Abstract
In this review, we outline critical molecular processes that have been implicated by discovery of genetic mutations in autism. These mechanisms need to be mapped onto the neurodevelopment step(s) gone awry that may be associated with cause in autism. Molecular mechanisms include: (i) regulation of gene expression; (ii) pre-mRNA splicing; (iii) protein localization, translation, and turnover; (iv) synaptic transmission; (v) cell signaling; (vi) the functions of cytoskeletal and scaffolding proteins; and (vii) the function of neuronal cell adhesion molecules. While the molecular mechanisms appear broad, they may converge on only one of a few steps during neurodevelopment that perturbs the structure, function, and/or plasticity of neuronal circuitry. While there are many genetic mutations involved, novel treatments may need to target only one of few developmental mechanisms.
En esta revisión se describen importantes procesos moleculares que han surgido a partir del descubrimiento de mutaciones genéticas en el autismo. Estos mecanismos necesitan ser incluidos dentro de la(s) etapa(s) mal ejecutada(s) que puede(n) estar asociada(s) con la causa del autismo. Los mecanismos moleculares incluyen: 1) regulación de la expresión génica, 2) empalme pre-RNAm, 3) localización, traslación y recambio de proteínas, 4) transmisión sináptica, 5) señales celulares, 6) las funciones de las proteinas en el citoesqueleto y el esqueleto (del cromosoma) y 7) la función de moléculas de adhesión neuronal. Aunque son numerosos los mecanismos moleculares, ellos pueden convergir sola en una de las etapas que altere la estructura, función ylo plasticidad de los circuitos neuronales durante el neurodesarrollo. Ya que hay muchas mutaciones genéticas involucradas, es posible que los nuevos tratamientos necesiten enfocarse sólo en alguno de los pocos mecanismos del desarrollo.
Dans cet article, nous décrivons les processus moléculaires critiques impliqués par la découverte de mutations génétiques dans l'autisme. Ces mécanismes pourraient être cartographies en étapes neurodéveloppementales mal achevées qui pourraient être associées à l'origine de l'autisme. Les mécanismes moléculaires comprennent: 1) la régulation de l'expression du gène ; 2) l'épissage du pré-ARNm ; 3) la localisation, la translation et le renouvellement des protéines ; 4) la transmission synaptique ; 5) la signalisation cellulaire; 6) les fonctions des protéines du cytosquelette et du squelette (du chromosome) ; et 7) la fonction des molécules d'adhésion cellulaire neuronales. Les mécanismes cellulaires semblent nombreux, mais peuvent confluer sur une seule des quelques étapes qui, au cours du neurodéveloppement, perturbe la structure, la fonction et/ou la plasticité du circuit neuronal. De nombreuses mutations génétiques sont en cause, mais les nouveaux traitements peuvent avoir besoin de cibler seulement un des mécanismes du développement.
Introduction
Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental disorders characterized by impaired social interaction, disrupted development of communication and language skills, and repetitive behaviors. Over an affected individual's lifetime, costs of care can reach about $3.2 million, while the annual cost to society is an estimated $35 billion.Citation1 Such burdensome costs combined with new high estimates in prevalence—the newest numbers place the developmental disorder at 1 in 88 childrenCitation2—call for a need to fully understand and to develop new treatments for autism. Treatment for ASD has shown uneven efficacy, and no treatment to date has demonstrated the ability to alleviate the core social deficits. While the high-functioning spectrum of ASD has shown promising and hopeful response to behavioral treatments, a sizable cohort, predominantly lower-functioning and/or with comorbid intellectual disability, has not demonstrated significant treatment gains.Citation3 For this latter group of patients, the need to develop new treatment paradigms is critical.
