
ABSTRACT
Acetylation of non-histone proteins plays important roles in regulating protein functions but the mechanisms of action are poorly understood. Our recent study uncovered a previously unknown mechanism by which C-terminal domain (CTD) acetylation of p53 serves as a “switch” to determine the interaction between a unique group of acidic domain-containing proteins and p53, as well as revealed that acidic domains may act as a novel class of “readers” for unacetylated p53. However, the properties of acidic domain “readers” are not well elucidated yet. Here, we identified that the charge effect between acidic domain “readers” and the p53 CTD is necessary for their interaction. Both the length and the amino acid composition of a given acidic domain contributed to its ability to recognize the p53 CTD. Finally, we summarized the characteristic features of our identified acidic domains, which would distinguish this kind of “readers” from other types of acidic amino acid-containing domains.
Introduction
Acetylation has been emerging as a general posttranslational modification (PTM) with a widespread distribution on lysine residues of both histone and non-histone proteins.Citation1 During past decades, over 3600 acetylated lysine sites have been identified in more than 1750 human proteins.Citation2 The status of protein acetylation is dynamically established by lysine acetyltransferases (KATs) called “writers” and removed by lysine deacetylases (KDACs) called “erasers”.Citation3 The most understood functions of lysine acetylation comes from the extensive studies of histone biology. Lysine acetylation on histone tails within the nucleosome is correlated with a relaxed chromatin conformation that facilitates transcription machinery recruitment and gene expression.Citation4 In addition, growing evidence indicates that acetylation on non-histone proteins plays more diverse roles beyond transcriptional regulation.Citation5-7 Acetylated lysine residues often act as a platform to recruit the bromodomain-containing proteins that are referred as the “readers” of lysine acetylation.Citation8,9 All of these “writers,” “erasers” and “readers” represent an important layer of an epigenetic regulatory network in chromatin-based events.Citation4 However, the precise mechanisms by which lysine acetylation regulates proteins, especially the non-histone proteins and their subsequent functions are still not completely understood.
p53 is a transcription factor that activates or represses a series of downstream targets, in physiologic conditions or in response to various stimuli, to participate in the regulation of multiple cellular processes including cell cycle arrest, apoptosis, senescence, DNA repair, oxidative response and metabolism.Citation10 More than 50% of human cancers have p53 mutations, and p53 knockout mice spontaneously develop tumors, demonstrating a bone fide role of tumor suppression by p53.Citation11-13 Considering the pivotal role in coordinating cellular responses to various stresses, the protein level, as well as the activity of p53 must be strictly modulated. In unstressed conditions, cellular p53 is maintained as a low level, predominantly by its negative regulator MDM2 that ubiquitinates p53 and promotes proteasome-dependent degradation of p53.Citation14,15 When cells encounter internal or external stresses (e.g. DNA damage, ribonucleotide depletion or oncogene activation), p53 can be rapidly stabilized and activated by various PTMs including phosphorylation and acetylation.Citation16 Actually, p53 is the first example of a non-histone protein that was found to be functionally regulated by acetylation and deacetylation.Citation17-19 It is known that acetylation occurring in distinct domains of p53 contributes to different aspect of p53 functions. For example, acetylation on the p53 DNA-binding domain (DBD) determines promoter-specific transcription of p53 target genes, whereas acetylation on its C-terminal domain (CTD) plays a general role in regulating p53 stability, DNA-binding affinity and transactivity.Citation20-22 Mechanistically, acetylation of p53 is considered to reduce the non-specific binding of p53 to DNA elements or recruit bromodomain-containing coactivators to facilitate p53-driven transcription.Citation23,24 However, it is still not clear whether other potential mechanisms underlying acetylation-mediated regulation of p53 exist.
Our recent study revealed that acidic domain-containing proteins including SET, VPRBP, DAXX and PELP1, represent a unique group of factors that interact with and regulate p53 in an acetylation-dependent manner.Citation25,26 Specifically, the acidic domain of these factors directly interacted with the CTD of p53; however, these interactions were completely blocked by acetylation, but not other types of modifications of the p53 CTD. These observations uncovered a new model of CTD acetylation-mediated regulation of p53 functions and provided a concept that the acidic domains may act as a class of “readers” for the unacetylated p53 CTD. However, the characteristics of these acidic domain “readers” are not clearly illustrated: 1) Why does the acidic domain specifically interact with the p53 CTD? 2) How can acidic domain “readers” be distinguished from other types of acidic amino acid-containing domains?
