
Abstract
The nucleolus is the hallmark of nuclear compartmentalization and has been shown to exert multiple roles in cellular metabolism besides its main function as the place of rRNA synthesis and assembly of ribosomes. Nucleolar proteins dynamically localize and accumulate in this nuclear compartment relative to the surrounding nucleoplasm. In this study, we have assessed the molecular requirements that are necessary and sufficient for the localization and accumulation of peptides and proteins inside the nucleoli of living cells. The data showed that positively charged peptide entities composed of arginines alone and with an isoelectric point at and above 12.6 are necessary and sufficient for mediating significant nucleolar accumulation. A threshold of 6 arginines is necessary for peptides to accumulate in nucleoli, but already 4 arginines are sufficient when fused within 15 amino acid residues of a nuclear localization signal of a protein. Using a pH sensitive dye, we found that the nucleolar compartment is particularly acidic when compared to the surrounding nucleoplasm and, hence, provides the ideal electrochemical environment to bind poly-arginine containing proteins. In fact, we found that oligo-arginine peptides and GFP fusions bind RNA in vitro. Consistent with RNA being the main binding partner for arginines in the nucleolus, we found that the same principles apply to cells from insects to man, indicating that this mechanism is highly conserved throughout evolution.
Abbreviations
| aa | = | amino acid |
| CPP | = | cell penetrating peptide |
| DDC | = | dense fibrillar compartment |
| FC | = | fibrlllar centers |
| FITC | = | fluorescein isothiocyanate |
| GBP | = | GFP binding protein |
| GC | = | granular compartment |
| GFP | = | green fluorescent protein |
| NoLS | = | nucleolar localization sequence |
| NLS | = | nuclear localization sequence |
| PI | = | propidium iodide |
| TAMRA | = | 5-carboxytetramethylrhodamine |
Introduction
Proteins inside the highly compartmentalized cell nucleus often localize in correlation with their function to discrete subnuclear regions also referred to as subnuclear bodies.Citation1,2 These nuclear substructures were identified as sites where specific biochemical reactions take place, e.g., DNA replication foci, or as sites of storage and modification for different proteins like in Cajal bodies and nuclear speckles.Citation3,4 The localization of proteins in the nucleus is often mediated by specialized signals, targeting or interaction motifs of different length and can change over time, cell cycle and with external stimuli.Citation5-8 The most prominent subnuclear structure is the nucleolus, which appears as a dark region in phase contrast microscopy due to its high density.Citation9-11 The nucleolus is the place of transcription and storage of the rDNA as well as the assembly and maturation site for ribosomes and, in translationally active cells, filled with pre-ribosomal particles and components. The nucleolar structure is very dynamic and forms around rDNA loci.Citation12,13 It has further substructures, which are connected to the different steps in ribosome biogenesis and can be best identified by antibody labeling or electron microscopy. The fibrillar centers (FC) harbor the rDNA in the nucleolar center and are surrounded by the dense fibrillar components (DFC) containing the nascent rRNA transcripts. The granular component (GC) is filled with pre ribosomal particles composed of rRNA and proteins, extends into the nucleus and is surrounded by chromatin.Citation11,14,15
An early study has shown that the nuclear import of several proteins is mediated by short sequences of 6–10 basic amino acid (aa) termed nuclear localization sequences (NLS), some of which also serve as nucleolar targeting sequence (NoLS).Citation16 Another nucleolar targeting sequence (NoLS) was described in 1990 and contains a minimum of 5 basic aa, often lysines and arginines.Citation17 The recent systematic analysis of 46 NoLS sequences for their amino acid composition and sequence features has shown greater sequence diversity including bipartite sequences as well as overlap with NLS. The study determined also a high content of basic amino acids (48%) and a predominant location in the termini of proteins.Citation18 Proteins smaller than 60 kDa or up to a diameter of 9 nm can passively diffuse into the nucleusCitation19 but this process becomes more and more inefficient with increasing size (reviewed inCitation20,21). The nucleus is separated from the cytoplasm by a membrane system and involves an active import mechanism.Citation22 In contrast, the localization of proteins to the nucleolus has never been shown to involve active transport mechanisms. Nuclear proteins without nucleolar function are often less concentrated or completely excluded from the nucleoli while nucleolar proteins are highly enriched in this nuclear compartment. The localization or exclusion of proteins from the nucleolus is subject to dramatic changes upon cellular stimuli, stress and complete reorganization during cell cycle progression.Citation23-25
In a previous study, we have shown that a reporter protein traverses nucleoli rapidly with less obstruction compared to nucleoplasmic regions rich in chromatin.Citation26 In that study, we described a structural feature of the nucleolus with channel like regions void of nucleic acids and proteins that facilitates a fast movement of non-nucleolar proteins through this compartment. Contrastingly, the neighboring densely packed structures in the nucleolus reduce the available space for the localization of non-nucleolar proteins.Citation26 Still it is unclear, what are the essential properties or requirements of proteins and peptides to accumulate in the nucleoli and what are the features of these molecules that prevent nucleolar accumulation. The systematic identification of NoLS sequences by an artificial neural network trained with confirmed NoLS sequences has found thousands of potential sequences that could serve to mediate nucleolar targeting. Several previously unknown NoLS have been experimentally confirmed thereafter.Citation18,27 However, a considerably false positive prediction rate especially among sequences from nuclear proteins raises the question of how NLS and NoLS can be distinguished. The difference in the composition of a standard NLS and the peptides that accumulate in nucleoli is rather subtle, with the NLS being a few basic charged amino acids shorter than, e.g., the nucleolar marker peptide of 10 arginines described by us.Citation16,28 This observation led to the following 2 questions: (a) what are the specific requirements in amino acid composition for peptides and proteins to localize to the nucleoli and (b) what are the key differences between NoLS and NLS sequences?
In this study, we systematically investigated the molecular properties of peptides and proteins that result in different distribution levels between nucleoplasm and nucleolus. For this purpose, we tested and analyzed the distribution of a series of fluorescently labeled short peptides with different charge and isoelectric properties between the cytoplasm, nucleoplasm and their accumulation in the nucleolus. The same peptide sequences were genetically fused to the 5′ end of the open reading frame coding for the tracer protein GFP, with no intracellular binding sites, to test the effect of the same short peptide sequences on the localization of proteins.
Materials and Methods
Peptides, plasmids and cell lines
All peptides were synthesized from L-amino acids except deca-arginine (R10), which was made as 2 versions one from L- and one from D-amino acids by Peptide Specialty Laboratories GmbH (Heidelberg, Germany). The peptides were coupled directly to fluorescein at the N-terminus, contain a C-terminal amide-group and were purified by HPLC. The TAT peptide was synthetized with D-amino acids and conjugated with TAMRA as described.Citation29 It can be excluded that different amounts of fluorescent labeling on the different peptides play a role in the intracellular distribution, since the in vitro synthesis of peptides coupled to a resin material allows only in line addition of amino acids and the fluorophore. The pI values of the peptides and proteins were calculated using the ProtParam tool on http://web.expasy.org/protparam/.
The GFP based mammalian expression constructs were generated by using oligonucleotides encoding the respective poly-D, poly-G and poly-R sequences (supplementary Table 1). The oligos with overhanging 3′ and 5′ AgeI sites were annealed and ligated into the AgeI site of an NLS-GFP construct based on the pEVRF vector.Citation30 This vector is identical to the one described before for the CMV-driven expression of GFP-tagged PCNA.Citation31 In addition, the pEGFP-C1 vector (Clontech, Heidelberg, Germany) without a NLS sequence was used.
Human HeLa and HEK 293-EBNA cells, as well as mouse C2C12 myoblasts were cultured as described before.Citation32,33 Pac2 zebrafish cells were grown at 28°C in Leibovitz L-15 medium supplemented with 15% fetal calf serum in presence of 1% penicillin-streptomycin. Sf9 insect cells were maintained in EX-CELL TM 420 Insect Serum Free (SAFC) medium with L-glutamine (Biosciences) and 10% fetal calf serum and cultured with shaking at 100 rpm and 28°C. The L40ccua yeast strain used for the current study has the following genetic background: (MATa his3_200 trp1–901 leu2–3,112 LYS2::(lexAop)4-HIS3 ura3::(lexAop)8- lacZ ADE2::(lexAop)8-URA3 GAL4 gal80 can1 cyh2).Citation34 Yeast cells were grown at 30°C in yeast extract/peptone/dextrose (YPD) and were supplemented with 2% glucose as a carbon source.
