, p < 0.05). We then conducted a flow cytometric analysis of brain cells after Aβ staining with methoxy-X04, in which depletion of intestinal bacteria remarkedly decreased both percentage of Aβ-positive CD11b+ brain cells and the mean fluorescence intensity (mFI) of CD11b+ cell population in APP-transgenic mice (; t test, p < 0.05), apparently indicating that microglial internalization of Aβ did not contribute to cerebral Aβ clearance in gut bacteria-depleted AD mice.
Figure 7. Depletion of gut bacteria reduces β-secretase activity in the brain of Il-17a-wildtype, but not Il17a-deficient APP-transgenic mice.
In the same APP-transgenic mouse strain as we used in this study, another group has reported that the absence of gut bacteria increased protein levels of Aβ-degrading enzymes neprilysin and Ide.Citation8 However, our study showed that depletion of intestinal bacteria altered neither Neprilysin nor Ide gene transcription in the brains of Il-17a-deficient and wildtype APP-transgenic mice (; t test, p > 0.05), which suggested that extracellular degradation of Aβ was not the mechanism mediating Aβ reduction in our AD mice.
Our previous study has shown that inhibition of neuroinflammation inhibits β- and γ-secretase activity in the brain of AD mice.Citation20 We found that depletion of intestinal bacteria slightly but significantly decreased β-, but not γ-secretase activity (; two-way ANOVA, antibiotic treatment vs. control, p < 0.05), as observed in a published study.Citation18 It was not surprising that the same inhibitory effects of antibiotic treatment on β- and γ-secretase activity could not be seen in Il-17a-deficient APP-transgenic mice (; two-way ANOVA, antibiotic treatment vs. control, p > 0.05), as the antibiotic treatment did not change the inflammatory activation in the brain of Il-17a-deficient AD mice (see ).
Depletion of intestinal bacteria potentially increases Aβ efflux through blood-brain-barrier in APP-transgenic mice, which is driven by Il-17a inhibition
The transportation of Aβ from brain parenchyma to peripheral circulation is an efficient pathway for the cerebral Aβ clearance.Citation56 LRP1 and ABCB1 are a couple of key transporters at the BBB that are responsible for Aβ efflux.Citation57,Citation58 We found that the protein levels of Abcb1 and Lrp1 were significantly increased in 5-month-old APP-transgenic mice, which had been treated with antibiotics for 2 months (; t test, p < 0.05). Remarkably, the up-regulation of both Lrp1 and Abcb1 in the brain homogenate by depletion of intestinal bacteria was again abolished by knockout of Il-17a gene (; t test, p > 0.05).
Figure 8. Depletion of gut bacteria increases Abcb1 and Lrp1 expression in the blood-brain-barrier of Il-17a-wildtype, but not Il17a-deficient APP-transgenic mice.
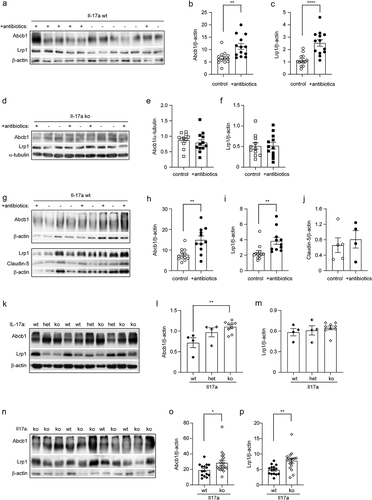
We also isolated blood vessels from 5-month-old APP-transgenic mice with and without treatments with antibiotics. We validated the results from the entire brain homogenate that treatments with antibiotics strongly elevated protein levels of Abcb1 and Lrp1 at the BBB (; t test, p < 0.05). Notably, the protein level of claudin-5 in cerebral blood vessels did not differ between APP-transgenic mice with and without antibiotic treatment (; t test, p > 0.05). Similarly, we did not detect any mouse IgG in brain homogenates of antibiotics-treated AD mice with Western blot (data not shown), which suggests that depletion of intestinal bacteria increased expression of Abcb1 and Lrp1 at the BBB, but did not significantly impair the BBB.
In further experiments, we detected Lrp1 and Abcb1 in the brain homogenates from 5-month-old APP-non-transgenic female mice with different expression of Il-17a. We observed that deficiency of Il-17a significantly increased the protein level of Abcb1, but not Lrp1, in a gene dose-dependent way (; one-way ANOVA, p < 0.05). Similarly, we isolated blood vessels from Il-17a-deficient and wild-type APP-transgenic mice and observed that deficiency of Il-17a significantly increased both Lrp1 and Abcb1 in the tissue lysate of blood vessels (; t test, p < 0.05), suggesting that depletion of intestinal bacteria potentially increases Lrp1/Abcb1-mediated Aβ efflux through inhibiting Il-17a signaling.