Understanding the neurodevelopment mechanisms gone awry may provide crucial insights into the underlying pathobiology of autism and identify novel, effective treatment methods. An essential step is to determine what aspects of brain development and function are impaired in autism. Forward genetics, a process that identifies putative genes or gene networks, allows researchers to identify mutations, sometimes specific molecules, and perhaps converging mechanisms involved in autism.Citation4 One important question researchers should attempt to answer is: Can mutation discoveries lead us to specific step(s) that are perturbed during neurodevelopment? By answering this question, researchers may be able to identify distinct neurodevelopmental processes responsible for autistic subtypes that may allow for targeted treatments of autistic symptoms. In this review, we will argue that genetic studies in particular have helped us pinpoint a small number of neurodevelopmental steps that are generally involved in autism to those of the late steps of neurodevelopment, that are primarily involved in the development of neurocircuitry, namely axon and dendrite growth and arborization, and also experience-dependent synapse modification.
Human brain development and structural brain differences in autism
In contrast to the >22 000 genes in the human genome and steep number of molecular mechanisms within a functioning and differentiating cell, the number of steps involved in human brain development are relatively few and finite. Eleven processes are conceptualized in Essentially, we can divide these developmental stages of brain development into two categories: fetal and postnatal. Fetal brain development is largely experience-independent and begins with neural tube formation and patterning, and neurogenesis whereby neural progenitor cells proliferate and give rise to neurons of the brain. These newborn neurons then must undergo migration from the fetal neurogenic niche (the ventricular zones or subcortical structures for γ-aminobutyric acid (GABA)-ergic cells) to their final position in the brain. Subsequently, the process of neuronal morphogenesis involves the formation of cellular polarization that leads to the development of axonal growth cones which begin traversing the brain, forming its complex circuitry. This period of development is marked by profound axon and dendrite branching and arborization that eventually determines the axons and dendrites of any given neuron.Citation5,Citation6
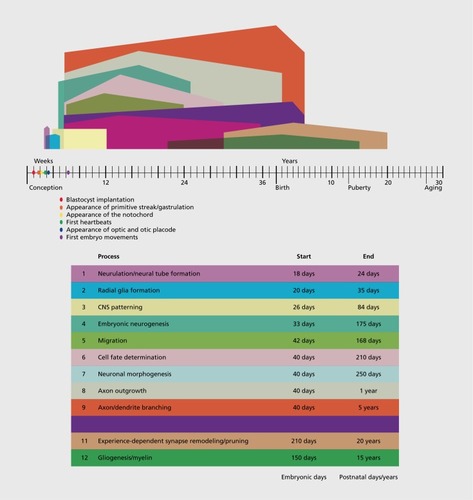
These aforementioned neurodevelopmental events are typically considered experience-independent processes. In other words, intrinsic genetic factors regulate each mechanism independent of sensory experience of the external world. Interestingly, there are many monogenic diseases that appear to affect one or various given stages above. For example, disorders that cause small brain size at birth, called primary microcephaly, result from a large number of single gene mutations that appear to affect neurogenesis.Citation7 Similarly, other monogenic disorders may result in abnormal patterning such as sonic hedgehog mutations or other mutations that may cause holoprosencephaly (failure of the forebrain to develop into two hemispheres).Citation8 Finally, there are a number of disorders of cortical migration that lead to abnormal layering of the brain or abnormal gyrus and sulcus formation.Citation9 Although there are exceptions, the above disorders have not been typically associated with autism symptoms; however, instead highly related conditions such as intellectual disability and epilepsy are more frequently described, along with the associated structural brain malformation. Interestingly, with respect to axon outgrowth, there are a number of monogenic disorders that may involve abnormalities of axon growth and/or targeting that have been associated with autistic symptoms. Joubert syndrome, for example, is a genetically heterogeneous condition that displays abnormalities in axon outgrowth and has been associated with autism symptoms.Citation10 Similarly, one neurodevelopmental abnormality in tuberous sclerosis (TSC) is also abnormal axon growth, and TSC is also recurrently although inconsistently associated with autism.Citation11 Joubert syndrome is generally associated with structural brain malformations. TSC is most frequently associated with a variety of morphologic abnormalities including tubers, but also with abnormalities of the corpus callosum. Growth of the corpus callosum has frequently been cited as an indicator of problems in the axon growth step of neurodevelopment. Indeed, isolated agenesis, hypogenesis, or dysgenesis of the corpus callosum have been associated with an increase in autism symptoms.Citation12,Citation13
In addition to the experience-independent neurodevelopmental processes, concomitant with the determination of axons and dendrites, neurons begin the process of forming connections via synaptogenesis commencing a series of experience-dependent developmental mechanisms. The process of synapse formation in the developing brain involves the production of a wide excess of synapses and a subsequent pruning back, perhaps strengthening of some and loss of others.Citation14,Citation15 In this case, neuronal activity thought to be mediating the processes of experience may result in chromatin modifications that lead to long-lasting effects on gene expression, brain development, and circuit architecture. This mechanism is most important for postnatal synaptic plasticity and during the synaptic pruning that begins at birth and becomes most widespread, continuing into adolescence.