Live cell peptide loading and transfections
All peptide experiments were performed exclusively with living cells plated on 8-well Ibidi chambers. The different poly-lysine (poly-K) and poly-arginine (poly-R) peptides were scratch loaded into living cells. For the scratch loading of peptides we used syringes and scratched over the chamber bottom in different directions.Citation35,36 The size of the peptides permitted passive diffusion into the cells and nuclei after delivery. After the scratch loading the cells were allowed to recover for about half an hour at 37°C before exchanging the medium to remove unloaded peptides. Microscopy and image acquisition was started immediately after the procedures described above to circumvent peptide shortening by proteases and, hence, mislocalization. Peptide concentrations from 10 to 100 µm were initially tested and no apparent changes in cell viability were observed during the course of the experiments. As low concentrations of 20 µM were sufficient for quantitative imaging, these conditions were used for the subsequent quantification. Scratch loaded cells did not show dramatic differences in the level of intracellular fluorescence, likely because the peptide concentration in the medium was the same in all cases. Only cells with normal cellular physiology comparable to non-scratch loaded cells were imaged. The criteria for the selection of cells that repaired after scratch loading were: i) cells are completely attached and stretched out on the chamber bottom surface; ii) no membrane lesions or leakage of cytoplasmic constituents visible in phase contrast; iii) round or oval shaped nuclei with smooth outline and nucleoli visible in phase contrast.
The transfection of cells with plasmid constructs was performed using the CaPO4-DNA co-precipitation protocol as described before,Citation37 except for HEK 293-EBNA cells, which were transfected using poly-ethylenimine (1 mg/mL in ddH2O, pH 7; Sigma-Aldrich, St. Louis, MO, USA) as described before.Citation38
Cell fixation and antibody staining
Cells grown on glass coverslips for 24 h were incubated with 10 µM FITC-R10 for 1 h. After washing with 1xPBS, cells were fixed in 3.7% formaldehyde in PBS for 10 min and permeabilized with 0.25% Triton X100 in PBS for 10 min. Immunostaining with anti-B23 mouse monoclonal antibody (clone FC82291; Sigma) was followed by detection with donkey anti-mouse IgG antibody conjugated with TexasRed (Jackson). Coverslips were mounted with Moviol (Sigma).
Microscopy, image acquisition and analysis
Live cell microscopy was performed with a Zeiss LSM510Meta confocal setup mounted on an Axiovert 200 M inverted microscope using a 63x phase contrast plan-apochromat oil objective NA1.4. The microscope was placed in an incubation chamber heated to 37°C to maintain the cell incubation conditions (Okolab, Italy). For the measurements of plasmid transfected cells, a Leica TCS SP confocal microscope was used. For all acquisition settings the main beam splitter was HFT UV/488/543/633. FITC was excited with the 488 nm line of an argon laser and fluorescence detected with a band-pass filter 500–530 nm. Phase contrast or differential interference contrast images were recorded simultaneously with FITC fluorescence in the transmission channel. The live-cell DNA dye DRAQ5 was used and imaged as described.Citation32
Cells loaded with peptides were chosen for imaging by having low but easily detectable intracellular fluorescence intensity. The same criteria were applied to cells transfected with plasmid constructs. Imaging of live cells was performed using as much as possible similar settings per instrument.
The SNARF 4F cell-permeant dye (Sigma) was loaded into cells by incubating for 30 min at a final concentration of 5 µM in growth medium. Fluorescence was excited at 561 nm and emission detected at 587 nm and 640 nm.
For image analysis, the nuclei were identified in the corresponding phase contrast images by means of the nuclear membrane delineation and nucleoli correspond to the dark dense structures inside the cell nucleus. Identification of nucleoli was confirmed in earlier experiments using a nucleolar marker for living cells.Citation28 The selection of regions (cytoplasm, nucleus, nucleolus) and measurements of mean signal intensity were performed in ImageJ. Measurements of mean fluorescence intensities were performed by excluding non-interest areas from the measurement, e.g. the nucleoli areas excluded from nucleoplasm and, correspondingly, for all other regions intensities were determined. In each experiment the average was calculated for 10 cells for each peptide and protein. Data were normalized to 1.0 (100%) with respect to the nucleoplasmic fluorescence (excluding the nucleoli) to directly illustrate the relative accumulation of fluorescence in the nucleoli. Cytoplasmic fluorescence levels were normalized correspondingly (excluding nucleus and nucleoli). Data analysis and statistical tests were performed and displayed with Origin 7 software.
The complete list of predicted nucleolar localization sequences was downloaded from http://www.compbio.dundee.ac.uk/www-nod/ and analyzed for mean length, mean numbers of amino acids and frequencies in Excel and Origin 7.
Protein preparation
GFP and NLS-R7-GFP were prepared from HEK cells 48 h post transfection using an ice-cold buffer composed of 20 mM Tris-HCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 M NaCl and 0.4% NP40. After homogenization by mechanical shearing through a 23 gauge syringe, lysates were incubated for 10 min on ice. Following centrifugation (14,000 rpm for 12 min at 4°C) supernatants were incubated for one hour with GFP binding protein (GBP) coupled to sepharose beads at 4°C on a rotary shaker as described.Citation39 GBP bound proteins were washed 3 times with ice-cold HEPES buffer (140 mM NaCl, 2.5 mM KCl, 5 mM HEPES, 5 mM glycine, pH 7.4) and used for the in vitro RNA pull-down assay. All buffers were supplemented with 1 mM of protease inhibitor PMSF (Carl Roth, Karlsruhe, Germany).
RNA preparation
Total RNA was isolated from HEK cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. To remove traces of genomic DNA, RNA was treated with RNase-free, recombinant DNaseI (Macherey Nagel, Dueren, Germany) for 30 min at 37°C and further purified with the Qiagen RNeasy Mini Kit. To assess the concentration and purity of RNA, the ratio of absorbance at 260 nm and 280 nm was measured on a TECAN infinite M200 plate reader (Tecan Group Ltd., Maennedorf, Switzerland). To further verify RNA quality, total RNA was denatured for 5 minutes at 99°C, separated on a 1.5% Tris-acetate-EDTA/agarose gel supplemented with 0.05 µL/mL Roti-Safe GelStain (Carl Roth, Karlsruhe, Germany) by electrophoresis and imaged on an Amersham Imager 600 (GE Healthcare, Freiburg, Germany).
In vitro RNA binding assays
For RNA slot blots, total RNA was mixed with RNase-free ddH2O at a final concentration of 500 ng/µL and blotted on pre-equilibrated (5 min methanol, 5 min 20x saline sodium citrate) PVDF membranes (0.45 µm pore size; Pall GmbH, Dreieich, Germany). After air-drying, membranes were blocked for 30 min with 5% milk in PBS supplemented with 1x ProtectRNA RNase inhibitor (Sigma-Aldrich, St. Louis, MO, USA) and incubated for 1 h with either TAT, D-R10, or L-R10 (all fluorescently labeled), respectively, on a rotary shaker. Following three washing steps (5 min each) with HEPES buffer, remaining fluorescent signals were detected on an Amersham Imager 600 (GE Healthcare, Freiburg, Germany). Quantities of blotted RNA were monitored in parallel by methylene blue (0.02%) staining (Carl Roth, Karlsruhe, Germany) in 0.3 M sodium acetate (Merck, Darmstadt, Germany) pH 5.5.
For the pulldown assay, immobilized GFP and NLS-R7-GFP, respectively, were incubated with equal amounts (3.4 µg) of total RNA in 20 µL HEPES buffer for 1.5 h at 4°C on a rotary shaker. After three washing steps with HEPES buffer, RNA was labeled with 3.3 µg/mL propidium iodide (Sigma-Aldrich, St. Louis, MO, USA). Following three washing steps in HEPES buffer, remaining fluorescent signals were measured on a TECAN infinite M200 plate reader (Tecan Group Ltd., Maennedorf, Switzerland) using excitation/emission at 488/555 nm and 515/617 nm for GFP and propidium iodide, respectively. To control for the amount of GFP and NLS-R7-GFP, fluorescent signals of propidium iodide were normalized to GFP signals. In addition, the same beads were imaged in an UltraVIEW VoX spinning disc confocal system (PerkinElmer), mounted on a Nikon TI microscope. Images were taken with a 20x/0.7 NA objective. GFP and propidium iodide or TAMRA were imaged with 488 and 561 nm laser excitation and 527 ± 55 and 612 ± 70 nm emission filters, respectively.
