
Abstract
It is well established that insulin-induced remodeling of actin filaments into a cortical mesh is required for insulin-stimulated GLUT4 exocytosis. Akt2 and its downstream effectors play a pivotal role in mediating the translocation and membrane fusion of GLUT4-storage vesicle (GSV). However, the direct downstream effector underlying the event of cortical actin reorganization has not been elucidated. In a recent study in Nature Communications,Citation1 Lim et al identify Tropomodulin3 (Tmod3) as a downstream target of the Akt2 kinase and describe the role of this pointed-end actin-capping protein in regulating insulin-dependent exocytosis of GSVs in adipocytes through the remodeling of the cortical actin network. Phosphorylation of Tmod3 by Akt2 on Ser71 modulates insulin-induced actin remodeling, a key step for GSV fusion with the plasma membrane (PM). Furthermore, the authors establish Tm5NM1 (Tpm3.1 in new nomenclature)Citation2 as the cognate tropomyosin partner of Tmod3, and an essential role of Tmod3-Tm5NM1 interaction for GSV exocytosis and glucose uptake. This study elucidates a novel effector of Akt2 that provides a direct mechanistic link between Akt2 signaling and actin reorganization essential for vesicle fusion, and suggests that a subset of actin filaments with specific molecular compositions may be dedicated for the process of vesicle fusion.
Actin remodeling is a fundamental yet complex process that involves numerous actin-binding proteins (ABPs), which regulate the dynamics of the actin cytoskeleton in response to signaling cascades elicited by growth stimuli or environmental cues.Citation3,4 The reorganization of actin filaments can be described in multiple discrete ways including de novo nucleation by Arp2/3 complex and formins, branching, elongation/termination by actin-capping proteins on the barbed- and pointed-ends, and the disassembly of actin filaments by actin-severing proteins.Citation4 In skeletal muscle cells and adipocytes, actin remodeling has been explicitly implicated in the context of insulin-dependent translocation of the glucose transporter, primarily, GLUT4, to the cell surface.Citation5,6 Previous studies have shown impaired GLUT4 vesicle exocytosis and glucose uptake when the actin cytoskeleton is disrupted by actin filament inhibitors such as Latrunculin B, Cytochalasin D, and Jasplakinolide.Citation7,8 However, detailed molecular process and mechanism regarding how the actin cytoskeleton and its remodeling participate GLUT4 vesicle fusion remain enigmatic. A consensus view is that the dynamic cortical actin rearrangement, but not the static actin barrier, is required for insulin-stimulated GLUT4 translocation as evidenced by the appearance of thickened cortical actin at the cell periphery under the condition of insulin stimulation.Citation5,6,9,10 Temporally enriched cortical actin in membrane ruffles as well as an increased rate of actin polymerization may accelerate the process of vesicle fusion,Citation11,12 thereby promoting GLUT4 insertion efficiency and the ensuing glucose uptake. Although it has become increasingly clear that protein kinase B or Akt2 is a key converging node of insulin action to direct the trafficking of GSVs to the PM via inactivation of RabGAP AS160,Citation13,14 whether and how Akt2 is directly involved in membrane fusion per se is largely unclear. Our latest studyCitation1 demonstrates that Tmod3 is a substrate of Akt2 and a critical regulator of cortical actin remodeling at the final stage of exocytosis, thus uncovering a missing link between GLUT4 exocytotic control governed by Akt2 signaling and cortical actin reorganization.
