
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Chromatin structures are transmitted to daughter cells through a complex system of nucleosome disassembly and re-assembly at the advancing replication forks. However, the role of replication pausing in the transmission and perturbation of chromatin structures has not been addressed. RRM3 encodes a DNA helicase, which facilitates replication at sites covered with non-histone protein complexes (tRNA genes, active gene promoters, telomeres) in Saccharomyces cerevisiae. In this report we show that the deletion of RRM3 reduces the frequency of epigenetic conversions in the subtelomeric regions of the chromosomes. This phenotype is strongly dependent on 2 histone chaperones, CAF-I and ASF1, which are involved in the reassembly of nucleosomes behind replication forks, but not on the histone chaperone HIR1. We also show that the deletion of RRM3 increases the spontaneous mutation rates in conjunction with CAF-I and ASF1, but not HIR1. Finally, we demonstrate that Rrm3p and CAF-I compete for the binding to the DNA replication clamp PCNA (Proliferating Cell Nuclear Antigen). We propose that the stalling of DNA replication predisposes to epigenetic conversions and that RRM3 and CAF-I play key roles in this process.
Introduction
The active or silent states of eukaryotic genes are determined by heritable chromatin structures, which are faithfully transmitted through multiple rounds of DNA replication.Citation1 Such epigenetic mechanisms warrant the continuity of tissue-specific gene expression programs. On the other hand, switches between the active and silent states (herein referred to as epigenetic conversions) confer the ability of the cells to differentiate and adapt.Citation2 The mechanisms of such epigenetic conversions remain largely unknown. Our limited understanding of these events is derived mostly from the analyses of Position-Effect Variegation (PEV1) in D. melanogaster Citation3 and Telomere Position Effect (TPE) in S. cerevisiae.Citation4 Both phenomena are characterized by random active→silent and silent→active switches of a variety of reporter genes.Citation2
Chromatin is completely reassembled during DNA replication. Several histone chaperones play a central role in this process to re-establish the pre-existing chromatin state on the newly synthesized DNA.Citation1 Two chaperones, ASF1 and FACT, move ahead of the fork via contacts with the replicative helicase CMG (CDC45-MCM-GINS).Citation5 These chaperons disassemble the existing nucleosomes and ferry them behind the fork. Another chaperone, Chromatin Assembly Factor I (CAF-I), travels behind the fork via contacts with the replication sliding clamp Pol30p (PCNA; Proliferating Cell Nuclear Antigen)Citation6,7 and reassembles nucleosomes using the “old” and “new” H3/H4 histones.Citation1 It has been shown that mutations in many replication factors and chaperones, including POL30, ASF1 and the MCM genes, lead to a significant reduction of gene silencing at the telomeres.Citation8-11 However, the deletion of the large subunit of CAF-I (CAC1) reduced the frequency of epigenetic switches at the telomeres.Citation11
Replication forks that are progressing through the subtelomeric regions encounter tightly-bound proteins and unusual DNA structures such as G-quadruplexes, t-loops or D-loops.Citation12,13 The presence of these structures contributes to the frequent pausing of replication forks,Citation14-17 but it is not clear how these events contribute to the transmission of chromatin. In mammalian stem cells, the transitions between active and silent states has been correlated to the promoter-proximal pausing of RNA polymerase II,Citation18,19 but it is not known if transcription pausing causes chromatin perturbation in S. cerevisiae. In this study we addressed these questions by analyzing the rates of epigenetic conversions in mutants that are known to increase the pausing of DNA replication or RNA pol II transcription complexes. We found that the RRM3 helicase, which associates with Pol30p (PCNA) and relieves stalled replication forks at protein-binding sites, also participates in the control of epigenetic conversions. We also show that CAF-I and Rrm3p compete for the association with Pol30p (PCNA).
Results
RRM3 regulates the frequency of epigenetic conversions
We used an established assay for epigenetic conversions at the telomeres of S. cerevisiaeCitation11 to address how the frequency of such conversions is influenced by stalled transcribing or replicating complexes. Briefly, we inserted URA3 in the VII-L telomere, selected the transformed cells on SC-ura plates and then grew them in non-selective medium to allow for silencing. We then selected for cells with silent (growth on 5-FOA-containing plates) or active (growth on SC- plates lacking uracil) URA3 and then again in non-selective medium to allow for unrestricted switches between the active and silent state of URA3. After 20 generations (as determined by the doubling of culture density at OD600) we calculate the proportions of cells with active/silent URA3 and use them to assess the rates of epigenetic conversions.Citation11 In “wild type” cells the rates of conversion are about 6–8% per generation. By the 20th generation after the release from selection these cells produce 60%/40% distribution of cells with active/silent URA3.Citation11 This proportion is maintained at an equilibrium as the cells continue to switch between the 2 states. A significant deviation from this distribution of the cells at the 20th generation is an indication for a loss in gene silencing (a reduction in the proportion of FOAR cells regardless of selection) or a loss of the conversion rates (the proportion of FOAR and ura+ cells depends on the selection).Citation11 We performed this assay in spt4Δ and dst1Δ strains, which lack genes that support the efficient elongation by RNA polymerase II Citation20,21 and in rrm3Δ, pif1Δ and tof1Δ strains, which lack genes with distinct roles at the DNA replication forks.Citation22-25 As a control we included the cac1Δ strain, which has previously displayed a substantial decline in the frequency of epigenetic conversions.Citation11 The sas3Δ strain served as a control with a moderately reduced levels of telomeric gene silencing but no known effects on epigenetic conversions.Citation26
Our experiments showed that, in comparison to the isogenic BY4742 strain, the cells with the deletions of SAS3, SPT4, DST1, PIF1 and TOF1 display a modest 1–2 fold reduction in the proportion of cells with silenced URA3 (% FOAR cells) regardless of the initial selection for active or silenced URA3 (). We did not further pursue these minor loss-of-silencing phenotypes. In the rrm3Δ strain the proportion of cells with silent URA3 was 92% when cells were initially selected on SC/FOA plates and 14.1% when initially selected on SC-ura plates (raw data and statistical evaluation is shown in Supplemental Table 2). Conversely, the proportion of cells with active URA3 was 6% when the initial selection was on SC/FOA plates, but 88.9% when the initial selection was on SC-ura plates (Supplemental Table 2). Albeit less severe than in cac1Δ, this phenotype clearly pointed to a reduction in the frequency of epigenetic conversions ().