Results
Short specific sequence motifs in proteins are well known to play a key role in localizing proteins to specific cellular and subcellular compartments. We have shown earlier that short peptides of 10 arginines with a strong basic charge have the potential to accumulate inside the nucleoli of living cells and can be used to label this subnuclear compartment.Citation28 Now, we wished to investigate the specific molecular requirements of peptides and proteins that are necessary and sufficient to reach and accumulate inside of the nucleolus.
Therefore, our first aim was to determine the properties of charge, length and composition of basic charged peptides necessary for nucleolar targeting and accumulation. We used short synthetic peptides labeled with FITC for visualization, which were scratch loaded into living C2C12 mouse myoblast cells. The peptides, either composed of arginines (R) or lysines (K) with a length varying from 5 to 12 amino acids, were allowed to enter the cells and distribute in the cytoplasm and nucleus. The observation of the cells by confocal microscopy started after a recovery time of 30 min after scratching loading. For each peptide at least 10 cells were selected and imaged with respect to similar intracellular fluorescence intensity levels. The microscopic observation of the cells shows for all peptides a homogeneous distribution in the cytoplasm, where only vesicular structures were free of labeled peptides. Similarly, in the nucleoplasm the peptides distributed without enriching at any foci. All L-amino acid peptides tested here, K5 to K12 and R5 to R12 accumulated predominantly in the nucleus compared to the cytoplasm, as shown by the fluorescence intensity in mouse C2C12 cells (). The peptides R6 to R12 clearly showed higher fluorescence intensity in areas inside the cell nucleus, which were identified as nucleoli correlating with the dark dense structures visible in the phase contrast images (). The peptide R5 did not accumulate in nucleoli (), which was also the case for all poly-K peptides (). This indicates that charge alone is not the only determinant but rather the isoelectric properties, which differ between K and R polymers. To quantitatively analyze the intensity and distribution patterns, we determined for 10 cells the mean fluorescence intensity in the cytoplasm, nucleus (excluding nucleoli) and in the nucleoli. The regions for fluorescence intensity measurements were chosen with reference to the phase contrast images and overlayed with the fluorescence image as shown in . Data were normalized to the nucleoplasmic levels to reflect the accumulation potential of molecules in the nucleolus over the nucleoplasmic levels (). The numerical values are given in supplementary Tables 2 and 3.
Figure 1. Distribution of poly-(R)and poly-(K)peptides in living cells. Intracellular distribution of poly-R peptides in (A) and poly-K peptides (B) in living C2C12 mouse cells. In each panel, the fluorescence image is on top of the corresponding phase contrast image. Nucleoli are clearly visible as dark round structures within the nuclei in the phase contrast images. The bar diagrams in (C) show the quantification of the poly-K and poly-R peptide mean fluorescence in cytoplasm, nucleoplasm and nucleoli averaged for 10 cells from 2 independent experiments. The nucleoplasmic values were used for normalization. Areas for quantification were defined as described in methods and overlayed with fluorescence images as shown. Intracellular distribution of R10 peptide in living cells of different species as indicated is shown in (D). In (E) living yeast cells were further stained with DRAQ5 for better visualization of the nucleus and a line intensity profile in arbitrary units (a.u.) of both DNA and peptide is shown. The intracellular distribution of (D) and L-R10 peptides in living C2C12 cells and the corresponding quantification of mean fluorescence intensities is shown in (F). Scalebars: 5 µm.
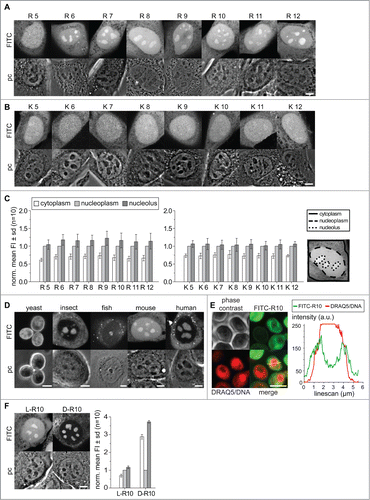
The comparison of the mean fluorescence intensity shows, for all poly-K peptides, an average cytoplasmic level of 0.74 fold less compared to the nucleoplasm. For poly-R peptides the cytoplasmic level is on average 0.68 fold less than in the nucleoplasm ( and supplementary Table 2). These data already demonstrate that poly-R peptides have the potential for stronger accumulation in the nucleus than poly-K peptides. Although all peptides tested are small enough to passively diffuse in and out of the nucleus and should distribute evenly, all of them accumulate in the nucleoplasm.
Strikingly, only the peptides R6 to R12 show a further visible accumulation inside nucleoli with an average of 1.16-fold over the nucleoplasmic level (). The statistical analysis with a non-parametric Kruskal-Wallis ANOVA test shows significance for the accumulation of R6 to R12 in the nucleolus. The poly-K peptides show only slightly higher level in the nucleoli with values between 1.02 and 1.06 fold over the nucleoplasm. An increase of the peptide length from R6 to R12 and thus the positive charge and isoelectric properties does not linearly increase the nucleolar accumulation level of the peptides. The highest level of nucleolar accumulation was found for the R9 peptide with 1.22 fold over the nucleoplasmic level, while the mean accumulation of R11 and R12 rather decreases slightly to 1.12 and 1.14-fold respectively ( and supplementary Table 2). This suggests that there is a saturation/plateau level at and above 9 arginine residues.
From yeast to human cells, main features of the nucleolus structure and function are conserved as it forms around clusters of rRNA genes and is the sites of ribosome biogenesis. We were now interested if the nucleolar localization and accumulation of proteins and peptides is similarly conserved. Therefore, we tested the nucleolar accumulation potential of the R10 peptide in cultured cells of different species. We scratch loaded the FITC labeled R10 into cells from yeast (S. cerevisiae), insects (D. melanogaster), fish (D. rerio), mouse (M. musculus) and human (H. sapiens) cells and analyzed the localization of the peptides by confocal microscopy (). In yeast cells the nucleus and nucleolus are more difficult to identify and clear accumulations in the cells are not as visible as for the cells of higher eukaryotes. Regions with less peptide inside the yeast cells could correspond to the vacuole. In the cells from insect, fish, mouse and human, on the other hand, the nucleoli are visible in the phase contrast images and show clearly an accumulation of the R10 peptide compared to the nucleus and cytoplasm of the cells (). To clarify the localization of the FITC-R10 in yeast we performed a co-labeling of yeast cells with the live cell DNA dye DRAQ5.Citation32 In this experiment the yeast nucleus is visible as intense labeled round compartment in the cells. In some cells we could identify a crescent shaped region in the periphery of the nucleus with higher FITC-R10 intensity compared to the nuclear interior and the cytoplasm. A corresponding fluorescence intensity linescan analysis confirms an accumulation of the peptide in a crescent shaped region in the nuclear periphery (). Early studies on the composition of yeast nuclei have shown that, depending on the orientation a round or crescent shaped peripheral nuclear compartment harbors the nucleolus.Citation40-42 All cell types of multicellular organisms tested here show a similar enrichment of the labeled poly-R peptides in the nucleoli. Some yeast cells show a similar accumulation in the nuclear periphery. The accumulation of peptides like R10 inside nucleoli is, thus, evolutionary conserved at least from insects to humans. Notably, HeLa cells in metaphase having no cell nucleus and with the nucleoli disassembled lack also the nucleolar accumulation of peptides found in interphase cells. Thus, the accumulation of poly-R peptides is directly linked to the structure and function of the nucleolus (supplementary Fig. 1D).