Depletion of intestinal bacteria potentially promotes synaptic plasticity in APP-transgenic mice, which is abolished by deficiency of Il-17a
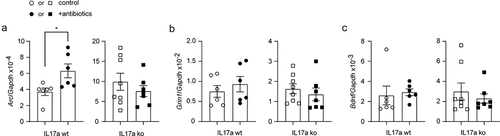
After finding that gut bacterial depletion attenuated inflammatory activation and amyloid pathology in the brains of APP-transgenic mice, we performed a preliminary analysis of the neuroprotective effect of antibiotic treatment. Our previous study has shown that upregulated transcription of the immediate early gene Arc and the gene Grin1, which encodes subunit 1 of the NMDA-type ionotropic glutamate receptor, is associated with the improvement of cognitive function in APP-transgenic mice.Citation35 BDNF plays an important role in the maintenance of synaptic plasticity in learning and memory,Citation59 the transcript of which is influenced by gut bacteria.Citation60 Therefore, we measured the transcripts of Arc, Grin1 and Bdnf genes in brains of 5-month-old APP-transgenic mice with and without 2-month treatment of antibiotics. As shown in , treatment with the antibiotic cocktail significantly increased the transcription of Arc, but not Grin1 and Bdnf genes in Il-17a-wildtype APP mice (t test, p < 0.05). Again, knockout of Il-17a gene abolished the neuroprotective effect of antibiotic treatment (; t test, p > 0.05). However, the neuroprotective effects of antibiotic treatment should be further investigated by other methods, such as behavioral tests and electrophysiological approaches, in older (e.g., 9-month-old) APP-transgenic mice as we did previously.Citation20,Citation28
Figure 9. Depletion of gut bacteria increases Arc transcription in the brain of Il-17a-wildtype, but not Il17a-deficient APP-transgenic mice.
Discussion
Gut bacteria contribute to AD development,Citation61 but how gut bacteria regulate brain pathology remains unclear. By using young female APP-transgenic mice with rapidly developing Aβ pathology, we found that depletion of gut bacteria decreased microglial inflammatory activation and Aβ levels, particularly soluble Aβ, and might promote synaptic plasticity in the brain. These effects were abolished by knockout of Il-17a gene. The attenuation of cerebral Aβ pathology by gut bacterial depletion may be attributed to the inhibition of β-secretase activity and the upregulation of Aβ efflux-related Lrp1 and Abcb1 expression in the brain, which was also abrogated by Il-17a deficiency.
Bacterial phenotypes are potentially transferred between contacting mice.Citation86 We co-housed 4–6 Il-17a-deficient and wild-type female AD mice in the same cage for treatments with and without antibiotics, which reduced the variability caused by different housing conditions. Depletion of gut bacteria in APP-transgenic mice inhibited proinflammatory activation in the brain tissue as evidenced by reduction of microglial number, down-regulation of Il-1β and Ccl-2 transcription and up-regulation of Il-10 transcription, consistent with published studies.Citation8,Citation10,Citation18 In individual microglia, depletion of gut bacteria decreased transcripts of both pro- (e.g., Tnf-α, Il-1β, Ccl-2) and anti-inflammatory (Il-10) genes, which is also in agreement with the published finding that the absence of gut bacteria leads to global defects in microglial activation.Citation17 The opposing regulation of Il-10 transcription in microglia and brain tissue suggests that brain cells other than microglia, such as astrocytes, may also respond to gut bacteriaCitation62 and produce Il-10 in the brain.
We noted that intestinal bacterial depletion in 5-month-old C57BL/6J mice did not have the same effect on the inflammatory modulation as in AD mice, and even promoted inflammatory activation (e.g., transcriptional up-regulation of Tnf-α, and down-regulation of Chi3l3) in the brain of 24-month-old C57BL/6J mice. In fact, the mechanism leading to inflammatory activation in the brain, and particularly in microglia, is not the same in AD and healthy aging. For example, inflammation in aging microglia may be due to a reduced ability to clear endogenous metabolites, whereas microglia in AD mice respond efficiently to exogenous Aβ. A transcriptomic studyCitation63 identified 85 aging-associated genes in female C57BL/6J mice, of which only 21 genes overlapped with DAM genes that were identified in APP-transgenic mouse model,Citation55 with the exception of the DAM-characteristic genes Trem2 and Apoe. Thus, it stands to reason that a significant inflammatory modulation caused by gut bacterial depletion depends on the preactivated status and pattern of inflammatory brain cells (e.g., microglia).
Depletion of gut bacteria resulted in a significant reduction of Il-17a-expressing CD4-positive cells in the gut and spleen as well as bacterial DNA in brain tissue, suggesting that gut bacteria regulate brain pathology in AD mice via at least two pathways: 1) regulating peripheral Il-17a-expressing T lymphocytes and 2) directly modulating cells in the brain with bacterial components (not necessary living cells). Very interestingly, we also observed that deficiency of Il-17a reduces bacterial DNA in the brain of APP-transgenic mice. Due to our limited research resource, we have not yet succeeded in the microbiome analysis of brain tissues. The recent study detected Streptococcus, Clostridium, Roseburia and Tyzzerella in the brain of APP/PS1 mice.Citation54 The bacteria from Cutibacterium acnes were found in the brain of AD patient and linked to AD pathogenesis.Citation64 How the bacteria or their structural components enter the brain is unclear. It has been reported that deficiency of Il-17a alters the composition of gut bacteria, i.e., increases the abundance of Barnesiella genus bacteria, and hinders the development of EAE.Citation65 Barnesiella bacteria are the main source of hypoxanthine in the gut. Intestinal epithelial cells utilize hypoxanthine for energy balance and nucleotide biosynthesis.Citation66 Thus, Il-17a deficiency may favor intestinal barrier function and lead to the reduction of bacterial DNA in the brain of APP-transgenic mice. Inhibition of Il-17a signaling, which can be achieved by administering Il-17a inhibitors or by depleting specific bacterial taxa in the gut that stimulate the development of Il-17a-producing T cells, therefore has the potential to avoid bacterial overload in the brain and prevent AD progression.