Figure 1. Frequency of epigenetic conversions at the VIIL telomere. (A) The URA3-tel construct was integrated in the VIIL telomere of the strains shown along the horizontal axis. Cells were selected in parallel on SC/5-FOA (upper graph) and SC-ura (lower graph) and single colonies were transferred to liquid YPD medium for 20 generations. Cultures were then serially (1:10) diluted and spotted on YPD, SC-ura and SC/5-FOA plates. Colonies were counted and the percentage of FOAR (black columns) and URA+ (open columns) was calculated. The data is from Supplemental Table 2. (B) Cells were selected on SC/ura− (gray fill), and SC/5-FOA (black fill) and released in non-selective YPD medium. Aliquots were taken at the indicated number of generations and the percent of FOAR cells was measured and plotted. The data is from Supplemental Table 2.
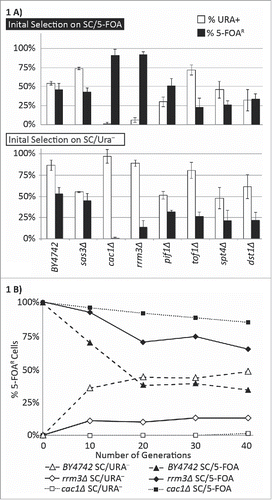
We revisited this phenotype by testing the proportions of active and silent URA3 at multiple time points after the release from selection (). As reported earlier, BY4742 cells reached a steady dynamic proportion of FOAR/Ura+ cells in about 20 generations after the release.Citation11 cac1Δ cells were far from FOAR/Ura+ equilibrium at the 40th generation. rrm3Δ cells were recovering faster than cac1Δ cells, but slower than BY4742 cells. These results corroborated the notion that the deletion of RRM3 causes a substantial reduction in the frequency of epigenetic conversions, but not as severe as in cac1Δ cells.
The deletion of CAC1 and ASF1 strongly suppress the loss-of-variegation in rrm3Δ cells
Both Rrm3p and Cac1p function at the advancing replication forks. The similar loss of variegation observed in rrm3Δ and cac1Δ () suggested that the observed epigenetic effects could be caused by the perturbation of histone turn-over at the fork and that Rrm3p and CAF-I operate through related mechanisms. We performed several experiments to address these issues. Firstly, we compared the variegation phenotypes of rrm3Δ and cac1Δ to that of strains with deletions of histone chaperones with well-defined roles in nucleosome turn-over. ASF1 encodes a histone chaperone that works along CAF-I in the replication-coupled reassembly of nucleosomes.Citation27 HIR1 is a histone chaperone believed not to participate in the replication-coupled nucleosome reassembly.Citation27 In agreement with earlier studies,Citation28 the deletion of HIR1 did not affect gene silencing. No significant alteration in the frequency of epigenetic conversions was observed in this strain (). The asf1Δ strain displayed characteristics that were consistent with a reduction of epigenetic conversions, but not as pronounced as in cac1Δ and rrm3Δ. In the asf1Δ the proportion of cells with silent URA3 was 82.2% after selection on SC/FOA plates and 40.3% when selected on SC-ura plates. The proportion of cells with active URA3 was 35% when selected on SC/FOA plates and 84.1% when the selection was on SC-ura plates (Supplemental Table 2).
Figure 2. The deletion of CAC1 or ASF1 suppress the variegation phenotype in rrm3Δ. The URA3-tel construct was integrated in the VII-L telomere of the strains shown along the horizontal axis. Cells were selected in parallel on SC/5-FOA (upper graph) and SC/ura− (lower graph) and single colonies were transferred to liquid YPD medium for 20 generations. Cultures were serially (1:10) diluted and spotted on YPD, SC/ura− and SC/5-FOA plates. Colonies were counted and the percentage of FOAR (black columns) and URA+ (open columns) was calculated. The data is from Table 2.