All amino acids incorporated into proteins by translation in living organisms are L-enantiomers and only post-translational enzymatic reactions in various organisms convert some to D-amino acids.Citation43,44 Besides that, free D-amino acids were discovered only in brain tissue.Citation45 We argue that a poly D-arginine peptide may not exhibit the specific binding to the same cellular target components, e.g., proteins or nucleic acids, as the L-enantiomer. In contrast, the peptide charge and isoelectric properties are not altered when composed of D-amino acids. This phenomenon is exploited in mirror image phage displays used in the search for therapeutic peptides with novel targets and improved properties.Citation46,47 The latter exploits the stability of the D-amino acid peptides to proteolytic degradation. Hence, we compared the distribution of FITC-R10 composed of D and L amino acids (). Inside living cells both D- and L-R10 peptides distribute homogenously in the cytoplasm and accumulated in the nucleoli. The main difference in the distribution is that D-R10 had a much lower concentration in the nucleoplasm compared to the L-R10, therefore the measurement of relative accumulation of D-R10 over the nucleoplasmic level appears much stronger (). However, the absolute detected fluorescence intensities were comparable between both types of peptides using the same microscopic detection settings. This finding indicates either a binding of the L-peptide to a nucleoplasmic component, which is not the case for the D-peptide, and/or the lack of degradation of the D-R10. Interestingly though, the mean intensity of fluorescence in the nucleoli is at similar levels for both types of peptides.
Next, we tested the role of various peptides in the targeting of proteins inside the cell nucleus. We used peptides fused to the neutral tracer protein GFP to determine localization patterns. For this purpose, we used GFP already fused to a NLS sequence (SV40 T antigen derived; PKKKRKVCitation48) and added downstream further basic, acidic and neutral peptides of different lengths followed by the enhanced GFP coding sequence. The different GFP fusions were expressed in C2C12 mouse myoblast cells () and in human HeLa cells (supplementary Fig. 1) and imaged with a confocal microscope. We also tested the distribution of an uncoupled GFP and the NLS-GFP alone as comparison and as controls for cytoplasmic and nucleoplasmic localization with no expected nucleolar localization and accumulation.
Figure 2. Intracellular distribution of peptide tagged GFP tracer proteins in living cells. (A) Representative microscopic images of live C2C12 cells transfected with GFP, NLS-GFP and a fusion of NLS-GFP with 8 glycines (8G) as example for the addition of neutral amino acids is shown in the first panel. The second panel shows images of cells with fusions of an increasing number of aspartates as examples for acidic amino acid fusions to NLS-GFP. The GFP fluorescence is depicted in the upper row with the corresponding differential interference contrast (DIC) images below. (B) Microscopic Images of the distribution of the NLS-GFP fused to 4 to 7 additional arginines. The graphs in (C) represent the mean distribution of the GFP constructs depicted in (A) and (B) in cytoplasm and nucleoli in relation to the nucleoplasm for an average of 10 cells from 2 independent experiments. Scalebars: 5 µm.
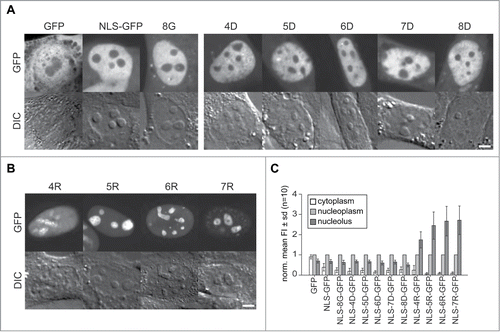
The microscopic images of the GFP versions in show for the GFP without NLS a homogeneous distribution throughout the cell with similar levels in cytoplasm and nucleus with significant reduction in the nucleoli (p = 0.01 see supplementary Table 2). The slightly lower mean intensity in the cytoplasm is the result of membranous compartments devoid of GFP protein (; supplementary Table 2). The homogeneous distribution between nucleus and cytoplasm is the result of passive diffusion into the nucleus. Molecules with a diameter of up to ∼9 nm are capable of entering the cell nucleus by passive diffusion, which has been measured for fluorescent molecules the size of GFP to occur within a few minutes.Citation19,49 The NLS coupled GFP, though, shows more than 2-fold accumulation in the nucleus over the cytoplasmic level but with a similar reduction in the nucleoli, visible as round elevated objects in the DIC images. This distribution pattern is similar for all NLS-GFP variants with additional acidic (aspartate) and neutral (glycine) amino acid series (). The nucleolar level of the different proteins in is significantly reduced in the range of 0.7 to 0.5-fold compared to the nucleoplasmic level as determined by measuring the mean fluorescence intensity ( and supplementary Table 2).
Contrastingly, all NLS-GFP versions coupled to poly-R peptides from R4 to R7 show a clear accumulation in the nucleoli around 2 to 3-fold over the nucleoplasmic level. The nucleolar accumulation is lowest for NLS-R4-GFP at 1.7-fold and increases with the growing number of arginine residues up to 2.7-fold for NLS-R7-GFP ( and C and supplementary Table 2). Interestingly, the comparison of the data for peptides and proteins shows that in contrast to the FITC-R5 peptide, we see already for the R4 fused to NLS-GFP a nucleolar accumulation. This may reflect the proteolytic degradation of the L-amino acids containing peptides versus the more stable corresponding GFP fusion proteins. On the other hand, the additional arginine within the NLS sequence could account for this apparent difference.
Noteworthy, peptides without nucleolar accumulation like K5 and R5 show no reduced concentration inside nucleoli. Already in an earlier study testing functional peptides not accumulating in nucleoli, we observed a similar homogenous nuclear and nucleolar distribution of a fluorophore labeled peptide.Citation50 These observations exemplify the dense structure of the nucleoli resulting in a size dependent sieving effect for molecules without potential for accumulation therein. Larger molecules like GFP (with dimensions of around 2.4 × 4.2 nm)Citation51 are significantly excluded compared to much smaller peptides. Interestingly, the R5 to R7 coupled NLS-GFP molecules show in addition an even higher efficiency of nuclear import with levels up to 10-fold over the cytoplasm compared to NLS-GFP alone ( and supplementary Table 2). This observation shows that the addition of arginine residues to a minimal NLS increases the efficiency of nuclear import largely exceeding the passive leakage efflux of small protein molecules through nuclear pores. Alternatively, their active retention in the nucleus by binding, e.g., to the nucleolus, prevents their exit from the nucleus.
The data presented here so far demonstrate that peptides and proteins with a strong positive charge and high isoelectric point have the potential to accumulate in the nucleolus of live cells. The threshold for strong nucleolar accumulation in peptides is R6, while in proteins bearing an NLS (with one additional R) already 4 additional consecutive arginines are sufficient. In addition, the nuclear import is enhanced by the addition of at least 5 arginine residues in proximity to a minimal NLS. We conclude that the charge and isoelectric properties of the peptides and protein domains responsible for nucleolar accumulation is key in this process because first, the difference between nuclear import and nucleolar accumulation has never been shown to rely on an active mechanism. Second, even peptides synthesized with D-amino acids show nucleolar accumulation, which may fail to specifically bind to the same cellular targets as their L-enantiomer counterparts.
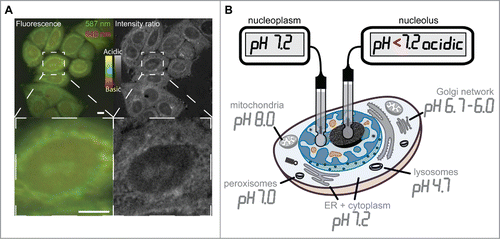
The evidence for a charge and isoelectric point dependency for peptide and protein accumulation in nucleoli led us to postulate the presence of an electrochemical component. Electrochemical interactions that would accumulate positively charged basic peptides should be mediated by highly abundant or strong negatively charged acidic nucleolar components. These have to be confined to nucleoli as are the accumulated basic peptides and proteins. Such molecules could be nucleic acids, which are highly abundant in the nucleoli in form of rRNA. Therefore, we wanted to determine the pH distribution inside the nucleus in living cells. We used the live cell permeable fluorescent dye SNARF-4F, which has 2 pH dependent ratiometric emission peaks at around 587 and 640 nm. The calculation of the ratio of the fluorescence intensity at the 2 emission peaks gives information on the pH of subcellular structures. The ratiometric images of HeLa cells show in the fluorescence channel view as well as in the intensity ratio view different cellular structures highlighted ( top row). The magnified image detail shows structures at the nuclear periphery and in the center of the nucleus highlighted as more acidic ( bottom row). Although the cytoplasm is generally considered to have a neutral pH, the more acidic regions in the cytoplasm do most likely represent parts of the Golgi network and organelles like peroxisomes.Citation52 The central nuclear region with more acidic pH is reminiscent of the nucleoli in HeLa cells shown in the phase contrast images in as well as the DIC images in and in supplementary Figure 1. Almost every cell in the overview image has the nucleoli highlighted as more acidic compared to the nucleoplasmic surrounding ( top row). Although precise measurements of subcellular pH values are not possible with the SNARF-4F dye, it allows us to conclude that the nucleolar environment is more acidic than the neutral pH of the nucleoplasm. The subcellular pH landscape was measured in various studies before and is reviewed in.Citation53 summarizes the reviewed data and was adapted to now incorporate the differentiated neutral pH of the nucleoplasm and more acidic pH of the nucleolus obtained from our measurements.
