
ABSTRACT
Eukaryotic DNA is packaged into regularly spaced nucleosomes, resembling beads on a string. Each bead contains ∼147 bp wrapped around a core histone octamer. Linker histone (H1) binds to the linker DNA to drive chromatin folding. Micrococcal nuclease (MNase) digestion studies reveal 2 mono-nucleosomal intermediates: the core particle (∼147 bp) and the chromatosome (∼160 bp; a core particle with additional DNA protected by H1). We have recently developed an improved method for mapping nucleosomes, using exonuclease III to remove residual linker (MNase-Exo-seq).Citation1 We discovered 2 new intermediate particles corresponding to core particles with ∼7 bp of linker protruding from one side (∼154 bp) or both sides (∼161 bp), which are formed in the absence of H1. We propose that these “proto-chromatosomes” are stabilized by core histone-DNA contacts in the linker, ∼7 bp from the nucleosome boundaries. These contacts may determine the topography of the H1 binding site.
DNA in eukaryotic cells is organized into chromatin, which is a complex between negatively charged DNA and positively charged histone proteins. The histones serve 2 functions: they drive the ordered condensation of the DNA to facilitate packaging into the nucleus and they regulate access to genetic information. There are 2 types of histone: core histones and linker histones. The core histones (H2A, H2B, H3 and H4) form an octamer composed of an (H3-H4)2 tetramer and 2 H2A-H2B dimers, which bind above and below the tetramer. When bound to DNA, the histone octamer coils 145–147 bp of DNA around itself in ∼1.7 negative superhelical turns to form a nucleosome core.Citation2,3 In cells, nucleosome cores are regularly spaced along the DNA, resembling beads on a string. This structure can be observed in the electron microscope after fixation at low ionic strength, which unfolds the chromatin, and is often called the 10-nm fiber because its width is about that of a nucleosome.Citation4 The linker DNA between nucleosome cores averages ∼15 – 95 bp in length, depending on the cell type (reviewed in ref.Citation5). The linker histone (H1 and its variants) binds to the DNA entry/exit point of the nucleosome core through its globular domain and to the linker DNA through its extremely positively charged C-terminal tail domain, forming a stem-loop structure.Citation6,7 Thus, H1 seals off the nucleosomal DNA turns and drives chromatin folding, primarily by partial neutralisation of the negative charge of the linker DNA.Citation8 For a review, see ref.Citation9. At physiological ionic strength and in the presence of H1, the beads-on-a-string structure folds spontaneously into a chromatin fiber about 30 nm wide (the “30-nm fiber”).Citation4 Exactly how the nucleosomal beads-on-a-string coils to form the 30-nm fiber is still controversial.Citation10
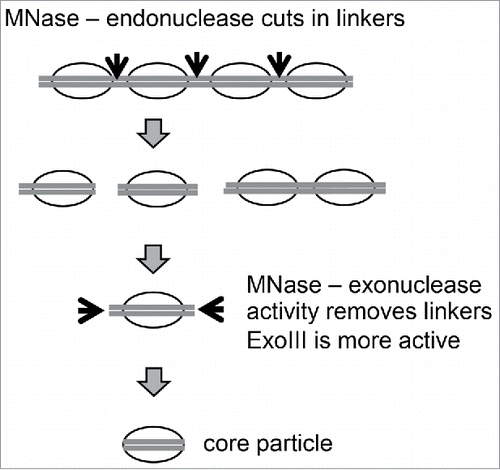
Chromatin studies rely heavily on the use of micrococcal nuclease (MNase), a calcium-dependent enzyme possessing both endo- and exo-nuclease activity. MNase digests protein-free DNA much faster than chromatin because the histones protect the DNA from digestion. Analysis of DNA purified from a mild MNase digest of chromatin reveals a “ladder” pattern in a gel, resulting from MNase cleavage of the linker DNA between regularly spaced nucleosomes, which is less protected than the nucleosome core. The bands in the ladder correspond to mono-nucleosomes, di-nucleosomes, tri-nucleosomes etc. As digestion proceeds, the longer oligo-nucleosomes are eventually reduced to mono-nucleosomes, as MNase cuts the remaining linkers through its endonuclease activity, and each band becomes progressively shorter as its exonuclease activity trims away residual linker DNA protruding from the nucleosome cores. The mono-nucleosome band is relatively broad because it represents a mixture of 2 particles: chromatosomes and core particles. Chromatosomes are mono-nucleosomes containing ∼160 bp and H1.Citation11 Further digestion destroys the linker DNA and displaces H1, resulting in nucleosome core particles, which contain ∼146 bp and the core histone octamer. The core particle represents a bead in the beads-on-a-string structure; it corresponds to the nucleosome structure solved by crystallography.Citation2 It is also the basis of a commonly used genome-wide nucleosome mapping method, MNase-seq, in which core particle DNA is purified and sequenced.