Here, by using the p53-SET interaction as a major example, we found that the charge effect between the acidic domain and the p53 CTD is essential for their interaction. In addition, 2 parameters including the length of the acidic domain and the composition of acidic amino acids within the acidic domain contributed to the ability of acidic domain interaction with the p53 CTD. Lastly, by comparing other kinds of acidic amino acidic-containing domains, we summarized, at least partially, the characteristic features of our identified acidic domain “readers.”
Results
The charge effect is required for the interaction between the p53 CTD and the acidic domain
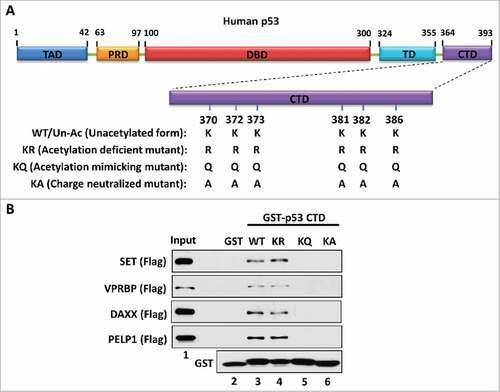
To dissect the mechanism by which the acidic domain specifically interacts with the p53 CTD, we initially focused on the p53-SET interaction. The p53 CTD is a lysine-rich domain with a positive charge while the acidic domain of SET is rich with acidic amino acids harboring a highly negative charge. Therefore, we postulated that the charge effect between p53 CTD and the SET acidic domain plays a key role in their interaction. To test this hypothesis, we generated a series of GST-fused constructs expressing a wild-type/unacetylated p53 CTD (WT/Un-Ac), an acetylation-deficient CTD mutant (lysine-to-arginine mutation, KR), an acetylation-mimicking CTD mutant (lysine-to-glutamine mutation, KQ) or a charge-neutralized CTD mutant (lysine-to-alanine mutation, KA) for an in vitro binding assay with SET (). As expected, both the WT/Un-Ac CTD and the acetylation-deficient KR mutant strongly interacted with SET, whereas the KQ mutant completely lost the ability to bind with SET (). More importantly, similar to the KQ mutant, the KA mutant also failed to interact with SET (). Of note, although the KR mutant loses all post-translational modifications that potentially occur on those wild-type lysine residues, the lysine-to-arginine mutations still maintain a positive charge within the CTD. In contrast, both KQ and KA mutants exhibit a charge-neutralized status of the CTD. Unlike the lysine-to-glutamine mutations in the KQ mutant, the lysine-to-alanine mutations in the KA mutant do not structurally mimic the sidechain of acetylated lysine residues. However, the KA mutant still gained the same property to that of the KQ mutant in respect of their interaction with SET. Similar interactive patterns between the p53 CTD and other acidic domain-containing proteins VPRBP, DAXX and PELP1 were also observed (), indicating that the charge effect between the p53 CTD and acidic domains are crucial for their interactions. This mechanism is reasonable and well correlated with our findings that acetylation disrupted p53 CTD interaction with acidic domains,Citation25 as acetylation neutralizes the positive charge of the lysine residues within the p53 CTD.