Figure 3. Intracellular pH landscape by ratiometric fluorescence microscopy toward the generation of an intracellular pH landscape. The figure in (A) displays fluorescence microscopy images of the pH sensitive dye SNARF-4F in live HeLa cells (color). The green channel shows the emission at 587 and the red channel the emission of the dye at 640 nm during excitation with the same wavelength. The resulting intensity ratio image (gray scale) shows ratiometric differences between the intensities of the 2 fluorescence emission peaks of the dye due to pH variations in subcellular structures. The fluorescence color scale and the ratio scale in gray indicate the relative range from more acidic to more basic pH. Panel (B) illustrates a map of the intracellular pH landscape by measuring the pH in various cellular compartments and substructures like nucleoplasm and nucleolus (modified fromCitation53). Scalebars: 5 µm.
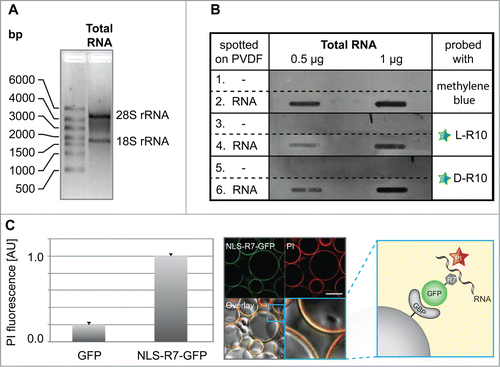
Next, we wanted to test whether the peptides and proteins with the sequence requirements sufficient to accumulate in nucleoli do bind RNA. To test our hypothesis, we purified total RNA () and performed slot blot analysis (). We blotted increasing amounts of total RNA on a PVDF membrane followed by incubation with either fluorescently labeled L-R10, or D-R10, respectively (). The fluorescently tagged D- and L-R10 were only detected on slots spotted with RNA (, rows 4 and 6) but not in control slots without RNA (, rows 3 and 5). As a loading control, membranes were stained in parallel with methylene blue (, rows 1 and 2). To test the RNA binding of basic peptides fused to GFP, we performed an in vitro RNA pulldown assay using NLS-R7-GFP immobilized to sepharose beads via the GFP binding protein (GBP)Citation39 (, scheme). This assay allowed us to observe (, confocal image) and quantify (, bar plot), the binding of RNA to NLS-R7-GFP. The accumulation of peptides in nucleoli is a common feature of arginine-rich cell penetrating peptides (CPPs).Citation54 Consistently, we found that the TAT peptide, one of the best known CPPs derived from HIV-1 TAT protein,Citation55 also binds to RNA (supplementary Fig. 2).
Figure 4. Analyses of RNA binding to poly-(R)peptides and GFP fusions. (A) Total RNA used for in vitro RNA binding assays separated by size showing the absence of genomic DNA contamination, as well as characteristic bands for the 28S and 18S rRNA, which are primarily synthesized and localized in nucleoli. The slot blot in (B) (representative from 2 independent experiments), rows 1 and 2, show a methylene blue stained PDVF membrane in the absence (1) and presence (2) of different amounts of total RNA (0.5 µg and 1 µg) and were used as a loading control. The rest of the blot shows the binding of fluorescently tagged L-R10 (3 and 4) and D-R10 (5 and 6) in the absence (3 and 5) and presence (4 and 6) of total RNA. Slots lacking RNA (rows 5 and 6) do not show binding of L- and D-R10, respectively. In contrast, slots probed with increasing amounts of RNA show increasing L- and D-R10 binding. (C) In vitro RNA pulldown assay using NLS-R7-GFP immobilized to sepharose beads via the GFP binding protein (GBP) (scheme). RNA was stained using propidium iodide (PI) and GFP alone was used as a negative control. RNA binding was measured on a fluorescent plate reader (bar plot) and imaged by confocal microscopy (microscopic images). Plots represent the average plus standard deviation of 2 independent experiments. Scalebar: 100 µm.
Defining experimentally the requirements of nucleolar peptide and protein accumulation has shown that several arginines in sequence are required. At least 6 arginine residues in a peptide and 4 arginines in combination with an NLS provide sufficient localized surface charge for nucleolar accumulation. Importantly, charge alone cannot explain the difference between poly-R and poly-K, which may additionally rely on the higher number of functional amino groups in arginines vs. lysines. Now we asked, how our findings relate to NoLS sequences found in the human proteome? A previous comprehensive study of nucleolar targeting sequences has systematically evaluated 46 nucleolar targeting sequences curated from the literature. Based on this analysis an algorithm was developed to predict nucleolar targeting based on peptide amino acid composition. The algorithm was then run against the International Protein Index database (IPI 3.4) and predicted >10,000 putative nucleolar targeting sequences. We have used this list to perform further analysis of the amino acid composition of predicted nucleolar targeting sequences. An overall statistics shows that the predicted NoLS have an average length of 27 amino acids with a most frequent length of 20 amino acids. The vast majority (83%) of predicted NoLS sequences has a length of 30 amino acids or less. For these NoLS the mean ratio of the number of basic (lysine, arginine and histidine) versus the total number of amino acids is 0.34. The shortest and most frequently predicted NoLS sequences of 20 amino acids therefore contain on average at least 7 basic amino acids. The 15 amino acids sequence containing NLS-R4 from the GFP coupled construct tested here has a ratio of 0.66 and contains 10 basic amino acids. A statistical analysis of the individual number of the amino acids for the 2001 predicted NoLS of 20 amino acid length most frequently contain at least 3 lysines, 3 arginines, one histidine, 2 cysteines, 2 prolines, and further each one aspartate, glutamate, glycine, alanine, threonine, valine, leucine, asparagine, glutamine. A peptide with the amino acid composition KKKRRRDEGATVLNQ would have a pKi value of 11.00, which is comparable to the FITC-K11 peptide tested in this study, which did not show significant nucleolar accumulation. But the combination of 4 arginines with an adjacent but separated NLS within 15 residues, as in NLS-R4-GFP, proved effective.