MNase-Exo-seq: An improved method for mapping nucleosome positions
Most transcription factors, though not all, have great difficulty binding to cognate sites assembled into a nucleosome. Their affinity for a nucleosomal site is typically >10-fold lower than for the same site in protein-free DNA.Citation12,13 The reduced affinity is due to steric hindrance by the histones, which block access to the inner surface of the nucleosomal DNA and, to a lesser extent, to the outer surface via the positively charged histone tail domains. In addition, the distortion of the DNA by tight bending around the histone octamer may also interfere with specific interactions of the transcription factor with the DNA. These considerations suggest models for gene regulation in which nucleosomes control access of transcription factors to their sites in promoters and enhancers, which is modulated by chromatin remodelling complexes.
To test such models, the precise position of each nucleosome relative to the DNA sequence must be determined. Ideally, this would involve the sequencing of populations of perfectly trimmed nucleosomal DNA of between 145 and 147 bp to give very accurate positions. The importance of accuracy is apparent when we consider that the linker DNA can be very short (typically ∼15 bp in yeast), such that an error of only 10 bp in the nucleosome position might be enough to incorrectly assign a transcription factor binding site as nucleosomal instead of in the accessible linker, or vice versa. Unfortunately, the uncertainty is of this order in typical MNase-seq experiments because of the mixture of chromatosomes and core particles. In fact, the distribution of DNA fragment lengths obtained after paired-end sequencing of mono-nucleosomal DNA indicates that it is a mixture not only of chromatosomes and core particles, but also includes chromatosomes with additional untrimmed linker DNA and sub-nucleosomal particles generated by internal digestion or invasion of core particles, as well as some genuine sub-nucleosomes (missing one or both H2A-H2B dimers) ().Citation14-17 In practice, it is virtually impossible to obtain a homogeneous population of nucleosome core particles using MNase-seq; a compromise must be reached in which the average length is close to 150 bp, with similar levels of over- and under-digestion. Of course, it can be achieved by cutting a narrow mono-nucleosome band from a gel, but this approach risks skewing the data, because different nucleosomes may be digested at different rates, depending on their DNA sequenceCitation18 and perhaps on their histone modifications or other factors.
Figure 1. Simultaneous digestion of chromatin with MNase and ExoIII reveals new intermediate mono-nucleosomal particles. DNA length distributions obtained by paired-end sequencing of nucleosomes from yeast cells. (A) MNase only. (B) MNase and ExoIII. Adapted from Fig. 1 in Citation1.
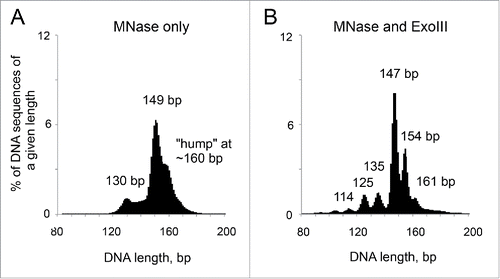
We reasoned that the major problem with MNase-seq is that the exonuclease activity of MNase is too slow, such that some mono-nucleosomes are not trimmed to core particles before others are digested internally. In an attempt to solve this problem, we added E. coli exonuclease III (ExoIII) to boost the exonuclease activity of MNase in a simultaneous digestion of reconstituted nucleosomes or of chromatin in budding yeast nuclei ().Citation1,19 In this situation, it should be possible to produce less digested mono-nucleosomes using MNase and to remove the residual linker DNA at a much faster rate, before internal digestion of nucleosomes by MNase becomes significant. Since ExoIII is a single-strand 3′->5′-exonuclease, mung bean nuclease was initially included to destroy the other strand. However, it turned out to be unnecessary, presumably because MNase cuts single-stranded DNA faster than double-stranded DNA and rapidly removes the residual DNA strand generated by ExoIII.
Figure 2. Digestion of chromain by MNase and ExoIII. Adapted from Fig. 1 in Citation1.
Our paired-end nucleosome sequencing data for yeast nucleosomes prepared from cell nuclei and for nucleosomes reconstituted with purified core histones and DNA reveal that the MNase-ExoIII method is a major improvement over MNase-seq, because it results in a remarkably sharp peak of core particle size ().Citation1 This population represents a set of very accurate nucleosome positions. However, other peaks are also detected, revealing an unexpected but interesting complication.