Figure 1. The charge effect is essential for the interaction between the p53 CTD and acidic domain-containing proteins. (A) Schematic diagram of WT/unacetylated p53 and 3 types of p53 mutants. TAD: transactivation domain; PRD: proline-rich domain; DBD: DNA-binding domain; TD: tetramerization domain; CTD: C-terminal domain. (B) Western blot analysis of the interaction between the p53 CTD and acidic domain-containing proteins. In vitro binding assay was performed by incubating GST alone or GST-p53 CTD with purified Flag-tagged acidic domain-containing proteins SET, VPRBP, DAXX or PELP1. The bound components were analyzed by western blot with α-Flag antibody. The purified GST or GST-fusion proteins used in this assay were analyzed in parallel by α-GST antibody
The length of the acidic domain contributes to its interaction with p53
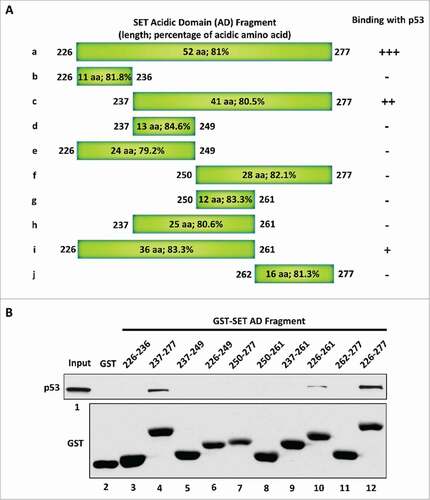
To directly interact with the p53 CTD via a charge effect, the strength of the negative charge within a given acidic domain has to reach a certain degree. Therefore, we considered that the length of the acidic domain could be one of the determinants for the ability of the acidic domain to interact with the p53 CTD. We chose the p53-SET interaction again, as the example to verify this point. To this end, we generated a series of truncated SET acidic domain fragments () and evaluated their interactions with p53 by performing an in vitro binding assay. As expected, the full-length SET acidic domain strongly bound with p53 (). Further analysis showed that p53 did not interact with the first 11 amino acids of the SET acidic domain (fragment b: 226–236) and loss of the first 11 amino acids did not dramatically affect the p53-SET interaction (fragment c: 237–277). Interestingly, loss of the next 13 amino acids completely abolished the p53-SET interaction (fragment f: 250–277), suggesting that these 13 amino acids (237–249) are critical for the interaction. However, the fragments d, e and h (237–249, 226–249, and 237–261, respectively) that retain these 13 amino acids still failed to interact with p53, whereas the 36 amino acid long fragment i (226–261) that includes these 13 amino acids showed weaker interaction with p53, suggesting a critical role of the length of the acidic domain for this interaction. Taken together, these data indicate that the 36 amino acid long fragment i (226–261) contain the minimal binding domain for interacting with p53; further, it is very likely that the length of the acidic domain is critical for this interaction.
Figure 2. The length of the acidic domain contributes to its interaction with the p53 CTD. (A) Schematic diagram of the full-length and sub-fragments of the SET acidic domain (AD). (B) Western blot analysis of the interaction between p53 and each fragment of the SET acidic domain. In vitro binding assay was performed by incubating purified p53 with purified GST alone or each of the GST-tagged fragments of the SET acidic domain. The binding components were analyzed by western blot with α-p53 antibody. The purified GST or GST-fusion peptide used in this assay was analyzed in parallel by α-GST antibody
The amino acid composition is crucial for the acidic domain binding with the p53 CTD
The amino acid composition is thought to be another factor that determines the strength of the negative charge within the acidic domain. Actually, more than 76% of the amino acids within our identified acidic domains (the acidic domain of SET, VPRBP, DAXX or PELP1) are acidic amino acids including glutamic acid and aspartic acid (). Moreover, the percentage of acidic amino acids within the SET acidic domain sub-fragments that interact with p53 is even higher ( and ). This characteristic may distinguish our identified acidic domains from other types of acidic amino acid-containing domains in the respect of their ability to bind with the p53 CTD. For example, numerous transcription factors including p53 and FOXO1 (), harbor an acidic amino acid-containing domain, also known as an acidic transactivation domain (TAD).Citation27 We therefore assessed the binding ability of p53-TAD or FOXO1-TAD to the p53 CTD. To this aim, we purified GST-tagged TADs for an in vitro binding assay. As shown in , we failed to detect an interaction between p53-TAD and the p53 CTD. In contrast, as a positive binding control, the SET acidic domain strongly bound the unacetylated p53 CTD. Similar result was observed when the interaction between the FOXO1-TAD and the p53 CTD was tested under the same conditions ().