There are also a limited number of monogenic disorders that appear to be associated with synaptic plasticity and autism. In particular, Fragile X syndrome (FXS) which is associated with a trinucleotide repeat expansion and loss-of-function mutation, is frequently associated with autism.Citation16 Interestingly, in some reports FXS is associated with an increase in cerebral volume.Citation17 Macrocephaly, increases in cerebral volume (generally greater than 2 standard deviations above the mean for age, ethnicity, and gender), has a longstanding association with autism.Citation18 Estimates suggest that approximately 30% of children with autism have macrocephaly.Citation19 However, there also appear to be a subset of children with autism who have microcephaly. Mutations in the gene PHOSPHATASE AND TENSIN HOMOLOG (PTEN) have been notably associated with autism and large head size,Citation20 while Rett syndrome (RTT) (due to mutations in MeCP2 gene) is also frequently associated with autistic symptoms and also generally with microcephaly. What are the underlying neurodevelopmental mechanisms that cause brain overgrowth or undergrowth? Of course, the timing of the emergence of this structural brain defect will greatly lead hypotheses regarding this question. For macrocephaly in idiopathic autism, there are proposals that the brain is generally normocephalic at birth and demonstrates a postnatal brain overgrowth. Assuming that relative timing of the different steps of human brain development are preserved (Figure 1), then this timing would rule out mechanisms such as neurogenesis, and would include an overabundance of dendrites and axons, and/or a failure to prune. Morphologic examination in mouse models have shown an excess of neuronal arborization in the Pten-mutant mouseCitation21 and a impoverishment of neuronal arbors in the Mecp2-null mouse.Citation22
Genomic programs underlying experience-dependent synapse plasticity utilize hundreds of genes
An experimental proxy for studying the processes of synaptic plasticity involves studying the gene networks that are regulated by neuronal activity or more specifically, neuronal membrane depolarization in cell culture systems. Genome -wide studies of the regulation of gene expression by neuronal activity have suggested that a large percentage of the genes in the genome may be regulated by neuronal activity and thereby, may play some role in synaptic plasticity. Is it conceivable that a large number of these genes may be mutated or play a role in autism? Gene variants at a significant number of these loci may contribute to autism in a complex genetic fashion. Regarding the vast genetic heterogeneity that may be at play in autism, it is worth considering the genetic architecture of intellectual disability (ID). Of course, ID may be related to autism in many cases, as approximately 38% of children with autism also have co-occurring ID.Citation2 ID is caused by a large variety of mutations, including chromosomal as well as many monogenic mutations such as X-linked loci. Indeed, greater that 10% of the genes on the X chromosome may be associated with ID.Citation23 By the lessons of ID, there are genetic mutations that would perturb just about all steps of neurodevelopment (Figure 1): however, if we restrict the clinical scope to “non-syndromic” intellectual disability (ie, cognitive effects without structural brain or medical effects), the mechanisms may be more refined to synaptic structure and in particular dendritic spine abnormalities.Citation24 Here, we also contend that those steps of neurodevelopment that are involved in autism are similarly constrained, and we will argue here that they are constrained to those steps that affect the formation of neuronal circuitry, ie axon and dendrite growth and arborization, and experience-dependent synaptic modification.