Exactly how the depletion of bacteria in the gut and subsequent Il-17a inhibition, in particular the reduction of Th17 cells, regulates microglial activity in our antibiotic-treated AD mice remains unclear. Il-17a inhibition itself does not appear to inhibit microglial activation. We have recently observed that knockout of Il-17a gene reduces microglial branches in APP-transgenic mice,Citation28 suggesting activation of microglial inflammatory response.Citation67 Our current study shows that Il-17a deficiency did not reduce the transcription of Tnf-α, Il-1β, Ccl-2 and Il-10 genes in isolated microglia (Supplementary Fig. S1, C). The inhibition of inflammation in both brain tissue and microglia in our antibiotic-treated AD mice may be due to the depletion of bacteria other than Il-17a-promoting taxa in the gut. In Il-17a-deficient APP-transgenic mice, antibiotic treatment did not inhibit inflammatory activation in brain tissue and microglia, for which there are perhaps several reasons. Deficiency of Il-17a reduced soluble Aβ (discussed later) and bacterial DNA, minimizing the inflammatory priming of microglia as in 5-month-old C57BL/6J mice, limiting the capacity to further inhibit inflammation by depleting gut bacteria. However, we believe that there are other unknown mechanisms by which Il-17a inhibition maintains and even enhances microglial activity in the brain, which we will investigate in the future studies. To answer this question, it is helpful to identify the Il-17a signaling-promoting bacteria or their metabolites in the gut of AD mice. Unfortunately, the available relevant results for today are very limited or controversial. For example, the bacterial product valeric acid was reported to increase the concentrations of Il-17, Il-1β, and Il-6 in the blood and brain of mice with experimental stroke.Citation68 However, treatment with valeric acid was also shown to reduce the concentrations of Tnf-α and Il-6 in the blood of mice after irradiation.Citation69 In addition, activation of the valeric acid receptor Gpr43 inhibited neuroinflammation and improved cognitive function in APP-transgenic mice.Citation21
It should be noted that microglial activation is a double-edged sword in AD pathogenesis: on the one side, it releases cytotoxic cytokines and reactive oxygen species, leading to neuronal damage; on the other side, it clears neurotoxic Aβ via microglial internalization, thereby safeguarding neurons.Citation70 In our recent study, p38α-MAPK deficiency in myeloid cells or Il-17a deficiency promotes microglial activation and Aβ clearance, which improves cognitive performance in APP-transgenic mice.Citation28 We and other groups have also shown that stimulation of TLR4 and TLR9 by injection of a low dose of LPS or synthetic oligodeoxynucleotides containing unmethylated CpG dinucleotides, similar to those found in bacterial DNA, induces mild and long-term microglial activation that facilitates the clearance of Aβ and hyperphosphorylated tau protein in human APP or tau -transgenic mice,Citation71–74 and in squirrel monkeys.Citation75 Our present study has shown that general depletion of gut bacteria reduces microglial phagocytosis of Aβ in APP-transgenic mice, which is consistent with previous observations that gut bacteria are essential for microglial maturation, activation and Aβ phagocytosis.Citation16–19 We would expect that specific depletion of Il-17a-promoting gut bacteria could maintain microglial activity and help clear the pathogenic Aβ molecules from the AD brain.
When studying the gut and brain, how gut bacteria modulate cerebral Aβ load is always an important question to answer. Our present study showed that microglia are not responsible for cerebral Aβ reduction in APP-transgenic mice after depletion of gut bacteria. Interestingly, we observed that depletion of gut bacteria increased the expression of Lrp1 and Abcb1 in the BBB of APP-transgenic mice, which was reversed by knockout of Il-17a gene. Knocking out Il-17a gene completely mimicked the effects of gut bacteria depletion on the expression of Lrp1 and Abcb1. Lrp1 and Abcb1 are two important transporters responsible for Aβ efflux across the BBB.Citation57,Citation58 Depletion or inhibition of endothelial Lrp1 and Abcb1 leads to Aβ accumulation in the AD mouse brain.Citation76,Citation77 Since transportation of Aβ from brain parenchyma to peripheral plasma might contribute 25% of total clearance of cerebral Aβ,Citation56 Aβ efflux represents a major pathway mediating the reduction of Aβ in the brain of AD mice following depletion of gut bacteria. It has been reported that the absence of ApoE retains less Aβ in the brain parenchyma and increases soluble Aβ in the interstitial fluid, possibly promoting the diffusion of soluble Aβ from the parenchyma into the perivascular space,Citation78 which favors Aβ efflux. Notably, depletion of gut bacteria decreased Apoe gene transcription in microglia, as observed in our study and by other scientists,Citation18 which may indicate the cooperation between microglia and BBB in the clearance of cerebral Aβ.