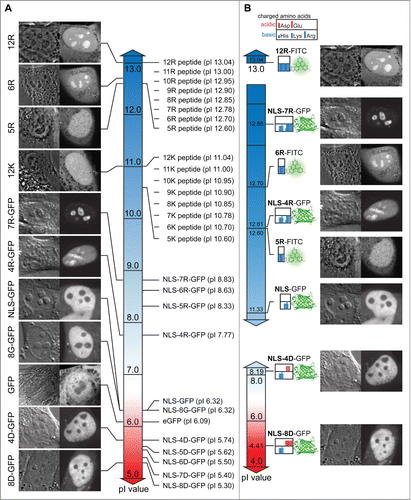
To gain a systematic overview over the peptide and protein localization dependent on charge, isoelectric properties and amino acid sequence composition, we created gradient charts depicting the pI values along with the respective nuclear and nucleolar accumulation as well as peptide composition maps (). The chart for peptide and total protein pI values shows a clear difference between all poly-K peptides (pI 10.6–11.04) exhibiting no nucleolar accumulation and the poly-R peptides with more basic pI values between 12.6 and 13.04 (). Although the pI values of the R5 and R6 peptides differ only slightly with 12.6 and 12.7 respectively, the charge driven threshold for nucleolar accumulation of peptides is found between these values (). When comparing the pI values of proteins it becomes apparent that the total protein pI does not represent specific charged targeting sequences or domains appropriately. All poly-R fused NLS-GFP versions with nucleolar accumulation range between pI 7.77 and 8.83, which is much less basic than any of the poly-R peptides. The pI value of GFP of 6.09 has a strong influence on the calculation of pI values for the fusion proteins with relatively short additions of basic amino acids (). Therefore, we also plotted the pI of the peptides tested and the protein N-terminal domains containing the NLS and the fused poly-R and poly-D sequences (). This representation shows that the pI values of the FITC-5R, which shows homogeneous nuclear distribution, and the NLS-4R, which shows nucleolar accumulation are virtually the same. The main difference between these sequences is the distribution of the charge over just 5 residues (5R) or 15 residues (NLS-4R) resulting in a different surface charge density. Therefore, we reason that in addition to the charge and isoelectric properties of peptides and protein localization sequences, also the charge distribution plays a key role in mediating nucleolar accumulation.
Figure 5. Chart of peptide and protein pI values, intracellular distribution and sequence composition. (A) Display of the pI value of FITC labeled peptides and full-length proteins coupled to different peptides along a continuous pI scale. Representative images illustrating the intracellular and intranuclear distribution are indicated along the scale. (B) Subsets of the pI scale with the values exclusively for the charged amino acid domains in the proteins and peptides tested combined with an illustrative representation of the domains. For the proteins the domain considered starts with the nuclear localization signal (NLS) and continues through the different motifs of charged amino acids (poly-R and poly-D) introduced into the GFP open reading frame. The peptides tested consist exclusively of basic amino acids. The clustering of the charged amino acids is displayed as color bars in the box adjacent to the respective fluorophore FITC or GFP. A legend for the charged amino acids is given above. Representative images for the distribution inside the cells and nuclei are displayed on the right for the respective peptide or protein. Scalebar: 5 µm.
Discussion
In this study, we have analyzed the requirements of peptides and proteins to accumulate in the nucleoli of living cells. It has been already established that specific sequences in proteins are responsible for the nuclear import (NLS) and further for the localization in the nucleoli (NoLS).Citation16,56 The systematic analysis of experimentally confirmed NoLS has shown that a high content of basic amino acids is common among nucleolar but also nuclear targeting signals, which makes it difficult to differentiate between NoLS and NLS. This difficulty is further aggravated by the fact that validated and predicted NoLS depict high sequence diversity.Citation18,57
Our results show first that all poly-K and poly-R peptides ranging from 5–12 amino acids accumulate in the cell nucleus compared to the cytoplasmic level and, therefore, match the criteria for nuclear localization sequences. Similarly, all NLS bearing GFP versions tested show a nuclear accumulation, which is generally a feature of nuclear proteins bearing a NLS.Citation57 In line with this interpretation, the GFP protein without NLS shows a homogeneous distribution between nucleus and cytoplasm. Interestingly, the mean cytoplasmic fluorescence intensity of all L-peptides tested (0.61 to 0.76 fold) is much higher than for any of the NLS containing GFP versions (0.08 to 0.39) showing that passive nuclear pore leakage is less efficient and slower for larger molecules. The distribution test for the same peptides with different chirality showed that FITC-R10 composed of D-amino acids has a reduced concentration in the nucleoplasm compared to the L-amino acid peptide. We interpret this observation as lack of specific binding to nuclear im/export factors together with a sequestration of the D-peptide in nucleoli. In addition, the potential degradation of L-amino acid peptides during the experiments, would result in freely diffusing FITC molecules not bound to peptides and increase their nucleoplasmic signal altogether.
The nucleolar accumulation of peptides is dependent on the amino acid composition as poly-R peptides, except R5, show strong accumulation whereas poly-K peptides accumulate only slightly and, in most cases, not significantly. A sequence of at least 6 arginines per peptide is required, while even 12 lysines show only very little effect. These findings indicate the existence of an isoelectric threshold to reach accumulation in nucleoli, although the pI difference between R5 and R6 is rather small with 12.6 and 12.7 respectively ().
Surprisingly, already the fusion of 4 arginines residues to NLS-GFP results in a strong protein accumulation in the nucleolus. The level of accumulation is even higher than for all FITC labeled poly-R peptides tested, while the pI of 12.61 for the domain containing the NLS-4R is virtually the same as for the non-accumulating R5 peptide. In this case, a cumulative effect of the poly-R sequence and the adjacent NLS within a short region of 15 amino acids, containing an additional arginine as well as further lysine residues, generates an effective motif that binds to nucleolar constituents. It has been shown a long time ago, that E. coli ribosomes contain at least 21 proteins with extremely high pI values >12.0, similar to the poly-R peptides and NLS-poly-R domains fused to GFP.Citation58
These data suggest that, in addition to the peptide and protein domain charge alone, the surface charge density is an important determinant of interaction with nucleolar constituents. A protein domain surface with a larger distribution of positive charges below a pI of 12.7 is also effective to promote sufficient interaction to accumulate in nucleoli. On the other hand, sequences with even more lysine residues or shorter examples with few lysines and arginines can also serve as NoLS as found in a previous systematic analysis and predicted by the Nucleolar Localization Sequence Detector webtool.Citation18 The other tracer proteins GFP, NLS-GFP and those in fusion with neutral or positively charged amino acids show no accumulation in nucleoli. In fact, they are rather largely “apparently” excluded from nucleoli. Nevertheless, it has been shown that even those molecules enter and rapidly traverse nucleoli.Citation26 On the contrary, even the non-accumulating poly-K peptides and others have been found to homogeneously distribute in nucleoplasm and nucleoli. These findings suggest a volume exclusion or molecular sieve effect of the nucleolar compartment similar to the size dependent passive diffusion through nuclear pore channels.
The requirement of a strong positive charge mediated by basic amino acids distributed over a certain molecular interaction surface seems to be a hallmark of the potential to accumulate proteins in the nucleolus. The nucleolar interaction partners are either more abundant than in the surrounding nucleoplasm or lead to a strong retention within this subnuclear compartment.Citation59 A charge and isoelectric properties driven accumulation has to be mediated by a countercharged nucleolar component. Highly abundant and integral functional determinant of nucleoli is rRNA, which can be considered as large negatively charged complexes. Several ribosomal proteins possess high pI values, thus, are positively charged and are very likely to electrostatically interact with RNA. We could indeed show the presence of a more acidic pH in the nucleoli by the live-cell pH sensitive dye SNARF-4F. The RNA transcribed in nucleoli is a prime candidate for the locally constrained proton donor that is also an ideal target for protein and peptide nucleolar localization sequences. Our suggestion is supported by a report on protein immobilization in the nucleolus by noncoding RNA via the nucleolar detention sequence (NoDS). In this study, the deletion of arginine motifs in the NoDS of Hsp70 prevents nucleolar accumulation as well as the precipitation of a peptide NoDS failed without arginines in the motif.Citation60 Sequence alignment of the NoLS for the human La protein with several other nucleolar localization domains of human proteins revealed a (R/K) (R/K) × (R/K) motif appearing frequently as single or multiple copies and similar motifs had been known before.Citation61,62 The importance of arginines has also been shown for a number of RNA binding proteins with arginine rich motifs.Citation63 In addition, a large-scale analysis of atomic contacts determined that the positively charged amino acid residues arginine and lysine are among the most frequent to interact with nucleic acids with high abundance of interaction with the phosphate backbone.Citation64,65 Protein domains with multiple functional arginines are common in RNA recognition motifs, e.g., RGG and RGX often found in a modular composition.Citation66,67 Furthermore, the existence of more functional amino groups in arginines relative to lysines explains their more stable electrostatic interaction with the phosphate groups in RNA.Citation68 The latter has been exploited as arginine-affinity chromatography to purify RNA.Citation69 Our in vitro binding analyses demonstrate that poly-R peptides and fusion proteins directly bind RNA, which is mostly abundant in the nucleolus due to massive ribosome production and much less in the nucleoplasm.Citation70-73 Finally, we could show, using cells from different species, that these principles of nucleolar targeting are evolutionarily conserved.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Data
Download Zip (3.7 MB)Acknowledgments
We gratefully acknowledge Jenny Völger, Petra Domaing and Franziska Witzel for their help and support. We are indebted to Dawid Grzela and Zsuzsanna Izsvak for the Pac2 cells and Anup Arumughan for the yeast cells. We thank Heinrich Leonhardt for many fruitful discussions.