Heterogeneous gene mutations in autism
Genetic studies in ASD have made substantial progress in the last decade. Numerous, individual mutations, largely corresponding to rare genetic variants, have been discovered.Citation4,Citation25 These studies have elucidated a variety of genetic loci and pathways regarding the genetic architecture of autism. No single locus in question appears to be found in greater than 1%, and the majority of loci are recurrent at a much lower rate, and some representative of private (single mutations). The nature of the rare mutations include gross chromosomal anomalies, copy number variants, single nucleotide variants, particularly de novo variants.Citation26-Citation30
These mutations have pinpointed a heterogeneous group of genes and loci that may contribute to the pathobiology of autism. These mutations appear to affect a range of mechanisms (Table I) including those that regulate: (i) gene expression; (ii) pre-mRNA splicing; (iii) protein localization, translation, and turnover; (iv) synaptic transmission, such as synaptic vesicle release and membrane excitability; (v) cell signaling; (vi) cytosketal and scaffolding proteins particularly at the postsynaptic membrane; and (vii) neuronal cell adhesions molecules. With each discovery of a new mutation, researchers are forced to ask: What stage of neurodevelopment is perturbed by this genetic mutation? Answering this question is essential to understanding the genetic contributions of autism. The purpose of this review is to critically address this question for the putative molecular mechanisms in autism that are outlined below. One major benefit to this approach is, while there are dozens of potential cellular mechanisms and hundreds of genes, the function of the mutation must converge on a given step of neurodevelopment of which there may be a more limited number of steps (Figure 1) and may be the ultimate target of treatment.
TABLE I. Molecular mechanisms and genes implicated in autism map onto a limited number of steps in neurodevelopment. *We refer readers to https://gene.sfari.org/autdb/Welcome.do for references regarding each gene mutation.
Gene expression and chromatin regulation
Increasing evidence suggests disruption of gene expression programs and/or expression of multiple genes at once can lead to cognitive disorders. We can make sense of how alterations in global gene expression may disrupt cognitive processes, as many facets of synapse maturation and function require the fine-tuned regulation of multiple genes. Perhaps most relevant to cognition, neural plasticity exercises activity-dependent modulation of gene expression and depends on the process of chromatin remodeling and coordinate gene transcription involving a large number of genes.Citation31-Citation33 A recent transcriptome analysis of post-mortem autistic brain tissue suggests a probable link between global dysregulation of transcription and autism.Citation34 Utilizing microarrays, the authors identified a little over four hundred genes showing differential expression between autistic and control cortex samples. Key developmental events like dendrite formation and synaptic pruning appear to require molecules involved in chromatin remodeling.Citation35,Citation36 Indeed, converging lines of genetic evidence suggest a role for such chromatin modification machinery in the pathogenesis of autism.
Perhaps the strongest insight into this mechanism comes from over a decade of research of the MECP2 gene, mutations of which lead to the development of RTT, which shares similarities to autism. MECP2 encodes methyl-CpG binding protein 2 (MeCP2) whose molecular function was originally believed to be a direct repressor of gene expression.Citation37 However, recent knockout and transgenic experiments reveal MeCP2 mediates both transcriptional activation as well as repression.Citation38 This most likely is a result of MeCP2 function on chromatin remodeling. For example, upon loss of MeCP2, histone H3 acetylation elevates globally and the levels of histone H1 double.Citation39 With regards to the protein's effect on cognition, neuronal activity leads to phosphorylation of MeCP2, and in vivo prevention of this phosphorylation results in neuronal and behavioral deficits characteristic of a loss in experience -dependent chromatin remodeling during nervous system development:Citation40 This link between MeCP2 and autism hints at a possible role for other methyl-CpG binding proteins in the pathogenesis of the disorder, and in fact, a study of 226 autistic individuals by Cukier et al identified 46 variants spread across four such proteins (MBD1-4).Citation41 In addition, a recent analysis of 2q23.1 microdeletion syndrome, which also shares similarities to autism, pinpointed MBD5 as the causative locus.Citation42 Further association of MBD5 with autism has been shown via sequencing of autistic individuals with chromosomal abnormalities.Citation43 Interestingly, three recent independent sequencing studies implicated another gene involved in chromatin remodeling: chromodomain-helicase-DNA-binding protein 8 (CHD8).Citation27,Citation28,Citation43 One study also identified de novo events in CHD3 and CHD7.Citation28 Individuals with mutations in CHD7 develop CHARGE syndrome, 68% of whom exhibit an autistic-like phenotype.Citation44 Furthermore, the histone methyltransferase EHMT1, which is responsible for another syndromic form of autism called Kleefstra's syndrome,Citation45 was identified in two of these studies.Citation28,Citation43 Another recent exome sequencing study of 343 simplex families identified 13 candidate genes involved in either transcription regulation or chromatin remodeling.Citation30 As a whole, these findings suggest that autism may arise as a result of impaired regulation of the chromatin state. Such dysregulation may result in improper synaptic wiring of brain circuitry and/or prevent the proper neuronal response from external stimuli necessary for the development of social cognition. Further analyses into the relationship between neuronal activity and chromatin remodeling are necessary to garner clues for how the two may orchestrate circuit formation.