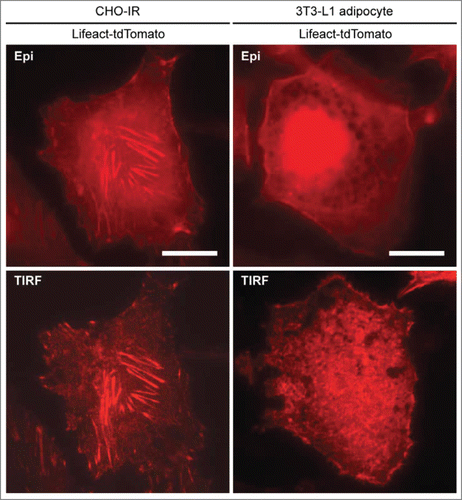
From an experimental point of view, it is not a trivial task for cell biologists to measure actin remodeling at the cellular level owing to the dynamic nature of the actin network either at local or global level under different conditions and a lack of a formalized index of actin remodeling for assessment and interpretation. Different optical imaging techniques and actin-labeling strategies have their own advantages and limitations. For example, epifluorescence microscopy is not useful in discerning individual actin filaments due to the poor z-resolution. Z-stack confocal imaging is suitable for visualizing the global actin cytoskeleton and has been widely used in the studies of actin remodeling in skeletal muscle.Citation6,15 However, an index of actin remodeling in these studies is often derived by measuring fluorescence intensities of phalloidin staining at different focal planes in fixed-cell samples at defined time intervals after treatment. Hence, results from fixed-cell experiments do not provide real-time kinetic information regarding the process of changes in actin behavior in cells receiving treatment at a given time. On the other hand, live-cell z-stack confocal imaging on actin is technically challenging due to the requirement for long period of imaging across different optical sections as well as the concerns over photostability and the expression level of the actin fluorescent protein. Unlike epifluorescence and confocal microscopy, total internal reflection fluorescence microscopy (TIRFM) is best suited for the visualization of cortical events with much improved axial resolution, even though the imaging zone is restricted to the ventral membranes. In our study,Citation1 we were able to show that actin remodeling in 3T3-L1 adipocytes could be visualized by expressing Lifeact-tdTomato, a fluorescent F-actin marker using TIRFM in live-cell conditions. Recording actin remodeling in real-time provides a much better perspective to appreciate the lateral dynamics of insulin-dependent actin remodeling and the successful application of the TIRFM-Lifeact-tdTomato approach has allowed us to investigate the relationship between Tmod3 phosphorylation, GLUT4 translocation and certain aspects of actin behavior. The examination in this system of the effects of Tmod3 knockdown and re-expression of phospho-mimetic or phospho-defective mutants has provided unequivocal evidence linking the observed Akt2-induced phosphorylation of Tmod3 to actin reorganization and GLUT4 exocytosis. Under insulin stimulation, there is a significant increase in cortical actin structures in the vicinity of the PM as well as increased ventral polymerized actin as shown by increased Lifeact-tdTomato fluorescence under TIRFM. It is yet to be determined the relative contributions of these pools of actin in promoting insulin-induced vesicle fusion events; cortical actin structures are condensed and relatively longer whereas ventral actin structures are shorter but significantly different from stress fibers as seen in typical fibroblasts (see for comparison). Taking into account the relative significance of these 2 distinct pools of actin on exocytosis, we analyzed the kinetics of TIRF intensities of Lifeact-tdTomato at the cell periphery and the ventral regions excluding the cell periphery, respectively. Results derived from both ways of measurement support a critical role for Tmod3 and its phosphorylation in insulin-dependent actin remodeling.
Figure 1. Comparison of actin structures between CHO-IR and 3T3-L1 adipocytes. CHO-IR (CHO cells stably expressing insulin receptor) and 3T3-L1 cells were imaged by using epifluorescence (Epi) or total internal reflection fluorescence (TIRF) microscopy. Cells expressing Lifeact-tdTomato were seeded in 35 mm glass-bottom dishes (MatTEK), incubated in imaging buffer and kept in a Tokai Hit temperature controlled incubation box at 37°C supplemented with 5% CO2. TIRFM setup (TIRF laser 561 nm) was based on Nikon Eclipse-Ti inverted microscope with EMCCD camera (1002 × 1002 pixels, 8 × 8 μm, 14-bit, Andor iXonEM + 885; Andor Technologies). Images were captured using a 100x NA/1.49 APO TIRF oil-immersion objective with immersion oil (nd = 1.515, Nikon) bridging the optical contact between the objective and the glass bottom dishes. The penetration depth of the evanescent field is estimated to be ∼200 nm. Images were acquired with no binning, at 27 MHz readout rate with average exposure times vary between 50∼100 ms. As revealed in TIRF microscopy, abundant stress fibers are present in the ventral section of the CHO-IR cell whereas cortical actin and short ventral actin filaments are enriched in the adipocyte. Scale bars, 20 μm.