Table 1. A summary of the characteristic features of acidic domain “readers” and other types of acidic amino acid-containing domains. The sequence, length, acidic amino acidic distribution pattern and composition, and the charge of each domain are listed. The acidic amino acids within the sequence of each domain are underlined
Figure 3. The amino acid composition is crucial for acidic domain binding with the p53 CTD. (A,B) Western blot analysis of the interaction between unacetylated p53 CTD and the acidic transactivation domain (TAD). In vitro binding assay was performed by incubating immobilized biotin-conjugated unacetylated p53 CTD with purified GST-p53-TAD (A) or GST-FOXO1-TAD (B), as indicated. The binding components were analyzed by western blot with α-GST antibody. The interaction between p53 CTD and SET acidic domain (AD) was analyzed in parallel as a positive binding control. (C) Schematic diagram of human MDM2 protein. (D) Western blot analysis of the interaction between p53 CTD and MDM2 central acidic domain (CAD). In vitro binding assay was performed by incubating immobilized biotin-conjugated unacetylated or acetylated CTD of p53 with purified GST or GST-MDM2-CAD. The binding components were assayed by western blot with α-GST antibody. (E) Western blot analysis of the interaction between each p53 fragment and the MDM2 central acidic domain. In vitro binding assay was performed by incubating GST or each GST-p53 fragment with purified GFP-Flag-MDM2-CAD. The binding components were assayed by western blot with α-GFP antibody. The purified GST or GST-fusion proteins used in this assay were analyzed in parallel by α-GST antibody
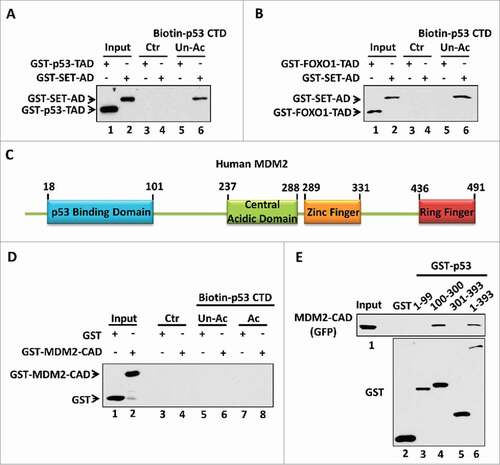
This importance of the amino acid composition was further proven using the p53-MDM2 interaction. MDM2 is a p53 downstream target which negatively regulates p53 stability and transactivity by forming a negative feedback loop.Citation14,15,28-30 Interestingly, MDM2 also has a central acidic domain (CAD; amino acids 237–288) (). Therefore, we evaluated whether the central acidic domain of MDM2 interacts with the p53 CTD in an acetylation-regulated manner. To this aim, we purified the recombinant GST-tagged MDM2 central acidic domain to test its interaction with unacetylated vs. acetylated p53 CTD. As shown in , we found that the central acidic domain of MDM2 did not bind either p53 peptide, no matter the acetylation status of the p53 CTD. Interestedly, using highly purified GFP- and Flag-tagged MDM2-CAD protein, our analysis revealed that the central acidic domain of MDM2 bound with the p53 DNA-binding domain (), suggesting an alternative mechanism rather than a charge effect to mediate the MDM2 central acidic domain interaction with p53. This observation is also consistent to a previous report suggesting that the central acidic domain of MDM2 may serve as a secondary platform to interact with p53.Citation31
When comparing acidic domain “readers” to the other types of acidic amino acid-containing domains (TADs of p53 and FOXO1 or MDM2-CAD), we found that despite comparable amino acid length, the ordering of the acidic amino acids within the acidic domain differ—the other types are separately distributed while the “readers” have a clustered distribution. Further, the percentage of acidic amino acids within the acidic domain of the other types is much lower compared with the “readers.” As shown in , the percentage of acidic amino acids within TADs and the MDM2-CAD is only about 20% and 34.6%, respectively. However, as mentioned above, this percentage of all acidic domain “readers” is all above 76%. The low percentage of acidic amino acid is correlated with the low strength of the negative charge within the other types of acidic domains. In fact, the theoretically calculated charge of p53-TAD, FOXO1-TAD and MDM2-CAD in pH 7.0 conditions is only −8.1, −7.6 and −17.8, respectively, which is markedly lower than the acidic domain “readers” (SET: −42; VPRBP: −35; DAXX: −41; PELP1: −66.9) (). These observations suggest that the low strength of the negative charge within the other types of acidic amino acid-containing domains is not sufficed to mediate protein-protein interaction in a similar mode as the interactions between our identified acidic domain “readers” and the p53 CTD. Taken together, these data indicate that the amino acid composition within a given acidic domain significantly contributes to its “reader” property.