The proto-chromatosome: A chromatosome without H1
Although there is a major sharp peak corresponding to perfectly trimmed core particles, weaker peaks corresponding to particles of sub-nucleosome size and 2 unexpected peaks corresponding to particles containing longer DNA (∼154 bp and ∼161 bp) are also observed. The smaller particles are those expected from invasion of the nucleosome by ExoIII, because they are related by ∼10 bp intervals (135, 125 and 114 bp). It may be possible to eliminate the smaller particles by fine adjustment of the ratio of MNase and ExoIII, but we have not yet found the appropriate digestion conditions. The 154-bp and 161-bp particles are more interesting. Initially, we supposed that they are chromatosomes, although yeast cells have much less H1 per nucleosome than most cells in higher organisms,Citation20 predicting that chromatosomes could account for only a minor fraction of the particles observed. We eliminated a role for H1 by mapping nucleosomes from yeast cells lacking H1 and by mapping nucleosomes reconstituted using purified recombinant yeast core histones and plasmid DNA.Citation1 Both approaches yielded the same set of particles. Therefore, these particles are not chromatosomes because they are stable without H1. That is, the protection of the extra DNA in these particles does not require H1, although H1 may enhance that protection.
It is clear from an MNase-ExoIII digestion series that the 161-bp and 154-bp particles are eventually digested to core particles and beyond, indicating that they are kinetic intermediates in the same digestion pathway. A comparison of the sequences of the 154-bp particles with those of the core particles (147 bp) reveals that 154-bp particles are core particles with a 7-bp extension on one side. This 7-bp extension has a relatively high G/C content and its 5′-end tends to be 5′-(A/T)(G/C), which is also observed in core particles prepared using MNase alone. Although the 154-bp particles are asymmetrical, the 161-bp particles have 7 or 8 bp extensions on both sides of the nucleosome core and are therefore almost symmetrical.
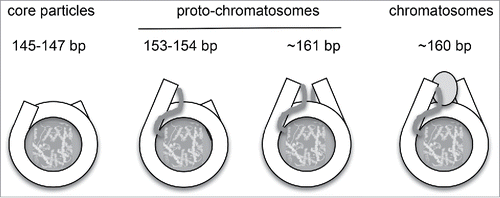
Although these particles are not chromatosomes, they are presumably related to them. Accordingly, we named these particles “proto-chromatosomes” to indicate their likely function in chromatin structure (). We propose that the complete proto-chromatosome is 161-bp, corresponding to a core particle with 7 or 8 bp extensions on both sides, with the appropriate topography to facilitate H1 binding, which results in increased protection of the same DNA extensions. That is, the H1 binding site is partially protected by the core histones, resulting in a pause in nuclease digestion similar to that at the boundaries of the core particle. These pauses in digestion probably reflect steric clashes between the nucleosome core and the nuclease, which must rotate around the DNA as it trims and invades the particle.Citation1 We have verified that proto-chromatosomes are not peculiar to yeast by MNase-ExoIII digestion of native and H1-depleted mouse liver chromatin, both of which generate the same set of particles.Citation1
Figure 3. Structural relationship between nucleosome core particles, proto-chromatosomes and chromatosomes. Views from above, drawn approximately to scale, based on the nucleosome structure. Citation2 The final 10 bp on each side of the nucleosome core are almost straight, projecting a short distance out of the particle. Proto-chromatosomes are shown with an extra 7 bp on one side (154 bp) or both sides (161 bp) with a continuing straight trajectory. We propose that the fundamental particle is the 161-bp proto-chromatosome, which has both extensions. We suggest that each extension is protected by a specific but unidentified core histone-DNA contact, most likely a histone tail (as indicated), to form the H1 binding site. The chromatosome is formed when H1 binds (shown here as a light gray oval in a symmetrical location although this is controversial). Adapted from Fig. 6 in Citation1.
Fine analysis of nucleosome positions using MNase-Exo-seq
The observation of unexpected particles in the MNase-ExoIII digest complicates the fine analysis of nucleosome positions. Perhaps the best and certainly the simplest option is to restrict the analysis to core particles (i.e., to ignore all the other particles). The famous dinucleotide patterns observed within nucleosomal DNA, such that A/T dinucleotides and G/C dinucleotides have 10-bp periodicities that are exactly out of phase with one another, reflecting the energetics of DNA bending during nucleosome formation,Citation21 are enhanced in the MNase-ExoIII core particle sequencesCitation1; shuffling the sequences to maximise the signal alignment is unnecessary. In addition, weaker signals are apparent that are not detectable in MNase-seq data, such as the preferential occurrence of A/T dinucleotides at ∼16, 37, 58 bp from the nucleosomal boundaries – that is, at exactly the same locations where the DNA is sharply bent into the minor groove.Citation3 This observation will be important for further development of structure-based models of nucleosome positioning.Citation22
On the other hand, if the distribution of nucleosomes between the various particle types is non-random, then the occupancies of specific nucleosome positions may be estimated incorrectly. It may be possible to include the proto-chromatosome data in the analysis by using the fact that they mostly represent 7-bp extensions of known positions. Most likely, this is a minor issue and will be important only when examining relatively minor positioning effects. Including the data from over-digested nucleosomes presents a more difficult problem, which might be solved technically by adjusting the MNase/ExoIII ratio, as discussed above.