Tropomodulins are a family of actin-capping proteins inclusive of 4 isoforms that cap the pointed end of actin filaments in a developmentally regulated and tissue/cell-type specific manner.Citation16 Tmod1 is predominantly expressed in terminally differentiated, post-mitotic cells such as RBCs, lens fiber cells. Tmod2 is neuron-specific, Tmod3 is expressed ubiquitously, and Tmod4 is restricted to skeletal muscle in mammals.Citation17 Similar to other isoforms, Tmod3 consists of 3 α helices at the N terminus: the 1st and 3rd helices are required for tropomyosin (TM)-binding, the 2nd helix is required for G-actin binding and TM-dependent F-actin capping.Citation18 The C-terminal domain of Tmod3 confers TM-independent F-actin capping, albeit with very low affinity, in the absence of TMs.Citation16 Conversely, Tmod3 caps the pointed ends of TM-decorated actin filaments with an affinity more than 1000-fold greater than for bare actin pointed ends.Citation19 However, the mechanism by which Tmod caps the pointed end is poorly understood. Previous studies in skeletal muscle and erythrocytes led to the proposal that 2 Tmod molecules cap the pointed end.Citation20,21 However, in in vitro conditions, one Tmod molecule is sufficient to block pointed-end elongation of TM-coated filaments.Citation22 Interestingly, a recent x- ray crystallography studyCitation23 has successfully resolved the crystal structures of actin complexes with the unstructured N-terminal and the LRR C-terminal domains of Tmod1. The crystallization was achieved by fusing the N- and C-terminal fragments of Tmod to gelsolin segment 1 via a stretch of amino acid flexible linker, respectively. Although it is not a crystal formed by full-length Tmod complexes with actin, the study revealed that one Tmod molecule interacts with 3 actin subunits at the pointed end, while also in contact with 2 tropomyosin molecules on each side of the filament. Moreover, the second α helix (64 to 77 residues) plays a crucial role in the interaction by forming a bridge across the nucleotide binding cleft on top of actin subdomains 4 and 2. It is important to note that the Ser71 residue is exclusively present in Tmod3, but absent in other isoforms. We demonstrate that phosphorylation of Tmod3 at Ser71 decreases actin-monomer binding, and potentiates the insulin-stimulated GSV exocytosis. It remains to be determined whether Tmod3 could function as an actin-monomer sequestering protein in the cellular context besides its well-known role in actin capping at the pointed end. We surmise that phosphorylation of Tmod3 at Ser71 may provide a regulatory switch to allow actin subunit exchange at the pointed-end (controlled addition or dissociation of actin subunits), and thereby determining the length of actin filaments under certain circumstances. For instance, in insulin-stimulated conditions, phosphorylation of Tmod3 may release G-actin to the local sites of exocytosis, thereby allowing the temporal and spatial buildup of cortical actin at the periphery to promote GLUT4 vesicle fusion. Furthermore, phosphorylation of Tmod3 may influence its actin-capping activity possibly through modulating the interaction between Tmod3 and its TM partner, Tm5NM1. In fact, there is an observed increased level of Tm5NM1 being co-precipitated by Tmod3 in insulin-stimulated conditions.Citation1 Since pointed-end capping activity of Tmod3 is known to increase dramatically in the presence of its cognate TM partner, regulating the interaction between Tmod3 and Tm5NM1 may be a plausible way to control the capping activity in the process of actin remodeling. Further studies are required to address this possibility.
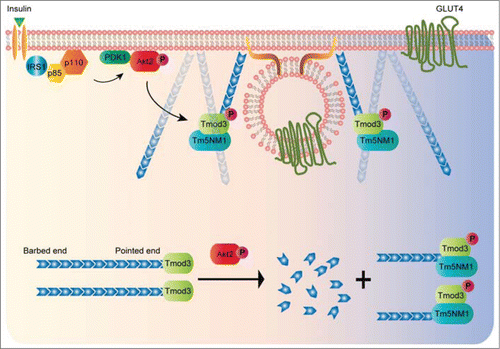
The discovery of Tm5NM1 as the cognate TM partner for Tmod3 and their interaction is implicated in the insulin-stimulated GLUT4 exocytosis suggests a possibility that a subset of actin filaments with specific molecular compositions (Tm5NM1-coatedCitation24-26) may be harnessed to promote the events of vesicle fusion. Notably, Tm5NM1 increases its presence significantly on the cell surface of 3T3-L1 adipocytes in response to insulin stimulation.Citation30 The fact that Tmod3 preferentially binds to Tm5NM1-decorated actin filaments suggests that there may be a dramatic increase in pointed-end capping activities of Tmod3 in insulin-stimulated condition, leading to formation and stabilization of de novo actin filaments at the cell periphery. Furthermore, the selective pairing of Tmod3 with Tm5NM1 may facilitate a specific actin-based myosin motor to target the vesicles to the exocytotic site with high precision,Citation27,28 given the fact that TM5NM1 has been shown to recruit the myosin motor MyoIIA to actin filaments.Citation29 In summary, the study by Lim et al (2015) provides a molecular link between Akt2 activation and actin remodeling (see for proposed model), and paves the way for many future investigations on the unprecedented active role of different actin-binding proteins in the process of membrane actin reorganization to facilitate the final stage of exocytosis.