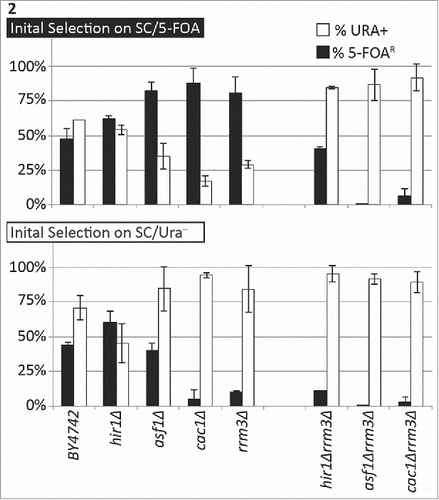
Next, we generated strains with double cac1Δrrm3Δ, asf1Δrrm3Δ and hir1Δrrm3Δ deletions and compared their variegation phenotypes to that of the single mutants (). The hir1Δrrm3Δ strain showed a higher proportion of cells with active URA3 regardless of the initial selection thus pointing to a loss of gene silencing in this double mutant. At the same time, the proportion of FOAR cells was higher in the cells that have been initially selected for the repressed state of URA3. In the cac1Δrrm3Δ, asf1Δrrm3Δ mutants we observed very low proportions of FOA-resistant cells regardless of the initial selection (). These cells clearly show a loss-of-silencing phenotype, which has completely masked the variegation loss. Together, these results indicated that the deletions of ASF1 or CAC1 dramatically suppress the loss-of-variegation phenotype in rrm3Δ and produce a significant loss of silencing net effect. In contrast, the deletion of HIR1 caused only a partial suppression of this phenotype. We concluded that the effect of RRM3 on epigenetic conversions is strongly linked to the replication-coupled assembly of nucleosomes by ASF1 and CAF-I and is only moderately affected by replication-independent histone exchange.
Rates of spontaneous mutations in the analyzed mutants
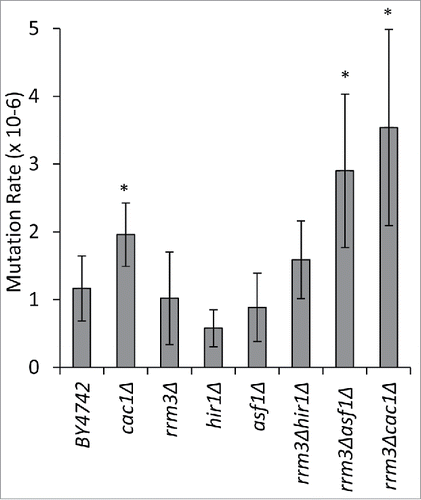
It has been previously shown that cac1Δ, but not rrm3Δ cells, display a moderate increase in spontaneous mutation rates.Citation11,14,29,30 We considered the possibility that the deletion of RRM3 in combination with genes that regulate the histone turn-over during replication can exacerbate these rates. We used a routine assay for the detection of forward mutations in the CAN1 gene.Citation31 Raw data and statistical evaluation is shown in Supplemental Table 3. This assay is completely independent from the positional variegation at the telomeres. In addition, we are not aware of any evidence suggesting that replication forks stall in the vicinity of CAN1. We observed no significant increase in the mutation rates in rrm3Δ, asf1Δ and hir1Δ cells relative to the isogenic BY4742 cells (). As reported previously,Citation11 in cac1Δ cells we detected about twice more spontaneous mutations relative to BY4742 (). In the double mutant cac1Δrrm3Δ the mutations rates were significantly higher than in each single mutant (). Similar rates were observed in asf1Δrrm3Δ, but not in hir1Δrrm3Δ (). We concluded that the deletion of RRM3 increases the mutation rates in strains that lack histone chaperones engaged in replication-coupled nucleosome assembly, but not with HIR1.
Figure 3. The deletion of RRM3 exacerbates mutation rates in cac1Δ and asf1Δ cells. Five independent colonies from the strains shown along the horizontal axis were inoculated in liquid YPD cultures and grown for 20 generations. 2.5–4.5 ×107 cells were plated on non-selective or SC/arg−can+ plates, respectively, and grown at 30°C for 4–5 d. The mutation rates in CAN1 were calculated as number of colonies on SC/arg−can+ divided by the number of plated cells. The asterisk indicates statistically significant values at p≤0.05. The data is from Supplemental Table 3.
Rrm3p and Cac1p compete for binding to PCNA
Both Cac1p and Rrm3p contain a PIP (PCNA Interacting Peptide) consensus sequence and directly interact with the replication sliding clamp PCNA.Citation32,33 It is conceivable that these 2 proteins exert their effect on the frequency of epigenetic conversions through competition or cooperation in their interaction with PCNA. We addressed this question by a routine 2-hybrid interaction assay.Citation34 The ORFs of RRM3 or CAC1, respectively, were fused to the LexA-DNA binding domain and used as baits. PCNA was fused to the activation domain of Gal4p and co-expressed with LexA-Cac1p or LexA-Rrm3p. The interaction between prey and bait proteins activates the expression of a LexA-driven LacZ reporter. It is important that in this set-up the bait-prey interaction is challenged by the endogenous Cac1p and Rrm3p. This interference provides an opportunity to test if these 2 proteins compete or cooperate for their interaction with PCNA. The two-hybrid assays were performed in cac1Δ, rrm3Δ and the isogenic BY4742 strains ().