Large recurrent copy number variants (CNVs) have been associated with autism.Citation46 Careful consideration of the molecular effects of such a genetic locus is warranted. On first consideration, it is likely that the majority of such loci alter the dosage or gene expression level of a number of contiguous genes. Is one gene involved in these loci or is it a combination of genes? For the majority of CNVs, it seems most likely that the latter model will prevail, that CNVs lead to a complex interaction of the effects of perturbed gene expression from multiple contiguous genes. In some ways, the loss of a gene that modulates gene expression such a chromatin modifying gene may have similar effects, ie perturbation of dosage of a collection of genes.
Pre-mRNA splicing
Disruption of A2BP1/FOX1, a gene involved in mediating RNA splicing, has been noted in two autistic individuals.Citation47-Citation48 This is especially intriguing in that another category of genes implicated in autism—cell adhesion molecules (CAMs)—exhibit numerous alternatively spliced transcripts that appear crucial for cell-cell recognition.Citation49 The aforementioned transcriptome analysis not only revealed A2BP1 to be downregulated in comparison to control tissue but also determined many of the protein's targets were genes involved in synaptic function.Citation34 In line with the CNV data, this suggests that the disorder may come about not from the perturbation of a single gene whose function is localized to the synapse but rather a disruption of several components of the synaptic machinery. This is a multigenic model again, even though the primary genetic effect may have a single major effect locus.
Thus, with regard to the molecular mechanism of global regulation of gene expression, multiple studies demonstrate that neurodevelopmental processes are sensitive to the dosage of a wide variety of genes, likely contributing to autism. Such processes most likely include experience-dependent modulation of neural networks via synaptogenesis and synaptic plasticity because such events appear to rely on a large and dynamic array of genes rather than some other genetically preprogrammed response that may be more confined in gene usage and thereby show more Mendelian inheritance. This also may explain why more overt signs of autism do not manifest until a later “critical period” of cognitive development and perhaps why there is a period of normal development in RTT patients followed by a regression in development. Such a regression may reflect an inability of neurons and neuronal circuits to properly adapt to environmental stimuli.
