
ABSTRACT
Replication stress, defined as the slowing or stalling of cellular DNA replication forks, represents a serious threat to genome stability. Numerous cellular pathways protect against replication stress and maintain genomic integrity. Among these, the Fanconi Anemia/homologous recombination pathways are critical for recognizing and repairing stalled replication forks. Members of these pathways play a vital role in protecting damaged forks from uncontrolled attack from cellular nucleases, which would otherwise render these irreparable. Recent studies have begun to shed light on the protective factors necessary to suppress nucleolytic over-processing of nascent DNA, and on the different cellular nucleases involved. Here, we review our recent identification of a novel fork protection factor, BOD1L, and discuss its role in preventing the processing of stalled replication forks within the context of current knowledge of the replication fork ‘protectosome’.
Replication stress and fork protection
Perturbation of cellular DNA replication by both endogenous and exogenous factors leads to replication fork slowing, stalling and/or collapse, termed replication stress. An inability to deal with such replication stress contributes to the development of human disease.Citation1 Notably, unresolved replication stress leads to genomic instability, a frequent hallmark of tumors. A plethora of cellular proteins are involved in the cellular response to replication stress. Since these components are vital in maintaining genome stability, they therefore represent critical obstacles to the initiation/progression of cellular transformation.
Over the past decade, much progress has been made in understanding the function of proteins involved in the replication stress response. These studies have underlined a crucial role for proteins involved in the Fanconi Anaemia (FA)/homologous recombination (HR) pathway in maintaining genome stability upon replication damage. Components of this pathway have classically been associated with the HR-dependent repair of inter-strand crosslinks (ICLs), and mutations in these genes give rise to Fanconi Anaemia, a rare disorder characterized by severe developmental abnormalities, bone marrow failure, tumor predisposition and a hypersensitivity to agents that induce ICLs, such as mitomycin C (MMC) and cisplatin (for a review seeCitation2). To date, mutations in 18 different genes (designated as complementation groups FANCA-T) have been identified in FA patients.Citation3,4
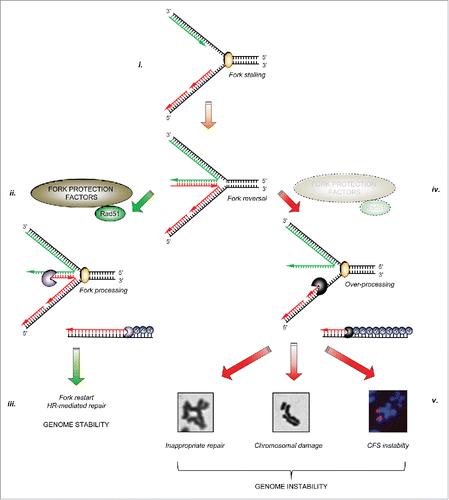
In addition to their importance in promoting the repair of ICLs, it is now apparent that several FA/HR proteins also play a role in protecting stalled/collapsed replication forks from uncontrolled nucleolysis. If left unprotected, excessive nucleolytic processing renders such forks unrecoverable, and may perturb replication to such an extent that stretches of under-replicated DNA accumulate, particularly around common fragile sites (CFS), which ultimately manifests as severe chromosome breakage. Furthermore, loss of fork protection may also lead to inappropriate repair of the stalled/collapsed replication fork by alternative pathways such as non-homologous end-joining (NHEJ), resulting in chromosomal fusions or translocations. These fork protection factors therefore represent important barriers to prevent genome instability ().
Figure 1. The importance of fork protection in maintaining genome stability: i. Cellular replication forks can stall for a variety of reasons. In certain circumstances, stalled forks may reverse to aid repair or restart. ii. A number of fork protection factors including RAD51 (described in the text) act to protect nascent DNA from over-processing by cellular nucleases (denoted by ‘pacman’ symbols). iii. This allows subsequent fork restart and/or repair by homologous recombination, and prevents genome instability. iv. In the absence of these protective factors, excessive nucleolytic processing of stalled/reversed forks leads to common fragile site and chromosomal instability, or to inappropriate repair giving rise to chromosomal fusions and radial chromosomes, ultimately leading to genomic instability (v).