Based on these observations, we propose that the acidic domain-containing proteins identified here are referred to a group of proteins that harbor a long, clustered distribution of acidic amino acids (including aspartic acid and glutamic acid). All these proteins contain a unique region with a highly negative charge serving as a physical platform to recognize the p53 CTD with a positive charge. Specifically, our identified acidic domain “readers” have at least 2 characteristic features: 1) the acidic domain is about 36 amino acids long or longer; 2) the acidic amino acids are clustered and the percentage of acidic amino acids within the acidic domain reaches more than 76%.
Discussion
Unlike bromodomain “readers” that recognize acetylated lysine residues by structurally forming a hydrophobic pocket,Citation32 our identified acidic domain “readers” recognize clustered unacetylated lysine residues by a charge effect. To evaluate a potential acidic domain “readers” of the p53 CTD, both the length and the amino acid composition within a given acidic domain should be simultaneously taken into consideration. For example, the sub-fragments b, d, e, f, g, h and j of the SET acidic domain have more than 76% of acidic amino acids; however, they cannot bind with the p53 CTD as these fragments are too short (). In addition, the p53-TAD, FOXO1-TAD and MDM2-CAD, which are all longer than 36 amino acids, are also unable to interact with the p53 CTD due to their low level of acidic amino acid composition (). Along with the identification of more acidic domains that interact with the p53 CTD in an acetylation-dependent manner, more detail of the characteristic features of the acidic domain “readers” will be further understood in the future.
In general, our identified acidic domain “readers” belong to one type of acidic amino acid-containing domains. In actuality, p53 can be regulated by multiple factors through protein-protein interaction mediated by different kinds of acidic amino acid-containing domains (). Firstly, our identified acidic domain-containing proteins including SET, VPRBP, DAXX and PELP1, regulate p53 activity through their acidic domain “readers” binding with the p53 C-terminus. Secondly, the central acidic domain of MDM2, which fails to act as a “reader” of unacetylated p53 CTD due to the low strength of the negative charge, specifically binds with the p53 DNA-binding domain and facilitates p53 degradation.Citation31 Thirdly, the p53 N-terminal transactivation domain per se is another type of acidic amino acid-containing domain that plays a key role in p53 transactivation via the recruitment of co-activators (e.g., p300/CBP and TAFs), mediators (e.g., MED17) or general transcription factors (e.g., TBP).Citation33-42 It will be interesting, in the future, to investigate whether a crosstalk exists among different kinds of acidic amino acid-containing domains to mediate p53 regulation.
Figure 4. Different types of acidic amino acid-containing domain-mediated regulation of p53. Acidic domain “readers” regulate p53 by interacting with the p53 C-terminus. MDM2 central acidic domain serves as a secondary platform to bind with the p53 DNA-binding domain and promote p53 degradation. The N-terminal transactivation domain of p53 is another acidic amino acid-containing domain responsible for the recruitment of co-activators, mediators or general transcription factors (GTFs) upon p53 activation

In addition to the C-terminal acetylation, several lysine residues (e.g., lysine residues 120 and 164 in human p53; lysine residues 98, 117, 161 and 162 in mouse p53) within the p53 DNA-binding domain are also found to be acetylated by multiple acetyltransferases under DNA damage conditions.Citation20,21,43-45 In contrast to the C-terminal domain that contains a lysine cluster, the lysine residues within the DNA-binding domain are separately distributed. Thus, it is probably not that acetylation on these lysine residues works through the same mechanism to that observed in the CTD acetylation-mediated p53 regulation. However, acetylation within the DNA-binding domain contributes to gene-specific transcription of p53 targets, which determines the cellular fate to undergo cell cycle arrest, apoptosis or ferroptosis.Citation20,21,46 Therefore, it will be worthwhile to dissect the mechanisms by which the separately distributed lysine-based acetylation regulates p53-dependent gene-specific transcription.
Bioinformatic analysis suggested that acetylation-mediated blockade between an acidic domain and a lysine-rich domain may represent a general mechanism to regulate protein-protein interaction in nature.Citation25 Our in vitro analysis strongly supports this mechanism; however, more efforts are still required to evaluate the significance of this regulatory paradigm under physiologic and in vivo conditions. Taken together, our study describe the characteristic features of acidic domain “readers” that specifically recognize unacetylated p53 CTD and, potentially, other lysine-rich domains similar to the p53 CTD.