Proto-chromatosomes, linker DNA length and the 30-nm fiber
The size of the complete proto-chromatosome, 161 bp, suggests 2 interesting corollaries. The first is that 161 bp is about the amount of DNA required to form 2 complete superhelical turns around the histone octamer, since a full superhelical turn in the core particle is ∼80 bp. This is an intriguing observation, but there is no evidence at this point for nucleosomes with 2 complete turns within the intact chromatin fiber. In fact, the crystal structure of a tetra-nucleosome does not support this idea,Citation23 although fibers with different linker lengths are predicted to form different 30-nm fiber structuresCitation24-26 and so a tetra-nucleosome with a different linker length might form a structure that is quite different.
The second interesting corollary is that the proto-chromatosome represents the core particle plus 15 bp of linker DNA. There is some evidence that the linker length follows a (10n +5) bp rule, where ‘n’ is an integer.Citation5,27 That is, linkers are quantised with lengths of 5, 15, 25 bp etc. This rule has crucial implications for the structure of the 30-nm fiber, because it determines the orientation of the second nucleosome relative to the first and therefore dictates the structure of the 30-nm fiber.Citation24-26 Assuming that proto-chromatosomes cannot overlap, then the minimum linker length is 15 bp, which fits the rule with n = 1 (n cannot be 0 unless they overlap). Consequently longer linkers would separate proto-chromatosomes in units of 10 bp (if n > 1), preserving their relative orientation because the B-DNA helix has 10.5 bp/turn. The nucleosome repeat length (measured in a gel) in yeast is short (∼165 bp) and practically coincides with a nucleosome spacing of 161 bp derived from genomic auto-correlation analysis of nucleosome sequences,Citation1,28 suggesting that yeast chromatin may be composed of a series of juxtaposed proto-chromatosomes with a spacing of ∼161 bp.
The most important issue to be resolved in future work is to determine which histone-DNA contacts protect the 7-bp extensions in the proto-chromatosome from digestion. There is evidence for the binding of histone tail domains to the linker DNA, particularly that of H3.Citation29,30 We are currently investigating whether the core histone tail domains are required to form the proto-chromatosome. The crystal structure of the proto-chromatosome would certainly shed light on its putative role in chromatin structure.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sergei Grigoryev and Răzvan Chereji for illuminating discussions.
Funding
This work was funded by the Intramural Research Program of the National Institutes of Health (NICHD and NCI) and by a grant from the National Institute of General Medical Sciences (R15GM116102 to FC).
References
- Cole HA, Cui F, Ocampo J, Burke TL, Nikitina T, Nagarajavel V, Kotomura N, Zhurkin VB, Clark DJ. Novel nucleosomal particles containing core histones and linker DNA but no histone H1. Nucleic Acids Res 2016; 44:573-81; PMID:26400169; http://dx.doi.org/10.1093/nar/gkv943
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997; 389:251-60; PMID:9305837; http://dx.doi.org/10.1038/38444
- Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent-mediated interactions in the structure of the nucleosome core particle at 1.9Å resolution. J Mol Biol 2002; 319:1097-113; PMID:12079350; http://dx.doi.org/10.1016/S0022-2836(02)00386-8
- Thoma F, Koller T, Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol 1979; 83:403-27; PMID:387806; http://dx.doi.org/10.1083/jcb.83.2.403
- van Holde KE. Chromatin. 1988; Springer-Verlag, New York.