Aβ is produced by β- (BACE1) and γ-secretases after serial digestions of APP. The published studies by our group and others have shown that the activity or protein level of β- and γ-secretases is regulated by inflammatory activation in the brain.Citation20,Citation79,Citation80 Not surprisingly, depletion of gut bacteria inhibited β-secretase activity in correlation with inflammatory inhibition in our APP-transgenic mice, which is consistent with the observation in germ-free AD mice.Citation18 We did not detect an increase in the expression of neprilysin and Ide in our APP-transgenic mice after antibiotic treatment, suggesting that extracellular degradation of Aβ is not the key mechanism for Aβ clearance after gut bacterial depletion. This result differs from the observation in a study on germ-free AD mice.Citation8
Antibiotic therapy for AD patients has gained interest.Citation37 In several studies, germ-free APP-transgenic mice show better cognitive function and lower cerebral Aβ levels than specific pathogen-free (SPF) AD mice.Citation18,Citation81 Our study supports antibiotic therapy, as gut bacterial depletion reduced Aβ burden in AD mice and, in particular, increased Aβ efflux-associated Abcb1 and Lrp1 across the BBB. Depletion of gut bacteria also up-regulated the transcription of Arc gene in the brain, which is associated with synaptic plasticity and cognitive improvement in APP-transgenic mice.Citation35,Citation82 However, as discussed above, the general depletion of gut bacteria inhibits the activation of microglia, which decreases the efficiency of Aβ clearance. In addition, gut bacteria have also been shown to promote the development of cognitive performance.Citation83 Adult germ-free mice transplanted with fecal microbiota from 2–3-month-old mice perform better cognitively than mice transplanted with fecal microbiota from 18–20-month-old mice.Citation84 Treatment with broad-spectrum antibiotics can deplete as well the taxa of gut bacteria that favor learning and memory. Our study also indicated that bacteria belonging to the genera Escherichia-Shigella and Parasutterella were more resistant to antibiotic treatment. The possible expansion of these bacteria, especially Escherichia-Shigella bacteria, after long-term antibiotic treatment could increase inflammatory activation in the brain and accelerate the progression of AD. The combination of our results and the literature indicates that it is important to manipulate a specific bacterial taxon, e.g. by reducing the bacteria that stimulate immune cells to produce Il-17a. Currently, antibiotic therapy targeting specific bacterial taxa is still a major challenge for AD patients.
A limitation of our study is that the direct anti-inflammatory effects of antibiotics orally used to deplete gut bacteria could not be excluded. Although oral vancomycin, neomycin, streptomycin and ampicillin have low systemic absorption,Citation60 the oral bioavailability of metronidazole is high (98.9%).Citation46 Metronidazole must not be omitted from the antibiotic cocktail as it decimates anaerobic bacteria in the gut. Metronidazole has the potential to suppress the activity of both innate and acquired immune systems.Citation85 We observed that intraperitoneal injection of the antibiotic cocktail inhibited inflammatory gene transcription in the brain; however, the affected genes were mainly limited to anti-inflammatory Il-10 and Mrc1 genes (see Supplementary Figure S2), with a pattern different from the inflammatory regulation caused by oral treatment of antibiotics. We also observed that oral antibiotic treatment decreased Il-17a transcription but increased transcription of Ifn-γ and Il-4 genes in CD4-positive spleen cells in APP-transgenic mice and promoted inflammatory activation in the brains of 24-month-old C57BL/6J mice. Indeed, oral antibiotic (amoxicillin/clavulanate) treatment of mice with antibiotic-sensitive and -resistant bacteria in the gut has shown that depletion of sensitive gut bacteria inhibits the development of Il-17a-expressing γδ T cells.Citation25 Therefore, we have reason to believe that the inhibition of neuroinflammation and Th17 development in our AD animal models was not due to the direct effects of antibiotics, but was a consequence of the depletion of gut bacteria. The potential inhibitory effect of antibiotics on immune cells is a common pitfall in antibiotic-treated mice in many gut and brain studies.Citation7,Citation17,Citation19
In summary, our results are consistent with published observations that depletion of gut bacteria attenuates microglial inflammatory activation and Aβ pathology in the brain of APP-transgenic mice; however, we further elucidated the possible mechanisms mediating the gut-brain axis in AD pathogenesis: 1) depletion of gut bacteria reduces peripherally circulating Il-17a-expressing T lymphocytes and translocation of bacterial components from the gut to the brain. Il-17a deficiency inhibits this bacterial translocation; 2) inhibition of Il-17a possibly maintains and even enhances microglial activity; 3) inhibition of Il-17a upregulates the expression of Lrp1 and Abcb1 in the BBB, possibly promoting Aβ efflux and Aβ clearance in the brain; and 4) depletion of gut bacteria has the potential to improve neuronal plasticity. Our study supports antibiotic therapy for AD patients; however, precise therapy targeting specific bacterial taxa is still a huge challenge. New methods and future work are needed to characterize the AD-specific microbiome profile in the gut. Particular attention should be paid to how Il-17a-associated intestinal bacteria can be manipulated.
Author contributions
Y.L. conceptualized and designed the study, acquired funding, conducted experiments, acquired and analyzed data, and wrote the manuscript. W.H., Q.L., I.T., and W.Q. conducted experiments, acquired data and analyzed data. T.H. provided resource and discussed study design. M.M. provided an animal facility and supervised animal experiments. K.F. provided a research laboratory and supervised the laboratory work. All authors contributed to the article and approved the submitted version.