Materials and methods
Construct generation
The GST-related constructs were obtained by PCR-cloning or sub-cloning the original cDNA (including p53, SET, FOXO1 and MDM2) into the PGEX-2TL vectors. The GFP-Flag-MDM2-CAD construct was generated by PCR-cloning the fragment into the pcDNA3.1 construct. The GST-p53 CTD constructs with point-mutations were generated by using a site-directed mutagenesis Kit (Stratagene, 200521).
Antibodies
α-Flag: Sigma F7425; α-p53: Santa Cruz sc-126; α-GST: LifeTein LT0423; α-GFP: Santa Cruz sc-9996.
GST pull-down assay
The E. coli containing construct expressing GST or GST-fusion protein was cultured at 37 °C for 4 hours, and then the protein expression was induced by 0.1 mM IPTG at 25 °C for another 4 hours. The bacteria were lysed in BC500 buffer (20 mM Tris-HCl pH 7.9, 500 mM NaCl, 10% Glycerol, 0.2 mM EDTA, 1% Triton X-100, fresh protease inhibitor) with sonication for cellular extract preparation. After purification by GST·Bind™ Resin (Novagen, 70541), equal amount of immobilized GST or GST-fusion protein was incubated with other purified proteins for 1 hour at 4 °C. After extensive washes by BC100 buffer (20 mM Tris-HCl pH 7.9, 100 mM NaCl, 10% Glycerol, 0.2 mM EDTA, 0.1% Triton X-100, fresh protease inhibitor), the binding components were analyzed by western blot with indicated antibodies.
Peptide pull-down assay
Control beads or immobilized biotin-conjugated peptide was incubated with purified proteins in BC100 buffer at 4 °C for 1 hour. After washes by BC100 buffer, the binding components were analyzed by western blot with indicated antibodies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The authors are grateful for funding from the National Cancer Institute of the National Institutes of Health under Award 5R01CA190477, 5RO1CA193890, 2RO1CA085533 and 5RO1CA216884 to W.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006; 23:607-18; PMID:16916647; https://doi.org/https://doi.org/10.1016/j.molcel.2006.06.026
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834-40; PMID:19608861; https://doi.org/https://doi.org/10.1126/science.1175371
- Kouzarides T. SnapShot: Histone-modifying enzymes. Cell 2007; 128:802; PMID:17320515; https://doi.org/https://doi.org/10.1016/j.cell.2007.02.018
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693-705; PMID:17320507; https://doi.org/https://doi.org/10.1016/j.cell.2007.02.005
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15-23; PMID:16289629; https://doi.org/https://doi.org/10.1016/j.gene.2005.09.010
- Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41:185-98; PMID:18804549; https://doi.org/https://doi.org/10.1016/j.biocel.2008.08.027
- Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther 2010; 10:935-54; PMID:20553216; https://doi.org/https://doi.org/10.1586/era.10.62
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999; 399:491-6; PMID:10365964; https://doi.org/https://doi.org/10.1038/20974
- Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol 2014; 6:a018762; PMID:24984779; https://doi.org/https://doi.org/10.1101/cshperspect.a018762
- Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell 2009; 137:413-31; PMID:19410540; https://doi.org/https://doi.org/10.1016/j.cell.2009.04.037
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; https://doi.org/https://doi.org/10.1038/35042675
- Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2-8; PMID:23263379; https://doi.org/https://doi.org/10.1038/ncb2641
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215-21; PMID:1552940; https://doi.org/https://doi.org/10.1038/356215a0
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387:299-303; PMID:9153396; https://doi.org/https://doi.org/10.1038/387299a0
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296-9; PMID:9153395; https://doi.org/https://doi.org/10.1038/387296a0
- Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137:609-22; PMID:19450511; https://doi.org/https://doi.org/10.1016/j.cell.2009.04.050
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001; 107:149-59; PMID:11672523
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001; 107:137-48; PMID:11672522
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997; 90:595-606; PMID:9288740; https://doi.org/https://doi.org/10.1016/S0092-8674(00)80521-8