Fork protection factors
Several FA/HR proteins are involved in suppressing replication fork over-processing by cellular nucleases.Citation5-11 It seems that the DNA recombinase RAD51 is crucial for fork protection, and defects in RAD51-mediated fork protection underlie the uncontrolled fork resection observed in the absence of many components of the FA/HR pathway. The most well-characterized function of RAD51 is its ability to promote strand exchange during HR, by displacing the single-stranded DNA binding protein RPA to form helical nucleofilaments. These filaments search for homologous sequences and catalyze strand invasion prior to the initiation of repair DNA synthesis and resolution of the joined molecules (for a review, see refCitation12). However, more recently it has been shown that the loading of RAD51 at replication forks also functions to stabilize replication fork intermediates and prevent deleterious nucleolytic over-processing.Citation5,6,13 Moreover, it seems that this protective function of RAD51 is similarly dependent on its ability to form nucleofilaments at replication forks.Citation5,6
In addition to RAD51, the FA proteins FANCA, FANCB, FANCD2, PALB2 (FANCN), BRCA1 (FANCS) and BRCA2 (FANCD1) also suppress genomic instability upon replication fork stalling, by preventing the degradation of nascent DNA.Citation6,11,14 Since BRCA1, PALB2 and BRCA2 are required to load RAD51 onto ssDNA at stalled replication forks, it is natural to assume that their ability to protect replication forks from degradation is due to their role in this process. However, studies of a BRCA2 mutant that lacks a conserved C-terminal RAD51-binding site revealed that this region is essential for fork protection but dispensable for loading RAD51 onto DNA, and for HR.Citation5 Therefore, subsequent to its loading, the stabilization of RAD51 at replication forks by BRCA2 is crucial in preventing excessive nucleolytic processing. Indeed, the artificial stabilization of RAD51 nucleofilaments on ssDNA by preventing its ATP-dependent dissociation protects against the over-processing of forks when BRCA2 is missing. In analogous fashion, the helicase/nuclease WRN is thought to protect nascent DNA by stabilizing RAD51 nucleofilaments, through a mechanism independent of its enzymatic activity.Citation7,8 Similarly, the TLS polymerase REV1 has recently been reported to prevent fork resection, again through its ability to promote RAD51 loading/foci formation.Citation9 Interestingly, PARP-1 has also been implicated in facilitating RAD51-mediated protection of stalled replication forks,Citation10 although it is unclear whether this is mediated through the poly-ADP ribose (PAR) chain-dependent recruitment of protective factors to stalled forks or via an alternative mechanism.
As mentioned above, FANCD2 stabilises and protect damaged forks from promiscuous nucleolytic attack.Citation6,15 However, deficiencies in FANCD2 do not lead to defects in the recruitment/stabilization of RAD51 to stalled forksCitation16,17; this suggests that an inability to load/stabilize RAD51 may not account for all instances of fork instability. In addition, although depletion of the FA core factors FANCA and FANCB also causes increased fork processing, it is unclear whether this is due to a subsequent failure to promote FANCD2 mono-ubiquitylation/relocalisation,Citation18 or via another mechanism.
It is plausible that, in certain contexts, RAD51 filament dissolution may also be crucial in maintaining replication fork stability. The RECQL5 helicase acts to suppress HR via the dissolution of RAD51 nucleofilaments.Citation19 Intriguingly, loss of RECQL5 alone leads to replication fork degradation, which is exacerbated by the loss of the FA pathway.Citation11 Therefore, it seems that both the de-stabilization and over-stabilization of RAD51 nucleofilaments are deleterious to replication fork integrity. Moreover, since RECQL5 has been linked to the regulation of transcriptional stress,Citation20 it is tempting to speculate that the role of RAD51 at replication forks may differ between different genomic contexts e.g. replication forks that have stalled due to DNA lesions versus those that have stalled upon collision with transcription bubbles. Importantly, the protective function of many of the proteins described in this section is likely distinct from their role in promoting HR-mediated repair of double-strand breaks (DSBs).