Supplementary Figures 220424.docx
Download MS Word (3.9 MB)Acknowledgments
We thank Prof. M. Jucker (Hertie Institute for Clinical Brain Research, Tübingen) for providing APP/PS1-transgenic mice, Prof. Y. Iwakura (Tokyo University of Science) for Il-17a knockout mice and Prof. R. Flavell (Yale University) for Il-17a-eGFP reporter mice. We appreciate Elisabeth Gluding and Kati Jordan for their excellent technical assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Our study has not generated new datasets. All data generated or analyzed during this study are included in this published article and its supplementary information files.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2024.2363014
Additional information
Funding
References
- Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K. et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537. doi:10.1038/s41598-017-13601-y.
- Dunham SJB, McNair KA, Adams ED, Avelar-Barragan J, Forner S, Mapstone M, Whiteson KL, Huffnagle GB. Longitudinal analysis of the microbiome and metabolome in the 5xfAD Mouse Model of Alzheimer’s Disease. mBio. 2022;13(6):e0179422. doi:10.1128/mbio.01794-22.
- Meyer K, Lulla A, Debroy K, Shikany JM, Yaffe K, Meirelles O, Launer LJ. Association of the gut microbiota with cognitive function in midlife. JAMA Netw Open. 2022;5(2):e2143941. doi:10.1001/jamanetworkopen.2021.43941.
- Zhang T, Gao G, Kwok LY, Sun Z. Gut microbiome-targeted therapies for Alzheimer’s disease. Gut Microbes. 2023;15(2):2271613. doi:10.1080/19490976.2023.2271613.
- Ma C, Li Y, Mei Z, Yuan C, Kang JH, Grodstein F, Ascherio A, Willett WC, Chan AT, Huttenhower C. et al. Association between bowel movement pattern and cognitive function: Prospective cohort study and a metagenomic analysis of the gut microbiome. Neurology. 2023;101(20):e2014–27. doi:10.1212/WNL.0000000000207849.
- Grabrucker S, Marizzoni M, Silajdzic E, Lopizzo N, Mombelli E, Nicolas S, Dohm-Hansen S, Scassellati C, Moretti DV, Rosa M. et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain. 2023;146(12):4916–4934. doi:10.1093/brain/awad303.
- Dodiya HB, Kuntz T, Shaik SM, Baufeld C, Leibowitz J, Zhang X, Gottel N, Zhang X, Butovsky O, Gilbert JA. et al. Sex-specific effects of microbiome perturbations on cerebral Abeta amyloidosis and microglia phenotypes. J Exp Med. 2019;216(7):1542–1560. doi:10.1084/jem.20182386.
- Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, Neher JJ, Fak F, Jucker M, Lasser T. et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. 2017;7(1):41802. doi:10.1038/srep41802.
- Kaur H, Nookala S, Singh S, Mukundan S, Nagamoto-Combs K, Combs CK. Sex-dependent effects of intestinal microbiome manipulation in a mouse model of alzheimer’s disease. Cells. 2021;10(9):2370. doi:10.3390/cells10092370.
- Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, Oyler-Castrillo P, Zhang X, Musch MW, Shen X. et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of Alzheimer’s disease. Sci Rep. 2017;7(1):10411. doi:10.1038/s41598-017-11047-w.
- Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, Musch MW, Liao F, Ward JF, Holtzman DM. et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep. 2016;6(1):30028. doi:10.1038/srep30028.
- Liu P, Wu L, Peng G, Han Y, Tang R, Ge J, Zhang L, Jia L, Yue S, Zhou K. et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav Immun. 2019;80:633–643. doi:10.1016/j.bbi.2019.05.008.
- Murray ER, Kemp M, Nguyen TT. The microbiota-gut-brain axis in alzheimer’s disease: A review of taxonomic alterations and potential avenues for Interventions. Arch Clin Neuropsychol. 2022;37(3):595–607. doi:10.1093/arclin/acac008.
- Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, Ferrari C, Guerra UP, Paghera B, Muscio C. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–68. doi:10.1016/j.neurobiolaging.2016.08.019.
- Vital M, Karch A, Pieper DH, Shade A. Colonic Butyrate-Producing Communities in Humans: an overview using omics data. mSystems. 2017;2(6). doi:10.1128/mSystems.00130-17.
- Erny D, Dokalis N, Mezo C, Castoldi A, Mossad O, Staszewski O, Frosch M, Villa M, Fuchs V, Mayer A. et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021;33(11):2260–2276 e2267. doi:10.1016/j.cmet.2021.10.010.
- Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–977. doi:10.1038/nn.4030.
- Colombo AV, Sadler RK, Llovera G, Singh V, Roth S, Heindl S, Sebastian Monasor L, Verhoeven A, Peters F, Parhizkar S. et al. Microbiota-derived short chain fatty acids modulate microglia and promote Abeta plaque deposition. Elife. 2021;10:e59826. doi:10.7554/eLife.59826.