Figure 2. A proposed model of Akt2-mediated phosphorylation of Tmod3 in the regulation of cortical actin remodeling and GSV-PM fusion. In insulin-stimulated adipocytes, activated Akt2 functions as a signaling hub to activate a number of downstream effectors that are necessary for multiple steps of GLUT4 exocytosis. Release of G-actin from Tmod3 upon phosphorylation by Akt2 may provide a temporal and spatial supply of actin monomers to the local sites of exocytosis in order to facilitate the transient buildup of cortical actin at the periphery. The pairing of Tmod3 and Tm5NM1 may help define a specific population of actin filaments at the cell periphery to enable the process of GLUT4 vesicle fusion with the PM.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Lim C-Y, Bi X, Wu D, Kim JB, Gunning PW, Hong W, Han W. Tropomodulin3 is a novel Akt2 effector regulating insulin-stimulated GLUT4 exocytosis through cortical actin remodeling. Nat Commun 2015; 6:5951; PMID:25575350; http://dx.doi.org/10.1038/ncomms6951.
- Geeves MA, Hitchcock-DeGregori SE, Gunning PW. A systematic nomenclature for mammalian tropomyosin isoforms. J Muscle Res Cell Motil 2014; PMID:25369766; http://dx.doi: 10.1007/s10974-014-9389-6
- Stossel TP. Cell surface actin remodeling. J Cell Sci 2006; 119:3261-4; PMID:16899816; http://dx.doi.org/10.1242/jcs.02994.
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003; 112:453-65; PMID:12600310; http://dx.doi.org/10.1016/S0092-8674(03)00120-X.
- Kanzaki M, Pessin JE. Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem 2001; 276:42436-44; PMID:11546823; http://dx.doi.org/10.1074/jbc.M108297200.
- Tong P, Khayat ZA, Huang C, Patel N, Ueyama A, Klip A. Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J Clin Invest 2001; 108:371-81; PMID:11489930; http://dx.doi.org/10.1172/JCI200112348.
- Tsakiridis T, Vranic M, Klip A. Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J Biol Chem 1994; 269:29934-42; PMID:7961991.
- Wang Q, Bilan PJ, Tsakiridis T, Hinek A, Klip A. Actin filaments participate in the relocalization of phosphatidylinositol3-kinase to glucose transporter-containing compartments and in the stimulation of glucose uptake in 3T3-L1 adipocytes. Biochem J 1998; 331 ( Pt 3):917-28; PMID:9560323.
- Khayat ZA, Tong P, Yaworsky K, Bloch RJ, Klip A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J Cell Sci 2000; 113 Pt 2:279-90; PMID:10633079
- Kanzaki M. Insulin receptor signals regulating GLUT4 translocation and actin dynamics. Endocr J 2006; 53:267-93; PMID:16702775; http://dx.doi.org/10.1507/endocrj.KR-65
- Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A, Xu P, James DE, Xu T. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab 2007; 5:47-57; PMID:17189206; http://dx.doi.org/10.1016/j.cmet.2006.11.013
- Koumanov F, Jin B, Yang J, Holman GD. Insulin signaling meets vesicle traffic of GLUT4 at a plasma-membrane-activated fusion step. Cell Metab 2005; 2:179-89; PMID:16154100; http://dx.doi.org/10.1016/j.cmet.2005.08.007
- Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 2003; 278:14599-602; PMID:12637568; http://dx.doi.org/10.1074/jbc.C300063200
- Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab 2007; 5:293-303; PMID:17403373; http://dx.doi.org/10.1016/j.cmet.2007.03.001
- Chiu TT, Patel N, Shaw AE, Bamburg JR, Klip A. Arp2/3-and cofilin-coordinated actin dynamics is required for insulin-mediated GLUT4 translocation to the surface of muscle cells. Mol Biol Cell 2010; 21:3529-39; PMID:20739464; http://dx.doi.org/10.1091/mbc.E10-04-0316