Cellular nucleases involved in fork degradation
While it is clear that uncontrolled nucleolytic activity is clearly detrimental to genome stability,Citation7,8,21-24 a certain degree of regulated processing by these nucleases is necessary to allow the repair/restart of damaged replication forks. Years of study have implicated the nucleases MRE11, CtIP, DNA2, and EXO1 in the nucleolytic processing of DSBs, and have uncovered complex mechanisms regulating DNA end resection. It is thought that MRE11 and CtIP act together to perform short-range resection, while EXO1 and DNA2/BLM act independently to execute 5′-3′ long-range processing to facilitate RAD51 loading and strand invasion.Citation25
Unrestricted nucleolytic processing by these proteins also underlies excessive fork resection. MRE11 has been implicated in the uncontrolled resection observed in the absence of many factors, including a functional FA/HR pathway: inhibition of MRE11 in the absence of FANCA, FANCB, BRCA1, BRCA2, FANCD2, RECQL5, REV1 and PARP1 prevents fork resection.Citation5,6,9,11,15 Moreover, MRE11-dependent fork resection underlies the increased chromosome breakage exhibited by BRCA2 null cells.Citation5 Thus it seems that these FA/HR factors specifically restrict the activity of MRE11 at replication forks to prevent over-processing. However, many of these studies (including our own) have made use of the MRE11 inhibitor Mirin. Therefore, it is conceivable that this inhibitor targets other cellular nucleases in a non-specific fashion, thus perhaps masking their importance in replication fork processing.
In addition to MRE11, DNA2 and EXO1 also play important roles in fork processing. DNA2 knockdown, but not depletion of EXO1 or MRE11, has been shown to alleviate fork processing after hydroxyurea.Citation26 Furthermore, RNAi-mediated depletion of DNA2 in FANCD2-deficient cells rescues their hypersensitivity to ICLs.Citation24 Finally, EXO1 has also been directly implicated in fork over-resection: EXO1 knockdown reduced fork over-processing in WRN exonuclease mutant cells.Citation7 In addition, yeast Exo1 is recruited to stalled forks where it mediates resection of newly synthesized DNA.Citation27 It therefore seems that different fork protection factors act to antagonize the actions of specific nucleases.
BOD1L protects replication forks from Dna2-dependent resection
Recently, we described the identification of a novel fork protection factor, BOD1L, which associates with the replication machinery.Citation28 BOD1L-depleted cells exhibit increased degradation of stalled/damaged forks in a manner comparable to FANCD2/BRCA1/BRCA2 deficient cells.Citation6 Furthermore, BOD1L-deficient cells were defective in their ability to form RAD51 foci at stalled forks, similar to cells lacking BRCA1/BRCA2/PALB2.Citation29-31 However, further analysis revealed that this was not due to an inability to load RAD51 at these sites; instead, BOD1L was required to stabilize RAD51 by preventing the anti-recombinase activities of the RECQ-like helicases BLM and FBH1. Down-regulation of either BLM or FBH1 suppressed both the aberrant fork processing and the genomic instability observed in BOD1L-deficient cells, presumably due to their ability to suppress RAD51-mediated HR by disassembling RAD51 nucleofilaments.Citation32-34 In this regard, BOD1L can be considered to function in an analogous manner to the C-terminus of BRCA2 i.e. by promoting RAD51 nucleofilament stability.Citation5,35 However, despite our observations that loss of BLM/FBH1 could not restore the loading of RAD51 in BRCA2 deficient cells, it is unclear whether these helicases also play a role in antagonizing BRCA2-dependent RAD51 stabilization.