- Xie J, Bruggeman A, De Nolf C, Vandendriessche C, Van Imschoot G, Van Wonterghem E, Vereecke L, Vandenbroucke RE. Gut microbiota regulates blood-cerebrospinal fluid barrier function and Abeta pathology. Embo J. 2023;42(17):e111515. doi:10.15252/embj.2022111515.
- Quan W, Luo Q, Hao W, Tomic I, Furihata T, Schulz-Schaffer W, Menger MD, Fassbender K, Liu Y. Haploinsufficiency of microglial MyD88 ameliorates Alzheimer’s pathology and vascular disorders in APP/PS1-transgenic mice. Glia. 2021;69(8):1987–2005. doi:10.1002/glia.24007.
- Zhou Y, Xie L, Schroder J, Schuster IS, Nakai M, Sun G, Sun YBY, Marino E, Degli-Esposti MA, Marques FZ. et al. Dietary fiber and microbiota metabolite receptors enhance cognition and alleviate disease in the 5xFAD Mouse model of alzheimer’s disease. J Neurosci. 2023;43(37):6460–6475. doi:10.1523/JNEUROSCI.0724-23.2023.
- Dupraz L, Magniez A, Rolhion N, Richard ML, Da Costa G, Touch S, Mayeur C, Planchais J, Agus A, Danne C. et al. Gut microbiota-derived short-chain fatty acids regulate IL-17 production by mouse and human intestinal γδ T cells. Cell Rep. 2021;36(1):109332. doi:10.1016/j.celrep.2021.109332.
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–139. doi:10.1016/j.immuni.2013.12.007.
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108 Suppl 1(supplement_1):4615–4622. doi:10.1073/pnas.1000082107.
- Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG. et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. 2016;22(5):516–523. doi:10.1038/nm.4068.
- Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, Tsitsou-Kampeli A, Sarel A, Cahalon L, Schwartz M. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun. 2015;6(1):7967. doi:10.1038/ncomms8967.
- Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, Cagnard N, Holzenberger M, Piaggio E, Aucouturier P. et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. 2016;139(Pt 4):1237–1251. doi:10.1093/brain/awv408.
- Luo Q, Schnoder L, Hao W, Litzenburger K, Decker Y, Tomic I, Menger MD, Liu Y, Fassbender K. p38alpha-MAPK-deficient myeloid cells ameliorate symptoms and pathology of APP-transgenic Alzheimer’s disease mice. Aging Cell. 2022;21(8):e13679. doi:10.1111/acel.13679.
- Marizzoni M, Mirabelli P, Mombelli E, Coppola L, Festari C, Lopizzo N, Luongo D, Mazzelli M, Naviglio D, Blouin JL. et al. A peripheral signature of Alzheimer’s disease featuring microbiota-gut-brain axis markers. Alzheimers Res Ther. 2023;15(1):101. doi:10.1186/s13195-023-01218-5.
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C. et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. doi:10.1126/sciadv.aau3333.
- Fassbender K, Walter S, Kuhl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ. et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2004;18(1):203–205. doi:10.1096/fj.03-0364fje.
- Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T. et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. 2012;188(3):1098–1107. doi:10.4049/jimmunol.1101121.
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128(Pt 8):1778–1789. doi:10.1093/brain/awh531.
- O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–460. doi:10.1038/nri3446.
- Hao W, Liu Y, Liu S, Walter S, Grimm MO, Kiliaan AJ, Penke B, Hartmann T, Rube CE, Menger MD. et al. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer’s disease-related symptoms and pathology. Brain. 2011;134(Pt 1):278–292. doi:10.1093/brain/awq325.
- Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W. et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007;20(6):947–956. doi:10.1159/000110455.
- Panza F, Lozupone M, Solfrizzi V, Watling M, Imbimbo BP. Time to test antibacterial therapy in Alzheimer’s disease. Brain. 2019;142(10):2905–2929. doi:10.1093/brain/awz244.
- Rakusa E, Fink A, Tamguney G, Heneka MT, Doblhammer G. Sporadic use of antibiotics in older adults and the risk of dementia: a nested case-control study based on German health claims data. J Alzheimers Dis. 2023;93(4):1329–1339. doi:10.3233/JAD-221153.
- Drummond RA, Desai JV, Ricotta EE, Swamydas M, Deming C, Conlan S, Quinones M, Matei-Rascu V, Sherif L, Lecky D. et al. Long-term antibiotic exposure promotes mortality after systemic fungal infection by driving lymphocyte dysfunction and systemic escape of commensal bacteria. Cell Host Microbe. 2022;30(7):1020–1033 e1026. doi:10.1016/j.chom.2022.04.013.
- Nebel RA, Aggarwal NT, Barnes LL, Gallagher A, Goldstein JM, Kantarci K, Mallampalli MP, Mormino EC, Scott L, Yu WH. et al. Understanding the impact of sex and gender in Alzheimer’s disease: A call to action. Alzheimers Dement. 2018;14(9):1171–1183. doi:10.1016/j.jalz.2018.04.008.
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S. et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7(9):940–946. doi:10.1038/sj.embor.7400784.
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17(3):375–387. doi:10.1016/S1074-7613(02)00391-6.
- Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O’Connor W Jr., Rongvaux A, Van Rooijen N, Haberman AM. et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475(7357):514–518. doi:10.1038/nature10228.
- Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, Bry L, Comstock LE, Gandhi R, Weiner HL. The host shapes the gut microbiota via fecal MicroRNA. Cell Host Microbe. 2016;19(1):32–43. doi:10.1016/j.chom.2015.12.005.
- Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J, Deng Y, Blennerhassett P, Macri J, Kd M. et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology. 2011;141(2):599–609, 609 e591–593. doi:10.1053/j.gastro.2011.04.052.
- Jensen JC, Gugler R. Single- and multiple-dose metronidazole kinetics. Clin Pharmacol Ther. 1983;34(4):481–487. doi:10.1038/clpt.1983.201.
- Lau SF, Wu W, Seo H, Aky F, Ip NY. Quantitative in vivo assessment of amyloid-beta phagocytic capacity in an Alzheimer’s disease mouse model. STAR Protoc. 2021;2(1):100265. doi:10.1016/j.xpro.2020.100265.
- Couter CJ, Surana NK. Isolation and flow cytometric characterization of murine small intestinal lymphocytes. JoVE (J Visualized Exp). 2016;111:e54114.
- Liu Y, Liu X, Hao W, Decker Y, Schomburg R, Fulop L, Pasparakis M, Menger MD, Fassbender K. IKKbeta deficiency in myeloid cells ameliorates Alzheimer’s disease-related symptoms and pathology. J Neurosci. 2014;34(39):12982–12999. doi:10.1523/JNEUROSCI.1348-14.2014.
- Hao W, Decker Y, Schnoder L, Schottek A, Li D, Menger MD, Fassbender K, Liu Y. Deficiency of IkappaB kinase beta in myeloid cells reduces severity of experimental autoimmune encephalomyelitis. Am J Pathol. 2016;186(5):1245–1257. doi:10.1016/j.ajpath.2016.01.004.
- Benjamin JL, Sumpter R Jr., Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. 2013;13(6):723–734. doi:10.1016/j.chom.2013.05.004.
- Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479–489. doi:10.1038/nri2800.
- Hao W, Luo Q, Menger MD, Fassbender K, Liu Y. Treatment with CD52 antibody protects neurons in experimental autoimmune encephalomyelitis mice during the recovering phase. Front Immunol. 2021;12:792465. doi:10.3389/fimmu.2021.792465.
- Thapa M, Kumari A, Chin C-Y, Choby JE, Jin F, Bogati B, Chopyk DM, Koduri N, Pahnke A, Elrod EJ. et al. Translocation of gut commensal bacteria to the brain. bioRxiv. 2023;2023.08.30:555630. doi: 10.1101/2023.08.30.555630.
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B. et al. A unique microglia type associated with restricting development of alzheimer’s disease. Cell. 2017;169(7):1276–1290 e1217. doi:10.1016/j.cell.2017.05.018.
- Roberts KF, Elbert DL, Kasten TP, Patterson BW, Sigurdson WC, Connors RE, Ovod V, Munsell LY, Mawuenyega KG, Miller-Thomas MM. et al. Amyloid-β efflux from the central nervous system into the plasma. Ann Neurol. 2014;76(6):837–844. doi:10.1002/ana.24270.
- Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, Cascorbi I, Walker LC, Kroemer HK, Warzok RW. et al. MDR1-P-Glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-beta peptides–implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007;17(4):347–353. doi:10.1111/j.1750-3639.2007.00075.x.
- Shinohara M, Tachibana M, Kanekiyo T, Bu G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: evidence from clinical and preclinical studies. J Lipid Res. 2017;58(7):1267–1281. doi:10.1194/jlr.R075796.
- Gao L, Zhang Y, Sterling K, Song W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl Neurodegener. 2022;11(1):4. doi:10.1186/s40035-022-00279-0.
- Frohlich EE, Farzi A, Mayerhofer R, Reichmann F, Jacan A, Wagner B, Zinser E, Bordag N, Magnes C, Frohlich E. et al. Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immun. 2016;56:140–155. doi:10.1016/j.bbi.2016.02.020.
- Chandra S, Sisodia SS, Vassar RJ. The gut microbiome in Alzheimer’s disease: what we know and what remains to be explored. Mol Neurodegener. 2023;18(1):9. doi:10.1186/s13024-023-00595-7.
- Chandra S, Di Meco A, Dodiya HB, Popovic J, Cuddy LK, Weigle IQ, Zhang X, Sadleir K, Sisodia SS, Vassar R. The gut microbiome regulates astrocyte reaction to Abeta amyloidosis through microglial dependent and independent mechanisms. Mol Neurodegener. 2023;18(1):45. doi:10.1186/s13024-023-00635-2.
- Li X, Li Y, Jin Y, Zhang Y, Wu J, Xu Z, Huang Y, Cai L, Gao S, Liu T. et al. Transcriptional and epigenetic decoding of the microglial aging process. Nat Aging. 2023;3(10):1288–1311. doi:10.1038/s43587-023-00479-x.
- Mone Y, Earl JP, Krol JE, Ahmed A, Sen B, Ehrlich GD, Lapides JR. Evidence supportive of a bacterial component in the etiology for Alzheimer’s disease and for a temporal-spatial development of a pathogenic microbiome in the brain. Front Cell Infect Microbiol. 2023;13:1123228. doi:10.3389/fcimb.2023.1123228.