Figure 4. Cac1p and Rrm3p compete for binding to PCNA. The strains shown along the X axis were transformed with a LexAOP-LacZ reporter plasmid and bait and prey plasmids as indicated. Cell extracts were prepared and β-galactosidase activity (U/mg of protein) was measured. Average values from 2 independent experiments (3 biological replicates each) are plotted on the Y axis. The asterisk indicates statistically significantly difference from wildtype BY4742 cells at p≤0.05. Data is from Supplemental Table 4. (A) Cac1p-PCNA interaction. Cells transformed with pEG202-LexADBD-CAC1 and pBL240. (B) Rrm3p-PCNA interaction. Cells transformed with pEG202-LexADBD-RRM3 and pBL240. (C) Differential interaction of Rrm3p and Cac1p with PCNA. BY4724 cells transformed with pEG202-LexADBD-CAC1 or pEG202-LexADBD-RRM3 and pBL240 or pBL240-Y133A, respectively as indicated.
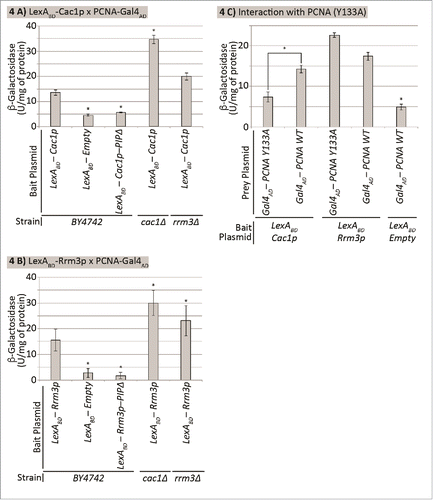
PCNA-Cac1p and PCNA-Rrm3p interactions were readily detectable in the BY4742 strain. Mutations in their PIPs reduced the β-galactosidase activity to the baseline of an empty bait plasmid (). Hence, the observed β-galactosidase activity is largely dependent on the PIP-mediated interactions. In cac1Δ and rrm3Δ cells we noticed an apparent increase in the β-galactosidase reporter signals relative to BY4742 (). These results showed that Rrm3p and Cac1p compete for their interaction with PCNA. The consistent differences in the increase of signal in cac1Δ and rrm3Δ cells for both the LexA-Cac1p and LexA-Rrm3p baits most likely reflects the abundance of these proteins in vivo.
The experiments in reiterated that the interactions of Cac1p and Rrm3p with PCNA depend on their PIPs, but did not show the extent of overlap between these interactions. We addressed this issue by exploring a PCNA mutant (Y133A), which has a substitution in its IDCL (Inter-Domain Connectivity Loop).Citation35 It is believed that the IDCL and the carboxy-terminus of PCNA form the hydrophobic pocket to accommodate PIP.Citation35,36 Two-hybrid assays were performed with LexA-Cac1p or LexA-Rrm3 baits and PCNA or PCNA(Y133A) preys, respectively (). The results showed that the Y133A mutation significantly reduced the interaction between Cac1p and PCNA, while having the opposite effect on the Rrm3p-PCNA interaction (). This data demonstrated that that PIP is necessary but not sufficient for the interaction of Cac1p with PCNA and that Cac1p and Rrm3p differ in their contacts with PCNA.
Discussion
How can RRM3 affect epigenetic conversions?
Epigenetic conversions are key events in metazoan development.Citation1,2,19,37 They play a role in many syndromes and genetic disorders Citation38 and in the adaptation of single cell eukaryotes to changes in the environment.Citation2,37 However, the mechanisms of such conversions are poorly understood. Previously, we have shown that the histone chaperone CAF-I regulates the frequency of epigenetic conversions at the telomeres of S. cerevisiae.Citation11 In the current manuscript we report that RRM3 has a similar function. RRM3 is known to facilitate the passage of replication forks through pause sites at tightly bound proteinsCitation15-17 whereas CAF-I assembles nucleosomes behind the replication fork.Citation1 We have shown that the epigenetic effects of RRM3 are strongly linked to the function of fork-associated histone chaperones, CAF-I and ASF1, and only moderately influenced by the replication-independent chaperone HIR1. The same conclusions applies to spontaneous mutations rates. The deletion of RRM3 in combinations with deletions of CAC1 and ASF1, but not HIR1, elevates these rates.