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006; 24:827-39; PMID:17189186; https://doi.org/https://doi.org/10.1016/j.molcel.2006.11.021
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24:841-51; PMID:17189187; https://doi.org/https://doi.org/10.1016/j.molcel.2006.11.026
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 2003; 15:164-71; PMID:12648672
- Friedler A, Veprintsev DB, Freund SM, von Glos KI, Fersht AR. Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 2005; 13:629-36; PMID:15837201; https://doi.org/https://doi.org/10.1016/j.str.2005.01.020
- Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand Sanchez R, Zeleznik-Le NJ, Ronai Z, Zhou MM. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol Cell 2004; 13:251-63; PMID:14759370
- Wang D, Kon N, Lasso G, Jiang L, Leng W, Zhu WG, Qin J, Honig B, Gu W. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 2016; 538:118-22; PMID:27626385; https://doi.org/https://doi.org/10.1038/nature19759
- Wang D, Kon N, Gu W. Acidic domains: “converse readers” for acetylation code. Oncotarget 2016; 7:80101-02; PMID:27806316; https://doi.org/https://doi.org/10.18632/oncotarget.12989
- Blau J, Xiao H, McCracken S, O'Hare P, Greenblatt J, Bentley D. Three functional classes of transcriptional activation domain. Mol Cell Biol 1996; 16:2044-55; PMID:8628270
- Picksley SM, Lane DP. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? Bioessays 1993; 15:689-90; PMID:7506024; https://doi.org/https://doi.org/10.1002/bies.950151008
- Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J 1993; 12:461-8; PMID:8440237
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993; 362:857-60; PMID:8479525; https://doi.org/https://doi.org/10.1038/362857a0
- Kulikov R, Winter M, Blattner C. Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem 2006; 281:28575-83; PMID:16870621; https://doi.org/https://doi.org/10.1074/jbc.M513311200
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012; 149:214-31; PMID:22464331; https://doi.org/https://doi.org/10.1016/j.cell.2012.02.013
- Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature 1997; 387:819-23; PMID:9194564; https://doi.org/https://doi.org/10.1038/42972
- Scolnick DM, Chehab NH, Stavridi ES, Lien MC, Caruso L, Moran E, Berger SL, Halazonetis TD. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res 1997; 57:3693-6; PMID:9288775
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 1997; 89:1175-84; PMID:9215639
- Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997; 387:823-7; PMID:9194565; https://doi.org/https://doi.org/10.1038/42981
- Thut CJ, Chen JL, Klemm R, Tjian R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science 1995; 267:100-4; PMID:7809597
- Lu H, Levine AJ. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci U S A 1995; 92:5154-8; PMID:7761466
- Ito M, Yuan CX, Malik S, Gu W, Fondell JD, Yamamura S, Fu ZY, Zhang X, Qin J, Roeder RG. Identity between TRAP and SMCC complexes indicates novel pathways for the function of nuclear receptors and diverse mammalian activators. Mol Cell 1999; 3:361-70; PMID:10198638
- Truant R, Xiao H, Ingles CJ, Greenblatt J. Direct interaction between the transcriptional activation domain of human p53 and the TATA box-binding protein. J Biol Chem 1993; 268:2284-7; PMID:8428901
- Liu X, Miller CW, Koeffler PH, Berk AJ. The p53 activation domain binds the TATA box-binding polypeptide in Holo-TFIID, and a neighboring p53 domain inhibits transcription. Mol Cell Biol 1993; 13:3291-300; PMID:8497252
- Chen X, Farmer G, Zhu H, Prywes R, Prives C. Cooperative DNA binding of p53 with TFIID (TBP): a possible mechanism for transcriptional activation. Genes Dev 1993; 7:1837-49; PMID:8405994
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell 2008; 133:612-26; PMID:18485870; https://doi.org/https://doi.org/10.1016/j.cell.2008.03.025
- Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep 2016; 17:366-73; PMID:27705786; https://doi.org/https://doi.org/10.1016/j.celrep.2016.09.022
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269-83; PMID:22682249; https://doi.org/https://doi.org/10.1016/j.cell.2012.04.026
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015; 520:57-62; PMID:25799988; https://doi.org/https://doi.org/10.1038/nature14344