Funding
RMM was supported by a post-doctoral grant from Fundação para a Ciência e Tecnologia, Portugal (SFRH-BPD-66611–2009). This work was supported by grants of the German Research Council (DFG CA198/3) to MCC.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Cardoso MC, Schneider K, Martin RM, Leonhardt H. Structure, function and dynamics of nuclear subcompartments. Curr Opin Cell Biol 2012; 24:79-85; PMID:22227228; http://dx.doi.org/10.1016/j.ceb.2011.12.009
- Lamond AI, Earnshaw WC. Structure and function in the nucleus. Science 1998; 280:547-53; PMID:9554838; http://dx.doi.org/10.1126/science.280.5363.547
- Dundr M, Misteli T. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol 2010; 2:a000711; PMID:21068152; http://dx.doi.org/10.1101/cshperspect.a000711
- Spector DL. Nuclear domains. J Cell Sci 2001; 114:2891-3; PMID:11686292
- Bickmore WA, Sutherland HG. Addressing protein localization within the nucleus. Embo J 2002; 21:1248-54; PMID:11889031; http://dx.doi.org/10.1093/emboj/21.6.1248
- Gorski SA, Dundr M, Misteli T. The road much traveled: trafficking in the cell nucleus. Curr Opin Cell Biol 2006; 18:284-90; PMID:16621498; http://dx.doi.org/10.1016/j.ceb.2006.03.002
- Mekhail K, Khacho M, Carrigan A, Hache RR, Gunaratnam L, Lee S. Regulation of ubiquitin ligase dynamics by the nucleolus. J Cell Biol 2005; 170:733-44; PMID:16129783; http://dx.doi.org/10.1083/jcb.200506030
- Eilbracht J, Schmidt-Zachmann MS. Identification of a sequence element directing a protein to nuclear speckles. Proc Natl Acad Sci U S A 2001; 98:3849-54; PMID:11274404; http://dx.doi.org/10.1073/pnas.071042298
- Lam YW, Trinkle-Mulcahy L, Lamond AI. The nucleolus. J Cell Sci 2005; 118:1335-7; PMID:15788650; http://dx.doi.org/10.1242/jcs.01736
- Raska I, Shaw PJ, Cmarko D. Structure and function of the nucleolus in the spotlight. Curr Opin Cell Biol 2006; 18:325-34; PMID:16687244; http://dx.doi.org/10.1016/j.ceb.2006.04.008
- Scheer U, Hock R. Structure and function of the nucleolus. Curr Opin Cell Biol 1999; 11:385-90; PMID:10395554; http://dx.doi.org/10.1016/S0955-0674(99)80054-4
- Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature 2005; 433:77-83; PMID:15635413; http://dx.doi.org/10.1038/nature03207
- Grob A, Colleran C, McStay B. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev 2014; 28:220-30; PMID:24449107; http://dx.doi.org/10.1101/gad.234591.113
- Cheutin T, O'Donohue MF, Beorchia A, Vandelaer M, Kaplan H, Defever B, Ploton D, Thiry M. Three-dimensional organization of active rRNA genes within the nucleolus. J Cell Sci 2002; 115:3297-307; PMID:12140261
- Derenzini M, Pasquinelli G, O'Donohue MF, Ploton D, Thiry M. Structural and functional organization of ribosomal genes within the mammalian cell nucleolus. J Histochem Cytochem 2006; 54:131-45; PMID:16204224; http://dx.doi.org/10.1369/jhc.5R6780.2005
- Dang CV, Lee WM. Nuclear and nucleolar targeting sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and HIV tat proteins. J Biol Chem 1989; 264:18019-23; PMID:2553699
- Hatanaka M. Discovery of the nucleolar targeting signal. Bioessays 1990; 12:143-8; PMID:2108666; http://dx.doi.org/10.1002/bies.950120310
- Scott MS, Boisvert FM, McDowall MD, Lamond AI, Barton GJ. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res 2010; 38:7388-99; PMID:20663773; http://dx.doi.org/10.1093/nar/gkq653
- Paine PL, Moore LC, Horowitz SB. Nuclear envelope permeability. Nature 1975; 254:109-14; PMID:1117994; http://dx.doi.org/10.1038/254109a0
- Lusk CP, Blobel G, King MC. Highway to the inner nuclear membrane: rules for the road. Nat Rev Mol Cell Biol 2007; 8:414-20; PMID:17440484; http://dx.doi.org/10.1038/nrm2165
- Gorlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol 1999; 15:607-60; PMID:10611974; http://dx.doi.org/10.1146/annurev.cellbio.15.1.607
- Breeuwer M, Goldfarb DS. Facilitated nuclear transport of histone H1 and other small nucleophilic proteins. Cell 1990; 60:999-1008; PMID:1690602; http://dx.doi.org/10.1016/0092-8674(90)90348-I
- Dundr M, Misteli T, Olson MO. The dynamics of postmitotic reassembly of the nucleolus. J Cell Biol 2000; 150:433-46; PMID:10931858; http://dx.doi.org/10.1083/jcb.150.3.433
- Mekhail K, Rivero-Lopez L, Al-Masri A, Brandon C, Khacho M, Lee S. Identification of a common subnuclear localization signal. Mol Biol Cell 2007; 18:3966-77; PMID:17652456; http://dx.doi.org/10.1091/mbc.E07-03-0295
- Leung AK, Lamond AI. The dynamics of the nucleolus. Crit Rev Eukaryot Gene Expr 2003; 13:39-54; PMID:12839096; http://dx.doi.org/10.1615/CritRevEukaryotGeneExpr.v13.i1.40
- Grunwald D, Martin RM, Buschmann V, Bazett-Jones DP, Leonhardt H, Kubitscheck U, Cardoso MC. Probing Intranuclear Environments at the Single Molecule Level. Biophys J 2008; 94:2847-58; PMID:18065482; http://dx.doi.org/10.1529/biophysj.107.115014
- Scott MS, Troshin PV, Barton GJ. NoD: a Nucleolar localization sequence detector for eukaryotic and viral proteins. BMC Bioinformatics 2011; 12:317; PMID:21812952; http://dx.doi.org/10.1186/1471-2105-12-317
- Martin RM, Tunnemann G, Leonhardt H, Cardoso MC. Nucleolar marker for living cells. Histochem Cell Biol 2007; 127:243-51; PMID:17205309; http://dx.doi.org/10.1007/s00418-006-0256-4
- Lattig-Tunnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, Cardoso MC. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat Commun 2011; 2:453; PMID:21878907; http://dx.doi.org/10.1038/ncomms1459
- Matthias P, Muller MM, Schreiber E, Rusconi S, Schaffner W. Eukaryotic expression vectors for the analysis of mutant proteins. Nucleic Acids Res 1989; 17:6418; PMID:2549518; http://dx.doi.org/10.1093/nar/17.15.6418
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. Dynamics of DNA replication factories in living cells. J Cell Biol 2000; 149:271-80; PMID:10769021; http://dx.doi.org/10.1083/jcb.149.2.271
- Martin RM, Leonhardt H, Cardoso MC. DNA labeling in living cells. Cytometry A 2005; 67:45-52; PMID:16082711; http://dx.doi.org/10.1002/cyto.a.20172
- Agarwal N, Hardt T, Brero A, Nowak D, Rothbauer U, Becker A, Leonhardt H, Cardoso MC. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res 2007; 35:5402-8; PMID:17698499; http://dx.doi.org/10.1093/nar/gkm599
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell 2004; 15:853-65; PMID:15383276; http://dx.doi.org/10.1016/j.molcel.2004.09.016
- McNeil PL. Incorporation of macromolecules into living cells. Methods Cell Biol 1989; 29:153-73; PMID:2643758; http://dx.doi.org/10.1016/S0091-679X(08)60193-4
- Schermelleh L, Solovei I, Zink D, Cremer T. Two-color fluorescence labeling of early and mid-to-late replicating chromatin in living cells. Chromosome Res 2001; 9:77-80; PMID:11272795; http://dx.doi.org/10.1023/A:1026799818566