It is well established that the replication forks stall at multiple positions in the genome. These include tightly-bound DNA proteins, tRNA genes active gene promoters.Citation17,39 The Rrm3p helicase is believed to relieve the stalling and to facilitate the resumption of DNA replication. The RRM3-depndent epigenetic effects observed here suggest that the stalling of the fork predisposes to epigenetic changes. So, how can the pausing of replication forks affect epigenetic conversions? In our experimental set-up URA3 is transcribed in the direction of the advancing replication fork toward the telomere. Hence, we can not explain our observations by the collisions between replication and transcription complexes. In addition, if the paused fork prevents the spreading of histone deacetylation from the telomere, we would expect a reduction in gene silencing and not reduction in conversion rates. Even more, we have shown that the effect of RRM3 is not recaptured by the deletion of genes that are functionally related or homologous to RRM3 (PIF1, TOF1) or genes that regulate RNA pol II elongation (). For this reason we favor the idea that some specific events at the paused fork, but not the stalling of the fork per se, produce the observed epigenetic conversions. It is tempting to speculate that at stalled forks CAF-I does not receive feedback from “old” histones and assembles nucleosomes from only “new” histones. It is this activity that predisposes to epigenetic change. In the absence of CAF-I the assembly of nucleosomes from “new” histones is suppressed or delayed thus allowing for alternative modes of assembly and the preservation of the epigenetic state. In support to this idea, a recent study in Drosophila cells has demonstrated that promoters with the highest occupancy of RNA pol II, which in S. cerevisiae are detected as RRM3-dependent replication pause sites,Citation17,39 show the highest level of change in nucleosome occupancy upon replication.Citation40,41 Even more, the depletion of CAF-I significantly delayed the nucleosome assembly at these positions.Citation40,41
It is possible that other perturbations at stalled forks, and more specifically the lack of Rrm3p, could preclude the assembly of nucleosomes from “new” histones only. The actual mechanism of RRM3 action remains unknown. However, it is clear that the effect of RRM3 is not strictly dependent on CAF-I. For example, the reduction of epigenetic switches in rrm3Δ is reversed by the deletion of either CAC1 or ASF1. The same argument applies to the rate of mutations. While the deletion of RRM3 alone does not lead to elevated mutation rates, the additional deletions of CAC1 and ASF1 clearly enhance these rates. These observations suggest that the changes in the frequency of epigenetic conversions in cac1Δ and rrm3Δ cells are not caused by an identical mechanism.
Interactions between PCNA, Rrm3p and CAF-I
The coordination of DNA replication and the re-assembly of chromatin is achieved in part by PCNA, which orchestrates numerous interactions with DNA replication and chromatin remodeling factors.Citation33 Many of these proteins contain a PIP consensus sequence and are likely to compete for the same binding site on PCNA. However, PCNA forms a homo-trimer clamp, which can interact with 3 PIP-containing proteins at a time and thus support multiple associations with PIP-containing proteins. We tested the possibility that the similar epigenetic effects of RRM3 and CAF-I are linked by their simultaneous interaction with PCNA. Our experiments showed a clear evidence for competition. In addition, we have seen no interaction between Rrm3p and CAF-I in co-immunoprecipitation experiments (not shown) and no physical interaction between these proteins has been reported in genome-wide proteome analyses in the Saccharomyces Genome Database (http://www.yeastgenome.org). All these observations fuel the notion that the epigenetic effects of RRM3 and CAF-I could not be attributed to their PIPs and directly linked to PCNA or to an interaction between them. This conclusion is in tune with earlier publications. We know that PIPs have been identified on both Cac1p and Cac2p subunits of yeast CAF-I Citation7 and that mutations in the Cac1-PIP that severely impair the binding to PCNA display no effect on the telomeric silencing phenotypes.Citation8 Instead, these defects become visible only when another histone chaperone (HIR1 or ASF1) is deleted.Citation8 Hence, the loss of the Cac1p-PCNA interaction does not phenocopy the loss of CAC1. Our earlier studies concur with these findings and indicate that the epigenetic phenotypes in cac1Δ and rrm3Δ should not be attributed to the PCNA-PIP interactions alone.Citation42 Together, our observations suggest a complex PCNA-based control at replication forks. We need to consider that more than one PCNA ring can operate at the lagging strand and that PCNA can partner with different proteins at the lagging and the leading strands.Citation33,43,44 For this reason we do not entirely exclude the possibility of communication between Rrm3p and CAF-I via PCNA. These ideas need to be vigorously tested by in vivo assays that decipher the composition and the state of paused replication forks.
In summary, our results strongly suggest that the pausing of replication forks at protein-binding sites predisposes to alterations of chromatin structure and epigenetic conversions and that Rrm3p and CAF-I are specifically involved in these processes.
In summary, our results strongly suggest that the pausing of replication forks at protein-binding sites predisposes to alterations of chromatin structure and epigenetic conversions and that Rrm3p and CAF-I are specifically involved in these processes.
Materials and methods
Strains and growth conditions
The strains used in this study are listed in Supplemental Table 1. Strains with replacement deletion of 2 genes have been produced by routine mating and sporulation. The cac1Δ RFB::URA3 and rrm3Δ RFB::URA3 strains have been produced by mating the corresponding cac1Δ and rrm3Δ strains to YBH17.Citation23 These strains contain 2 Replication Fork Boundary (RFB) elements from the rRNA gene cluster cloned between the early-firing origins ARS305 and ARS306 (see andCitation23).
All strains were routinely grown in YPD or Synthetic Complete (SC) drop-out media at 30°C unless otherwise indicated.