Given that MRE11-dependent resection underlies many aspects of excessive fork processing, we hypothesized that MRE11 may also be responsible for fork resection observed in the absence of BOD1L. Surprisingly, inhibition of MRE11 activity using Mirin had no impact on nucleolytic fork processing in BOD1L-depeleted cells: instead, blocking DNA2-dependent resection restored normal fork processing and restored genome stability in the absence of BOD1L. This further supports the idea that different fork protection factors act to protect replication forks from individual nucleases (). In light of this, loss of two independently acting fork protection factors would be expected to further increase fork resection. However, although BOD1L and BRCA2 suppress the actions of different nucleases at stalled forks (namely DNA2 and MRE11 respectively), we observed an epistatic relationship. Currently, the underlying reason for this is unclear. However, we hypothesize that nucleolysis of a damaged fork by one nuclease may prevent further nucleolytic processing by another.
Figure 2. The specificity of factors counteracting ‘fork’ nucleases: Three main cellular nucleases have thus far been implicated in the over-processing of stalled forks: MRE11, DNA2 and EXO1. There have been no reports that the nuclease CtIP is involved in over-processing of stalled replication forks. Several protective factors act on specific cellular nucleases to supress their aberrant activity on stalled replication forks (dotted red lines): several FA/HR proteins, the TLS polymerase REV1 and PARP1 have all been reported to inhibit MRE11-dependent fork resection, whilst the WRN helicase/nuclease prevents EXO1-dependent fork degradation. Recently, we demonstrated that BOD1L is required to suppress DNA2-dependent strand degradation of stalled replication forks, alongside speculative reports of a similar role for FANCD2.
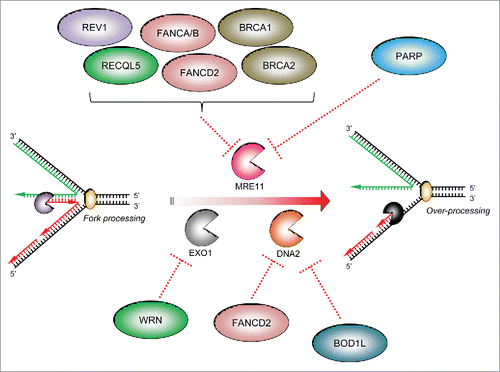
Together, our data suggests that the inability of BOD1L-deficient cells to stabilize RAD51 at stalled replication forks allows uncontrolled DNA2-dependent degradation of damaged forks. This is the first report of a fork protection factor that specifically acts to suppress DNA2-dependent processing of damaged replication forks.
A multi-protein fork protection ‘protectosome’ complex?
One intriguing finding from our studies is that BOD1L co-immunoprecipitates with at least two known fork protection factors: BRCA2 and FANCD2.Citation28 Moreover, unpublished data suggests that BOD1L may also interact with BRCA1. In addition, BOD1L loss is epistatic with depletion of BRCA1, BRCA2 and FANCA. We therefore suggest that a single complex containing multiple fork protection factors may exist. In support of this, several interactions between fork protection factors have been already reported in the literature, including between BRCA1, BRCA2, RAD51, FANCD2 and FANCA.Citation36 Alternatively, it may be that smaller sub-complexes of these proteins act in a lesion-specific or context-specific manner to counter specific nucleases. It is also possible that these proteins may be temporally regulated to prevent nucleolytic processing at different stages of replication fork repair/restart, by countering ‘early’ MRE11-dependent or ‘late’ DNA2/EXO1-mediated resection.
The physiological role of fork protection
Given the importance of replication fork protection in maintaining genome stability, this pathway likely plays a vital physiological role outside the laboratory. Firstly, during oncogene-induced replication stress, fork protection factors would be crucial to prevent chromosomal aberrations, which would otherwise promote cellular transformation. Since most fork protection factors also promote DNA replication and/or HR repair, which are important tumor suppressor functions, it is difficult to assess the impact of fork protection on tumourigenesis directly. Nevertheless, mutations affecting a CDK phosphorylation site in the C-terminus of BRCA2 that is important for regulating fork protection (but not HR) are found in individuals affected with breast cancer,Citation5,37 suggesting fork degradation-dependent, HR-independent mechanisms may contribute to tumorigenesis.