- Yamashiro S, Gokhin DS, Kimura S, Nowak RB, Fowler VM. Tropomodulins: Pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton 2012; 69:337-70; PMID:22488942; http://dx.doi.org/10.1002/cm.21031
- Cox PR, Zoghbi HY. Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics 2000; 63:97-107; PMID:10662549; http://dx.doi.org/10.1006/geno.1999.6061
- Fischer RS, Yarmola EG, Weber KL, Speicher KD, Speicher DW, Bubb MR, Fowler VM. Tropomodulin 3 binds to actin monomers. J Biol Chem 2006; 281:36454-65; PMID:17012745; http://dx.doi.org/10.1074/jbc.M606315200
- Krieger I, Kostyukova A, Yamashita A, Nitanai Y, Maéda Y. Crystal structure of the C-terminal half of tropomodulin and structural basis of actin filament pointed-end capping. Biophysj 2002; 83:2716-25; http://dx.doi.org/10.1016/S0006-3495(02)75281-8
- Moyer JD, Nowak RB, Kim NE, Larkin SK, Peters LL, Hartwig J, Kuypers FA, Fowler VM. Tropomodulin 1-null mice have a mild spherocytic elliptocytosis with appearance of tropomodulin 3 in red blood cells and disruption of the membrane skeleton. Blood 2010; 116:2590-9; PMID:20585041; http://dx.doi.org/10.1182/blood-2010-02-268458
- Fowler VM, Sussmann MA, Miller PG, Flucher BE, Daniels MP. Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J Cell Biol 1993; 120:411-20; PMID:8421055; http://dx.doi.org/10.1083/jcb.120.2.411
- Weber A, Pennise CR, Fowler VM. Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting ADP.P(i)-actin to ADP-actin at all pointed filament ends. J Biol Chem 1999; 274:34637-45; PMID:10574928; http://dx.doi.org/10.1074/jbc.274.49.34637
- Rao JN, Madasu Y, Dominguez R. Mechanism of actin filament pointed-end capping by tropomodulin. Science 2014; 345:463-7; PMID:25061212; http://dx.doi.org/10.1126/science.1256159
- Gunning P, O'Neill G, Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol Rev 2008; 88:1-35; PMID:18195081; http://dx.doi.org/10.1152/physrev.00001.2007
- Creed SJ, Bryce N, Naumanen P, Weinberger R, Lappalainen P, Stehn J, Gunning P. Tropomyosin isoforms define distinct microfilament populations with different drug susceptibility. Eur J Cell Biol 2008; 87:709-20; PMID:18472182; doi: 10.1016/j.ejcb.2008.03.004
- Gunning PW, Schevzov G, Kee AJ, Hardeman EC. Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol 2005; 15:333-41; PMID:15953552; http://dx.doi.org/10.1016/j.tcb.2005.04.007
- Bryce NS, Schevzov G, Ferguson V, Percival JM, Lin JJ-C, Matsumura F, Bamburg JR, Jeffrey PL, Hardeman EC, Gunning P, et al. Specification of actin filament function and molecular composition by tropomyosin isoforms. Mol Biol Cell 2003; 14:1002-16; PMID:12631719; http://dx.doi.org/10.1091/mbc.E02-04-0244
- Tojkander S, Gateva G, Schevzov G, Hotulainen P, Naumanen P, Martin C, Gunning PW, Lappalainen P. A molecular pathway for myosin II recruitment to stress fibers. Curr Biol 2011; 21:539-50; PMID:21458264; http://dx.doi.org/10.1016/j.cub.2011.03.007
- Woody S, Stall R, Ramos J, Patel YM. Regulation of myosin light chain kinase during insulin-stimulated glucose uptake in 3T3-L1 adipocytes. PLoS ONE 2013; 8:e77248; PMID:24116218; http://dx.doi.org/10.1371/journal.pone.0077248
- Kee AJ, Yang L, Lucas CA, Greenberg MJ, Martel N, Leong GM, Hughes WE, Cooney GJ, James DE, Ostap EM, Han W, Gunning PW, Hardeman EC. An actin filament population defined by the tropomyosin Tpm3.1 regulates glucose uptake. Traffic 2015; PMID:25783006; doi: 10.1111/tra.12282