Expression plasmids
pRS315-RRM3-MYC contains the RRM3 promoter and its ORF tagged with a 13 MYC-tags. The construct was produced by PCR from the genomic DNA of JZT200.Citation17 pRS313-CAC1-FLAG was described inCitation11. pSH18-34 (URA3, 2 µm, LexA-lacZ) was obtained from OriGene®. pEG202-LexADBD-CAC1 and pEG202-GAL4DBD-RRM3 were produced by cloning the open reading frames of CAC1 and RRM3 in frame with the LexA DNA Binding Domain (DBD) in pEG202 (OriGene®). The ORFs were produced by PCR from the genomic DNA of BY4742. pBL240 and pBL240-Y133A (LEU2, 2 µm, AmpR Gal4AD-POL30) express fusion PCNA-Gal4AD and PCNA(Y133A)-Gal4AD proteins, respectively.Citation35
Frequency of epigenetic conversions at the VII-L telomere
The assay is described in detail inCitation11. Briefly, individual colonies containing a URA3 reporter construct in the VII-L telomereCitation45 were grown in non-selective liquid medium (YPD) to allow for the establishment of both the silent and active state of the reporter, then re-selected in parallel on plates containing 5-FOA (SC/5-FOA) or lacking uracil (SC/ura−), respectively, and then returned to non-selective medium (YPD) to allow for unrestricted epigenetic switching. After 20 generations (measured as the doublings of OD600 the liquid cultures) the cells were serially diluted and plated in parallel on non-selective medium (YPD), SC/ura− and SC/5-FOA. The number of colonies were counted and the proportion of cells expressing URA3 (URA+) was calculated by dividing the number of colonies growing on SC/ura− by the number of colonies growing on the non-selective YPD plates. The proportion of cells with a silenced URA3 (FOAR) was calculated by dividing the number of colonies growing on SC/5-FOA by the number of colonies growing on the non-selective YPD plates. Average numbers from several independent colonies were then calculated and plotted. The calculations of epigenetic conversions per generation were conducted as inCitation11.
Yeast Two-hybrid Assay
Cells were transformed with the reporter plasmid pSH18-34 (URA3, 2 µm, LexA-lacZ), one of the bait plasmids (pEG202, pEG202-LexADBD-CAC1, pEG202-LexADBD-RRM3) and one of the prey plasmids pBL240 or pBL240-Y133A.Citation35 The cells were grown in SC/Leu−/Ura−/His−/2% Glucose at 30°C to OD600 = 0.8–1.0, then transferred to SC/Leu−/Ura−/His−/2% Galactose/1% Raffinose and incubated at 30°C for 4 hrs. The cells were collected, washed in ice-cold H2O, suspended in 4 mL Buffer P (50 mM sodium phosphate, pH 7.7, 300 mM sodium acetate, 10% v/v glycerol, 1 mM 2-Mercaptoethanol, 500 nM DTT, 1 mM PMSF and 1% v/v protease inhibitor cocktail (Sigma®) and split into 3 aliquots. The three replicas of protein extraction were processed as follows. Cells were pelleted, resuspended in 200 µL of Buffer P and 200 µL of 0.55 mm acid-washed glass beads were added. Cell lysis was by pulse-vortexing for 15 min at 4°C. The lysate was collected, kept on ice for 15 min, cleared by centrifugation and 50 µL were removed and stored at −20°C to measure total protein concentration. 200 uL of Buffer P + 4 mg/mL ortho-Nitrophenyl-β-galactoside (ONPG) was added to the remaining cell lysate and incubated at 30°C till “medium-yellow color” (OD420 between ∼0.3 and 0.7) was developed in the control tubes, then 500 µL of 1M Na2CO3 was added and OD420 and OD550 measurements were taken in triplicate, averaged and recorded. The units of β-galactosidase were calculated by the formula
Total protein levels were measured using the Bio-Rad® Protein Assay, and used to normalize the β-galactosidase assay, weighting the units of β-gal/mg of total protein.
Measurements of spontaneous mutation rates
The mutation rate for each strain (n = 5) was determined by the fluctuation analysis as in.Citation31 Briefly, 5 individual colonies were inoculated in liquid YPD medium and grown for 20–24 generations. The cells were washed once in sterile water and 2.5–4.5 × 107 cells were spread on both SC/arg− dropout plates containing canavanine (6 mg/mL) and non-selective YPD plates. The plates were grown at 30°C for 4–5 d and the colonies in SC/arg−can+ were counted and divided by the number of plated cells.
Abbreviations
| ASF1 | = | Anti-Silencing Factor 1 |
| CAF-I | = | Chromatin Assembly Factor I |
| ChIP | = | Chromatin Immuno-Precipitation |
| FACT | = | Facilitator of Activated Transcription on Chromatin templates |
| PCNA | = | Proliferating Cell Nuclear Antigen |
| PEV | = | Position-Effect Variegation |
| PIP | = | PCNA Interacting Peptide |
| RFB | = | Replication Fork Boundary |
| TPE | = | Telomere Position Effect |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
SUPPLEMENTAL_INFORMATION.docx
Download MS Word (29.6 KB)Acknowledgments
We thank Dr. Kaufman, Dr. Basrai, Dr. Labib, Dr. Zakian, Dr. Kolodner and Dr. Bielinsky for the generous donation of strains and plasmids.
Funding
This study is supported by a Discovery Grant to KY by the National Science and Engineering Research Council (NSERC) of Canada (grant # RGPIN-2015-06727). BW is supported by an Ontario Government Studentship (OGS).