Secondly, it is likely that the presence and function of these protective proteins likely influences an individual's response to chemotherapeutic treatment, particularly in response to agents (such as Hydroxyurea or Gemcitabine) that induce high levels of exogenous replication stress. In this instance, loss of fork protection likely contributes to tumor progression by permitting wide-ranging genomic rearrangements. Moreover, in cells lacking these components, transient treatment with chemotherapeutics that induce replication stress would likely induce further mutagenesis and genome instability.
Lastly, given that defects in replication stress response genes gives rise to developmental abnormalities and microcephaly,Citation1 it is likely that loss of the fork protection function of the encoded gene products contributes to the development of some of these clinical defects.
Concluding remarks and future perspectives
The cellular response to replication stress is integral to maintaining genome stability, and defects in proteins which deal with replication stress are frequently associated with increased genome stability.Citation1 Cellular components that protect against RS therefore act as anti-tumor barriers that must be surpassed/subverted before a tumor can develop. In particular, in the absence of these protective factors, unprotected replication forks are an important source of genomic instability, and in part underlie the severe chromosomal instability associated with loss of FA/HR components such as FANCD2, BRCA2 and BOD1L.Citation6,24,28
Although many fork protection factors have been identified, it is currently unclear how these factors suppress resection: they may interact with their partner nucleases directly, or may act indirectly to regulate nucleolysis through influencing the activity of anti-recombinases or by stabilizing replication fork components. In addition, it is possible that more novel factors remain to be discovered. Elucidation of the mechanisms underlying fork protection represents an exciting future area of research, and may provide new therapeutic avenues for the treatment of diseases associated with an aberrant replication stress response.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. John J. Reynolds and Ms. Rachel Jones for critical reading of the manuscript. We apologise to colleagues whose work has not been cited because of space restrictions.
Funding
M.R.H and G.S.S are funded by a Cancer Research UK (CR-UK) Senior Fellowship (C17183/A13030).
References
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID:24366029; http://dx.doi.org/10.1038/ncb2897
- Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Ann Rev Biophys 2014; 43:257-78; http://dx.doi.org/10.1146/annurev-biophys-051013-022737
- Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Shimamoto A, Tahara H, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genetics 2015; 96:1001-7; http://dx.doi.org/10.1016/j.ajhg.2015.04.022
- Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, Sougnez C, Gabriel SB, Elemento O, Chandrasekharappa SC, et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell Reports 2015; 12:35-41; PMID:26119737; http://dx.doi.org/10.1016/j.celrep.2015.06.014
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/10.1016/j.cell.2011.03.041
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106-16; PMID:22789542; http://dx.doi.org/10.1016/j.ccr.2012.05.015
- Iannascoli C, Palermo V, Murfuni I, Franchitto A, Pichierri P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res 2015; 43:9788-803; PMID:26275776
- Su F, Mukherjee S, Yang Y, Mori E, Bhattacharya S, Kobayashi J, Yannone SM, Chen DJ, Asaithamby A. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Reports 2014; 9:1387-401; PMID:25456133; http://dx.doi.org/10.1016/j.celrep.2014.10.025
- Yang Y, Liu Z, Wang F, Temviriyanukul P, Ma X, Tu Y, Lv L, Lin YF, Huang M, Zhang T, et al. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res 2015; 43:8325-39; PMID:26187992; http://dx.doi.org/10.1093/nar/gkv737
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 2012; 72:2814-21; PMID:22447567; http://dx.doi.org/10.1158/0008-5472.CAN-11-3417
- Kim TM, Son MY, Dodds S, Hu L, Luo G, Hasty P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res 2015; 43:893-903; PMID:25520194; http://dx.doi.org/10.1093/nar/gku1334