- Bednar J, Horowitz RJ, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci USA 1998; 95:14173-8; PMID:9826673; http://dx.doi.org/10.1073/pnas.95.24.14173
- Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, Bednar J, Dimitrov S. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci USA 2010; 107:9620-25; PMID:20457934; http://dx.doi.org/10.1073/pnas.1000309107
- Clark DJ, Kimura T. Electrostatic mechanism of chromatin folding. J Mol Biol 1990; 211:883-96; PMID:2313700; http://dx.doi.org/10.1016/0022-2836(90)90081-V
- Woodcock CL, Skoultchi AI, Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res 2006; 14:17-25; PMID:16506093; http://dx.doi.org/10.1007/s10577-005-1024-3
- Travers A. The 30-nm fiber redux. Science 2014; 344:370-2; PMID:24763580; http://dx.doi.org/10.1126/science.1253852
- Simpson RT. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochem 1978; 17:5524-31; http://dx.doi.org/10.1021/bi00618a030
- Imbalzano AN, Kwon H, Green MR, Kingston RE. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994; 370:481-5; PMID:8047170; http://dx.doi.org/10.1038/370481a0
- Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol 1995; 15:1405-21; PMID:7862134; http://dx.doi.org/10.1128/MCB.15.3.1405
- Cole HA, Howard BH, Clark DJ. The centromeric nucleosome of budding yeast is perfectly positioned and covers the entire centromere. Proc Natl Acad Sci USA 2012; 108:12687-92; http://dx.doi.org/10.1073/pnas.1104978108
- Cole HA, Howard BH, Clark DJ. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res 2011; 39:9521-35; PMID:21880600; http://dx.doi.org/10.1093/nar/gkr643
- Cole HA, Howard BH, Clark DJ. Genome-wide mapping of nucleosomes in yeast using paired-end sequencing. Methods Enzymol 2012; 513:145-68; PMID:22929768; http://dx.doi.org/10.1016/B978-0-12-391938-0.00006-9
- Cole HA, Ocampo J, Iben JR, Chereji RV, Clark DJ. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res 2014; 42:12512-22; PMID:25348398; http://dx.doi.org/10.1093/nar/gku1013
- Chereji RV, Kan T, Grudniewska MK, Romashchenko V, Berezikov E, Zhimulev IF, Guryev V, Morozov AV, Moshkin YM. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res 2016; 44:1036-51; PMID:26429969; http://dx.doi.org/10.1093/nar/gkv978
- Nikitina T, Wang D, Gomberg M, Grigoryev SA, Zhurkin VB. Combined micrococcal nuclease and exonuclease III digestion reveals precise positions of the nucleosome core/linker junctions: implications for high-resolution nucleosome mapping. J Mol Biol 2013; 425:1946-60; PMID:23458408; http://dx.doi.org/10.1016/j.jmb.2013.02.026
- Friedkin I, Katcoff D. Specific distribution of the Saccharomyces cerevisiae linker histone homologue Hho1p in the chromatin. Nucleic Acids Res 2001; 29:4043-51; PMID:11574687
- Satchwell SC, Drew HR, Travers AA. Sequence periodicities in chicken nucleosome core DNA. J Mol Biol 1986; 191:659-75; PMID:3806678; http://dx.doi.org/10.1016/0022-2836(86)90452-3
- Tolstorukov MY, Colasanti AV, McCandlish DM, Olson WK, Zhurkin VB. A novel roll-and-slide mechanism of DNA folding in chromatin: implications for nucleosome positioning. J Mol Biol 2007; 371:725-38; PMID:17585938; http://dx.doi.org/10.1016/j.jmb.2007.05.048
- Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 2005; 436:138-41; PMID:16001076; http://dx.doi.org/10.1038/nature03686
- Woodcock CL, Grigoryev SA, Horowitz RA, Whitaker N. A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc Natl Acad Sci USA 1993; 90:9021-25; PMID:8415647; http://dx.doi.org/10.1073/pnas.90.19.9021
- Correll SJ, Schubert MH, Grigoryev SA. Short nucleosome repeats impose rotational modulations on chromatin fibre folding. EMBO J 2012; 31:2416-26; PMID:22473209; http://dx.doi.org/10.1038/emboj.2012.80
- Norouzi D, Zhurkin VB. Topological polymorphism of the two-start nucleosome fibers. Biophys J 2015; 108:2591-600; PMID:25992737; http://dx.doi.org/10.1016/j.bpj.2015.04.015
- Brogaard K, Xi L, Wang JP, Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature 2012; 486:496-501; PMID:22722846
- Cui F, Cole HA, Clark DJ, Zhurkin VB. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res 2012; 40:10753-64; PMID:23012262; http://dx.doi.org/10.1093/nar/gks870
- Angelov D, Vitolo JM, Mutskov V, Dimitrov S, Hayes JJ. Preferential interaction of the core histone tail domains with linker DNA. Proc Natl Acad Sci USA 2001; 98:6599-604; PMID:11381129; http://dx.doi.org/10.1073/pnas.121171498
- Rhee HS, Bataille AR, Zhang L, Pugh BF. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 2014; 159:1377-88; PMID:25480300; http://dx.doi.org/10.1016/j.cell.2014.10.054