References
- Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 2012; 13:153-67; PMID:22358331; http://dx.doi.org/10.1038/nrm3288
- Yankulov K. Dynamics and stability: epigenetic conversions in position effect variegation. Biochem Cell Biol 2013; 91:6-13; PMID:23442136; http://dx.doi.org/10.1139/bcb-2012-0048
- Elgin SC, Reuter G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harbor Perspectives Biol 2013; 5:a017780; http://dx.doi.org/10.1101/cshperspect.a017780
- Moazed D. Mechanisms for the inheritance of chromatin states. Cell 2012; 146:510-8; http://dx.doi.org/10.1016/j.cell.2011.07.013
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G. Regulation of replication fork progression through histone supply and demand. Science 2007; 318:1928-31; PMID:18096807; http://dx.doi.org/10.1126/science.1148992
- Zhang Z, Shibahara K, Stillman B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 2000; 408:221-5; PMID:11089978; http://dx.doi.org/10.1038/35048530
- Rolef Ben-Shahar T, Castillo AG, Osborne MJ, Borden KL, Kornblatt J, Verreault A. Two fundamentally distinct PCNA interaction peptides contribute to chromatin assembly factor 1 function. Mol Cell Biol 2009; 29:6353-65; PMID:19822659; http://dx.doi.org/10.1128/MCB.01051-09
- Krawitz DC, Kama T, Kaufman PD. Chromatin assembly factor I mutants defective for PCNA binding require Asf1/Hir proteins for silencing. Mol Cell Biol 2002; 22:614-25; PMID:11756556; http://dx.doi.org/10.1128/MCB.22.2.614-625.2002
- Sharp JA, Fouts ET, Krawitz DC, Kaufman PD. Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing. Curr Biol 2001; 11:463-73; PMID:11412995; http://dx.doi.org/10.1016/S0960-9822(01)00140-3
- Rehman MA, Fourel G, Mathews A, Ramdin D, Espinosa M, Gilson E, Yankulov K. Differential requirement of DNA replication factors for subtelomeric ARS consensus sequence protosilencers in Saccharomyces cerevisiae. Genetics 2006; 174:1801-10; PMID:16980387; http://dx.doi.org/10.1534/genetics.106.063446
- Jeffery DC, Wyse BA, Rehman MA, Brown GW, You Z, Oshidari R, Masai H, Yankulov KY. Analysis of epigenetic stability and conversions in Saccharomyces cerevisiae reveals a novel role of CAF-I in position-effect variegation. Nucleic Acids Res 2013; 41:8475-88; PMID:23863839; http://dx.doi.org/10.1093/nar/gkt623
- Anand RP, Shah KA, Niu H, Sung P, Mirkin SM, Freudenreich CH. Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res 2011; 40:1091-105; PMID:21984413; http://dx.doi.org/10.1093/nar/gkr836
- Gilson E, Geli V. How telomeres are replicated. Nat Rev Mol Cell Biol 2007; 8:825-38; PMID:17885666; http://dx.doi.org/10.1038/nrm2259
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 2003; 12:1525-36; PMID:14690605; http://dx.doi.org/10.1016/S1097-2765(03)00456-8
- Ivessa AS, Zhou JQ, Schulz VP, Monson EK, Zakian VA. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev 2002; 16:1383-96; PMID:12050116; http://dx.doi.org/10.1101/gad.982902
- Makovets S, Herskowitz I, Blackburn EH. Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol Cell Biol 2004; 24:4019-31; PMID:15082794; http://dx.doi.org/10.1128/MCB.24.9.4019-4031.2004
- Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell 2009; 34:722-34; PMID:19560424; http://dx.doi.org/10.1016/j.molcel.2009.05.022
- Golob JL, Kumar RM, Guenther MG, Pabon LM, Pratt GA, Loring JF, Laurent LC, Young RA, Murry CE. Evidence that gene activation and silencing during stem cell differentiation requires a transcriptionally paused intermediate state. PLoS One 2011; 6:e22416; PMID:21886766; http://dx.doi.org/10.1371/journal.pone.0022416
- Young RA. Control of the embryonic stem cell state. Cell 2011; 144:940-54; PMID:21414485; http://dx.doi.org/10.1016/j.cell.2011.01.032
- Hartzog GA, Fu J. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim Biophys Acta 2013; 1829:105-15; PMID:22982195; http://dx.doi.org/10.1016/j.bbagrm.2012.08.007
- Kipling D, Kearsey SE. Function of the S. cerevisiae DST1/PPR2 gene in transcription elongation. Cell 1993; 72:12; PMID:8422673; http://dx.doi.org/10.1016/0092-8674(93)90044-Q
- Bairwa NK, Zzaman S, Mohanty BK, Bastia D. Replication fork arrest and rDNA silencing are two independent and separable functions of the replication terminator protein Fob1 of Saccharomyces cerevisiae. J Biol Chem 2010; 285:12612-9; PMID:20179323; http://dx.doi.org/10.1074/jbc.M109.082388
- Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev 2005; 19:1905-19; PMID:16103218; http://dx.doi.org/10.1101/gad.337205
- Ivessa AS, Zhou JQ, Zakian VA. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 2000; 100:479-89; PMID:10693764; http://dx.doi.org/10.1016/S0092-8674(00)80683-2
- Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013; 497:458-62; PMID:23657261; http://dx.doi.org/10.1038/nature12149