- Cardoso MC, Joseph C, Rahn HP, Reusch R, Nadal-Ginard B, Leonhardt H. Mapping and use of a sequence that targets DNA ligase I to sites of DNA replication in vivo. J Cell Biol 1997; 139:579-87; PMID:9348276; http://dx.doi.org/10.1083/jcb.139.3.579
- Herce HD, Deng W, Helma J, Leonhardt H, Cardoso MC. Visualization and targeted disruption of protein interactions in living cells. Nat Commun 2013; 4:2660; PMID:24154492; http://dx.doi.org/10.1038/ncomms3660
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 2008; 7:282-9; http://dx.doi.org/10.1074/mcp.M700342-MCP200
- Molenaar I, Sillevis Smitt WW, Rozijn TH, Tonino GJ. Biochemical and electron microscopic study of isolated yeast nuclei. Exp Cell Res 1970; 60:148-56; PMID:5424309; http://dx.doi.org/10.1016/0014-4827(70)90500-8
- Taddei A, Schober H, Gasser SM. The budding yeast nucleus. Cold Spring Harb Perspect Biol 2010; 2:a000612; PMID:20554704; http://dx.doi.org/10.1101/cshperspect.a000612
- Yang CH, Lambie EJ, Hardin J, Craft J, Snyder M. Higher order structure is present in the yeast nucleus: autoantibody probes demonstrate that the nucleolus lies opposite the spindle pole body. Chromosoma 1989; 98:123-8; PMID:2673672; http://dx.doi.org/10.1007/BF00291048
- Blackmond DG. The origin of biological homochirality. Cold Spring Harb Perspect Biol 2010; 2:a002147; PMID:20452962; http://dx.doi.org/10.1101/cshperspect.a002147
- Kreil G. D-amino acids in animal peptides. Annu Rev Biochem 1997; 66:337-45; PMID:9242910; http://dx.doi.org/10.1146/annurev.biochem.66.1.337
- Nagata Y, Yamamoto K, Shimojo T, Konno R, Yasumura Y, Akino T. The presence of free D-alanine, D-proline and D-serine in mice. Biochim Biophys Acta 1992; 1115:208-11; PMID:1346751; http://dx.doi.org/10.1016/0304-4165(92)90055-Y
- Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 1999; 99:103-15; PMID:10520998; http://dx.doi.org/10.1016/S0092-8674(00)80066-5
- Zhao L, Lu W. Mirror image proteins. Curr Opin Chem Biol 2014; 22C:56-61; http://dx.doi.org/10.1016/j.cbpa.2014.09.019
- Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell 1984; 39:499-509; PMID:6096007; http://dx.doi.org/10.1016/0092-8674(84)90457-4
- Dultz E, Huet S, Ellenberg J. Formation of the nuclear envelope permeability barrier studied by sequential photoswitching and flux analysis. Biophys J 2009; 97:1891-7; PMID:19804719; http://dx.doi.org/10.1016/j.bpj.2009.07.024
- Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F, Cardoso MC. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. Faseb J 2006; 20:1775-84; PMID:16940149; http://dx.doi.org/10.1096/fj.05-5523com
- Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996; 273:1392-5; PMID:8703075; http://dx.doi.org/10.1126/science.273.5280.1392
- Wu MM, Llopis J, Adams S, McCaffery JM, Kulomaa MS, Machen TE, Moore HP, Tsien RY. Organelle pH studies using targeted avidin and fluorescein-biotin. Chem Biol 2000; 7:197-209; PMID:10712929; http://dx.doi.org/10.1016/S1074-5521(00)00088-0
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 2010; 11:50-61; PMID:19997129; http://dx.doi.org/10.1038/nrm2820
- Herce HD, Garcia AE, Cardoso MC. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J Am Chem Soc 2014; 136:17459-67; PMID:25405895; http://dx.doi.org/10.1021/ja507790z
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988; 55:1189-93; PMID:2849510; http://dx.doi.org/10.1016/0092-8674(88)90263-2
- Schmidt-Zachmann MS, Nigg EA. Protein localization to the nucleolus: a search for targeting domains in nucleolin. J Cell Sci 1993; 105 ( Pt 3):799-806; PMID:8408305
- Kuusisto HV, Wagstaff KM, Alvisi G, Roth DM, Jans DA. Global enhancement of nuclear localization-dependent nuclear transport in transformed cells. Faseb J 2012; 26:1181-93; PMID:22155563; http://dx.doi.org/10.1096/fj.11-191585
- Kaltschmidt E. Ribosomal proteins. XIV. Isoelectric points of ribosomal proteins of E. coli as determined by two-dimensional polyacrylamide gel electrophoresis. Anal Biochem 1971; 43:25-31; PMID:4943259; http://dx.doi.org/10.1016/0003-2697(71)90103-5
- Audas TE, Jacob MD, Lee S. The nucleolar detention pathway: A cellular strategy for regulating molecular networks. Cell Cycle 2012; 11:2059-62; PMID:22580471; http://dx.doi.org/10.4161/cc.20140
- Audas TE, Jacob MD, Lee S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell 2012; 45:147-57; PMID:22284675; http://dx.doi.org/10.1016/j.molcel.2011.12.012
- Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep 2009; 10:231-8; PMID:19229283; http://dx.doi.org/10.1038/embor.2009.14
- Horke S, Reumann K, Schweizer M, Will H, Heise T. Nuclear trafficking of La protein depends on a newly identified nucleolar localization signal and the ability to bind RNA. J Biol Chem 2004; 279:26563-70; PMID:15060081; http://dx.doi.org/10.1074/jbc.M401017200
- Bayer TS, Booth LN, Knudsen SM, Ellington AD. Arginine-rich motifs present multiple interfaces for specific binding by RNA. Rna 2005; 11:1848-57.; PMID:16314457; http://dx.doi.org/10.1261/rna.2167605
- Treger M, Westhof E. Statistical analysis of atomic contacts at RNA-protein interfaces. J Mol Recognit 2001; 14:199-214; PMID:11500966; http://dx.doi.org/10.1002/jmr.534
- Lejeune D, Delsaux N, Charloteaux B, Thomas A, Brasseur R. Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins 2005; 61:258-71; PMID:16121397; http://dx.doi.org/10.1002/prot.20607
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 2007; 8:479-90; PMID:17473849; http://dx.doi.org/10.1038/nrm2178
- Rajyaguru P, Parker R. RGG motif proteins: modulators of mRNA functional states. Cell Cycle 2012; 11:2594-9; PMID:22767211; http://dx.doi.org/10.4161/cc.20716
- Woods AS, Ferre S. Amazing stability of the arginine-phosphate electrostatic interaction. J Proteome Res 2005; 4:1397-402; PMID:16083292; http://dx.doi.org/10.1021/pr050077s
- Martins R, Queiroz JA, Sousa F. New approach in RNA quantification using arginine-affinity chromatography: potential application in eukaryotic and chemically synthesized RNA. Anal Bioanal Chem 2013; 405:8849-58; PMID:24037617; http://dx.doi.org/10.1007/s00216-013-7334-y
- Baron-Benhamou J, Gehring NH, Kulozik AE, Hentze MW. Using the lambdaN peptide to tether proteins to RNAs. Methods Mol Biol 2004; 257:135-54; PMID:14770003
- Chen Y, Varani G. Protein families and RNA recognition. FEBS J 2005; 272:2088-97; PMID:15853794; http://dx.doi.org/10.1111/j.1742-4658.2005.04650.x
- Costa MC, Leitao AL, Enguita FJ. Biogenesis and Mechanism of Action of Small Non-Coding RNAs: Insights from the Point of View of Structural Biology. Int J Mol Sci 2012; 13:10268-95; PMID:22949860; http://dx.doi.org/10.3390/ijms130810268
- Lewis JD, Tollervey D. Like attracts like: getting RNA processing together in the nucleus. Science 2000; 288:1385-9; PMID:10827942; http://dx.doi.org/10.1126/science.288.5470.1385