- Kowalczykowski SC. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harbor Perspectives Biol 2015; 7:a016410; http://dx.doi.org/10.1101/cshperspect.a016410
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/10.1016/j.molcel.2010.01.021
- Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 2014; 206:493-507; PMID:25113031; http://dx.doi.org/10.1083/jcb.201404111
- Unno J, Itaya A, Taoka M, Sato K, Tomida J, Sakai W, Sugasawa K, Ishiai M, Ikura T, Isobe T, et al. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Reports 2014; 7:1039-47; PMID:24794430; http://dx.doi.org/10.1016/j.celrep.2014.04.005
- Ohashi A, Zdzienicka MZ, Chen J, Couch FJ. Fanconi anemia complementation group D2 (FANCD2) functions independently of BRCA2- and RAD51-associated homologous recombination in response to DNA damage. J Biol Chem 2005; 280:14877-83; PMID:15671039; http://dx.doi.org/10.1074/jbc.M414669200
- Godthelp BC, Wiegant WW, Waisfisz Q, Medhurst AL, Arwert F, Joenje H, Zdzienicka MZ. Inducibility of nuclear Rad51 foci after DNA damage distinguishes all Fanconi anemia complementation groups from D1/BRCA2. Mutation Res 2006; 594:39-48; PMID:16154163; http://dx.doi.org/10.1016/j.mrfmmm.2005.07.008
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007; 129:289-301; PMID:17412408; http://dx.doi.org/10.1016/j.cell.2007.03.009
- Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 2007; 21:3073-84; PMID:18003859; http://dx.doi.org/10.1101/gad.1609107
- Saponaro M, Kantidakis T, Mitter R, Kelly GP, Heron M, Williams H, Soding J, Stewart A, Svejstrup JQ. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 2014; 157:1037-49; PMID:24836610; http://dx.doi.org/10.1016/j.cell.2014.03.048
- Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genetics 2005; 14:693-701; http://dx.doi.org/10.1093/hmg/ddi065
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009; 11:753-60; PMID:19465922; http://dx.doi.org/10.1038/ncb1882
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing nonhomologous end joining suppresses DNA repairdefects of Fanconi anemia. Mol Cell 2010; 39:25-35; PMID:20598602; http://dx.doi.org/10.1016/j.molcel.2010.06.026
- Karanja KK, Lee EH, Hendrickson EA, Campbell JL. Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 2014; 13:1540-50; PMID:24626199; http://dx.doi.org/10.4161/cc.28476
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 2011; 25:350-62; PMID:21325134; http://dx.doi.org/10.1101/gad.2003811
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 2015; 208:545-62; PMID:25733713; http://dx.doi.org/10.1083/jcb.201406100
- Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 2005; 17:153-9; PMID:15629726; http://dx.doi.org/10.1016/j.molcel.2004.11.032
- Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, et al. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol Cell 2015; 59:462-77; PMID:26166705; http://dx.doi.org/10.1016/j.molcel.2015.06.007
- Costanzo V. Brca2, Rad51 and Mre11: performing balancing acts on replication forks. DNA Repair 2011; 10:1060-5; PMID:21900052; http://dx.doi.org/10.1016/j.dnarep.2011.07.009
- Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res 1999; 59:3547-51; PMID:10446958
- Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 2009; 7:1110-8; PMID:19584259; http://dx.doi.org/10.1158/1541-7786.MCR-09-0123
- Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell 2013; 50:589-600; PMID:23706822; http://dx.doi.org/10.1016/j.molcel.2013.04.032
- Xue X, Raynard S, Busygina V, Singh AK, Sung P. Role of replication protein A in double holliday junction dissolution mediated by the BLM-Topo IIIalpha-RMI1-RMI2 protein complex. J Biol Chem 2013; 288:14221-7; PMID:23543748; http://dx.doi.org/10.1074/jbc.M113.465609
- Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, Janscak P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem 2014; 289:27314-26; PMID:25122754; http://dx.doi.org/10.1074/jbc.M114.578823
- Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Structural Mol Biol 2007; 14:468-74; http://dx.doi.org/10.1038/nsmb1245
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006; 34:D535-9; PMID:16381927; http://dx.doi.org/10.1093/nar/gkj109
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434:598-604; PMID:15800615; http://dx.doi.org/10.1038/nature03404