- Power P, Jeffery D, Rehman MA, Chatterji A, Yankulov K. Sub-telomeric core X and Y' elements in S. cerevisiae suppress extreme variations in gene silencing. PLoS One 2011; 6:e17523; PMID:21437278; http://dx.doi.org/10.1371/journal.pone.0017523
- Rocha W, Verreault A. Clothing up DNA for all seasons: Histone chaperones and nucleosome assembly pathways. FEBS Lett 2008; 582:1938-49; PMID:18343227; http://dx.doi.org/10.1016/j.febslet.2008.03.006
- Kaufman PD, Cohen JL, Osley MA. Hir proteins are required for position-dependent gene silencing in Saccharomyces cerevisiae in the absence of chromatin assembly factor I. Mol Cell Biol 1998; 18:4793-806; PMID:9671489; http://dx.doi.org/10.1128/MCB.18.8.4793
- Myung K, Pennaneach V, Kats ES, Kolodner RD. Saccharomyces cerevisiae chromatin-assembly factors that act during DNA replication function in the maintenance of genome stability. Proc Natl Acad Sci U S A 2003; 100:6640-5; PMID:12750463; http://dx.doi.org/10.1073/pnas.1232239100
- Schmidt KH, Kolodner RD. Suppression of spontaneous genome rearrangements in yeast DNA helicase mutants. Proc Natl Acad Sci U S A 2006; 103:18196-201; PMID:17114288; http://dx.doi.org/10.1073/pnas.0608566103
- Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A 2003; 100:11529-34; PMID:12972632; http://dx.doi.org/10.1073/pnas.2035018100
- Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell 2007; 129:665-79; PMID:17512402; http://dx.doi.org/10.1016/j.cell.2007.05.003
- Mailand N, Gibbs-Seymour I, Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nat Rev Mol Cell Biol 2013; 14:269-82; PMID:23594953; http://dx.doi.org/10.1038/nrm3562
- Schmidt KH, Derry KL, Kolodner RD. Saccharomyces cerevisiae RRM3, a 5′ to 3′ DNA helicase, physically interacts with proliferating cell nuclear antigen. J Biol Chem 2002; 277:45331-7; PMID:12239216; http://dx.doi.org/10.1074/jbc.M207263200
- Das-Bradoo S, Ricke RM, Bielinsky AK. Interaction between PCNA and diubiquitinated Mcm10 is essential for cell growth in budding yeast. Mol Cell Biol 2006; 26:4806-17; PMID:16782870; http://dx.doi.org/10.1128/MCB.02062-05
- Gulbis JM, Kelman Z, Hurwitz J, O'Donnell M, Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996; 87:297-306; PMID:8861913; http://dx.doi.org/10.1016/S0092-8674(00)81347-1
- Wyse BA, Oshidari R, Jeffery DC, Yankulov KY. Parasite epigenetics and immune evasion: lessons from budding yeast. Epigenetics Chromatin 2013; 6:40; PMID:24252437; http://dx.doi.org/10.1186/1756-8935-6-40
- Almouzni G, Altucci L, Amati B, Ashley N, Baulcombe D, Beaujean N, Bock C, Bongcam-Rudloff E, Bousquet J, Braun S, et al. Relationship between genome and epigenome–challenges and requirements for future research. BMC Genomics 2014; 15:487; PMID:24942464; http://dx.doi.org/10.1186/1471-2164-15-487
- Rossi SE, Carotenuto W, Giannattasio M. Genome-wide localization of Rrm3 and Pif1 DNA helicases at stalled active and inactive DNA replication forks of Saccharomyces cerevisiae. Genom Data 2016; 7:162-5; PMID:26981397; http://dx.doi.org/10.1016/j.gdata.2015.11.024
- Ramachandran S, Henikoff S. Transcriptional regulators compete with nucleosomes Post-replication. Cell 2016; 165:580-92; PMID:27062929; http://dx.doi.org/10.1016/j.cell.2016.02.062
- Ramachandran S, Henikoff S. Nucleosome dynamics during chromatin remodeling in vivo. Nucleus 2016; 7:20-6; PMID:26933790; http://dx.doi.org/10.1080/19491034.2016.1149666
- Jeffery DC, Kakusho N, You Z, Gharib M, Wyse B, Drury E, Weinreich M, Thibault P, Verreault A, Masai H, et al. CDC28 phosphorylates Cac1p and regulates the association of chromatin assembly factor I with chromatin. Cell Cycle 2015; 14:74-85; PMID:25602519; http://dx.doi.org/10.4161/15384101.2014.973745
- Ulrich HD, Takahashi T. Readers of PCNA modifications. Chromosoma 2013; 122:259-74; PMID:23580141; http://dx.doi.org/10.1007/s00412-013-0410-4
- Pavlov YI, Mian IM, Kunkel TA. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr Biol 2003; 13:744-8; PMID:12725731; http://dx.doi.org/10.1016/S0960-9822(03)00284-7
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 1990; 63:751-62; PMID:2225075; http://dx.doi.org/10.1016/0092-8674(90)90141-Z
- Crotti LB, Basrai MA. Functional roles for evolutionarily conserved Spt4p at centromeres and heterochromatin in Saccharomyces cerevisiae. EMBO J 2004; 23:1804-14; PMID:15057281; http://dx.doi.org/10.1038/sj.emboj.7600161
- Heise F, Chung HR, Weber JM, Xu Z, Klein-Hitpass L, Steinmetz LM, Vingron M, Ehrenhofer-Murray AE. Genome-wide H4 K16 acetylation by SAS-I is deposited independently of transcription and histone exchange. Nucleic Acids Res 2012; 40:65-74; PMID:21908408; http://dx.doi.org/10.1093/nar/gkr649