- Regen T, Isaac S, Amorim A, Núñez NG, Hauptmann J, Shanmugavadivu A, Klein M, Sankowski R, Mufazalov IA, Yogev N. et al. IL-17 controls central nervous system autoimmunity through the intestinal microbiome. Sci Immunol. 2021;6(56):eaaz6563. doi:10.1126/sciimmunol.aaz6563.
- Lee JS, Wang RX, Alexeev EE, Colgan SP. Intestinal Inflammation as a dysbiosis of energy procurement: New Insights into an old topic. Gut Microbes. 2021;13(1):1–20. doi:10.1080/19490976.2021.1880241.
- Madry C, Kyrargyri V, Arancibia-Carcamo IL, Jolivet R, Kohsaka S, Bryan RM, Attwell D. Microglial Ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK-1. Neuron. 2018;97(2):299–312 e296. doi:10.1016/j.neuron.2017.12.002.
- Zeng X, Li J, Shan W, Lai Z, Zuo Z. Gut microbiota of old mice worsens neurological outcome after brain ischemia via increased valeric acid and IL-17 in the blood. Microbiome. 2023;11(1):204. doi:10.1186/s40168-023-01648-1.
- Li Y, Dong J, Xiao H, Zhang S, Wang B, Cui M, Fan S. Gut commensal derived-valeric acid protects against radiation injuries. Gut Microbes. 2020;11(4):789–806. doi:10.1080/19490976.2019.1709387.
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5.
- Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C. et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid a improves Alzheimer’s disease-related pathology. Proc Natl Acad Sci USA. 2013;110(5):1941–1946. doi:10.1073/pnas.1215165110.
- Qin Y, Liu Y, Hao W, Decker Y, Tomic I, Menger MD, Liu C, Fassbender K. Stimulation of TLR4 attenuates alzheimer’s disease-related symptoms and pathology in tau-transgenic mice. J Immunol. 2016;197(8):3281–3292. doi:10.4049/jimmunol.1600873.
- Scholtzova H, Chianchiano P, Pan J, Sun Y, Goni F, Mehta PD, Wisniewski T. Amyloid ß and Tau Alzheimer’s disease related pathology is reduced by toll-like receptor 9 stimulation. Acta Neuropathol Commun. 2014;2(1):101. doi:10.1186/PREACCEPT-2151623761356337.
- Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, Mehta PD, Spinner DS, Wisniewski T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29(6):1846–1854. doi:10.1523/JNEUROSCI.5715-08.2009.
- Patel AG, Nehete PN, Krivoshik SR, Pei X, Cho EL, Nehete BP, Ramani MD, Shao Y, Williams LE, Wisniewski T. et al. Innate immunity stimulation via CpG oligodeoxynucleotides ameliorates Alzheimer’s disease pathology in aged squirrel monkeys. Brain. 2021;144(7):2146–2165. doi:10.1093/brain/awab129.
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR. et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115(11):3285–3290. doi:10.1172/JCI25247.
- Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I. et al. Endothelial LRP1 transports amyloid-beta(1-42) across the blood-brain barrier. J Clin Invest. 2016;126(1):123–136. doi:10.1172/JCI81108.
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O’Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM. et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41(2):193–202. doi:10.1016/S0896-6273(03)00850-X.
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178(5):829–841. doi:10.1083/jcb.200705042.
- Hur J-Y, Frost GR, Wu X, Crump C, Pan SJ, Wong E, Barros M, Li T, Nie P, Zhai Y. et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature. 2020;586(7831):735–740. doi:10.1038/s41586-020-2681-2.
- Mezo C, Dokalis N, Mossad O, Staszewski O, Neuber J, Yilmaz B, Schnepf D, de Aguero MG, Ganal-Vonarburg SC, Macpherson AJ. et al. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun. 2020;8(1):119. doi:10.1186/s40478-020-00988-5.
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28(46):11760–11767. doi:10.1523/JNEUROSCI.3864-08.2008.
- Cerdo T, Ruiz-Rodriguez A, Acuna I, Torres-Espinola FJ, Menchen-Marquez S, Gamiz F, Gallo M, Jehmlich N, Haange SB, von Bergen M. et al. 1974. Infant gut microbiota contributes to cognitive performance in mice. Cell Host Microbe. 2023;31(12):1974–1988 e. doi:10.1016/j.chom.2023.11.004.
- Lee J, Venna VR, Durgan DJ, Shi H, Hudobenko J, Putluri N, Petrosino J, Ld M, Bryan RM. Young versus aged microbiota transplants to germ-free mice: increased short-chain fatty acids and improved cognitive performance. Gut Microbes. 2020;12(1):1–14. doi:10.1080/19490976.2020.1814107.
- Shakir L, Javeed A, Ashraf M, Riaz A. Metronidazole and the immune system. Pharmazie. 2011;66(6):393–398.
- Zhang Y, Shen Y, Liufu N, Liu L, Li W, Shi Z, Zheng H, Mei X, Chen CY, Jiang Z. et al. Transmission of Alzheimer’s disease-associated microbiota dysbiosis and its impact on cognitive function: evidence from mice and patients. Mol Psychiatry. 2023;28(10):4421–4437. doi:10.1038/s41380-023-02216-7.