Protein localization, translation, and turnover
The synapse plays host to a number of critical events for proper neuronal function including neurotransmitter release, synaptic vesicle recycling, and postsynaptic receptor activation and recycling. Such a dynamic environment poses a challenge for the cellular machinery responsible for protein synthesis and degradation because numerous molecules must work together in a precise manner to mediate these events and produce downstream effects like activity-dependent synaptic plasticity. Thus, it is conceivable that disruptions of any single one of these components could have a deleterious effect at the synapse. Alternatively, we can imagine a molecular mechanism whereby multiple features of the synaptic machinery are altered via the perturbation of an upstream regulator of these features, such as local protein regulation. Current genetic data seems to suggest both mechanisms contribute to the pathogenesis, however, they converge on neurodevelopmental processes dependent on the synapse. For example, mutations contributing to syndromic forms of autism have been discovered in fragile X mental retardation 1 (FMR1) and cytoplasmic FMR1-interacting protein 1 (CYFIP1), which are genes encoding for negative translational regulators.Citation50 Loss of function of such genes consequently enhances local protein translation altering synaptic plasticity. In fact, local translational regulation was first revealed as a central mechanism in proper neurodevelopment by studies of FXS, a disorder caused by hypermethylation of FMR1 and subsequent loss of fragile X mental retardation protein (FMRP) expression. FMRP represses translation at the synapse by stalling ribosomes on target mRNA transcriptsCitation51 and is critical for mGluR-mediated long-term depression.Citation52 Fmr1 knockout mice confirm the global upregulation of transcripts. In FXS, synaptic transcript products upregulate through FMRP's failure to recruit CYFIP1, a cytoplasmic FMRl-interacting protein that is also a eukaryotic translation initiation factor 4E (eIF4E) binding protein. Interestingly, loss of FMRP in both mice and humans results in abnormal dendritic spine morphology, a characteristic of many intellectual disability-associated disorders.
Case studies of nonsyndromic forms of autism have identified de novo variants in genes involved in translational control. Of note, eIF4E is downstream of several of these signaling pathways, and mutations directly in eIF4E have been discovered in three autistic individuals.Citation53 This study found de novo gene disruptions in 14 autism candidate genes and 13 CNVs that overlapped with FMRP target genes,Citation30 which supports the notion that FXS-associated autistic phenotypes may result from disrupted expression levels of specific gene products. It is hypothesized that disrupted protein translation may lead to abnormal neuronal morphology and, hence, abnormal synaptogenesis. This faulty brain connectivity may be responsible for the global impairment in learning and memory associated with disorders of intellectual disability like FXS. However, comorbid autism in these disorders could reflect a disruption of the same developmental mechanism but perhaps of more specialized circuits or synapses responsible for social learning.
Many candidate genes for ASD pathology map onto the endosomal pathway.Citation54 A family of protein exchangers localized on endosomes have recently been studied in syndromic autism. Christiansen syndrome, which presents like Angelman syndrome, has been associated with the functional loss of the endosomal Na+/H+ exchanger NHE6 (also known as SLC9A6).Citation55,Citation56 Many cases of nonsyndromic autism have been linked to deficits in cellular trafficking of proteinsCitation57; an autistic individual with a chromosomal inversion that disrupts receptor expression-enhancing protein 3 (REEP3), a putative regulator of vesicle trafficking between the ER and Golgi networkCitation58; two individuals with mutations in the small GTPase RAB39B and an individual with a haploinsufficiency of the small GTPase RAB39B Citation59; a translocation that disrupts the NEUROBEACHIN (NBEA) gene.Citation58 This evidence suggests mostly GTPases and their regulators of the recycling endosomes at the presynapse are affecting the transport of specific cargo. A recent study reported that collapse of the recycling endosome results in a decrease in spine density in an activity-dependent manner.Citation61
We can predict how mutations in genes involved in protein trafficking can directly affect neurite morphogenesis and synaptogenesis. Cellular trafficking of proteins is also indirectly critical for membrane dynamics underlying mechanisms of synaptic plasticity and neurotransmission. For example, the polarized expression of contactin-associated protein 2 (CAPSR2), a cell adhesion molecule in the neurexin family, relies on compartment-specific endocytosis.Citation62 A study by Bel et al showed how inhibition of endocytosis leaves CAPSR2 inserted in the somatodendritic compartment. Multiple studies found individuals with autism and/or related disorders with mutations in the CAPSR2 locus of CNTNAP2. Citation62-Citation64
Recent studies have implicated vesicular trafficking of brain-derived neurotrophic factor (BDNF) via secretory vesicles with reduced dendritic complexity, as well as significant differences in dendritic spine numbers and morphological spine types.Citation65 Whether BDNF mediates activity-dependent dendritic spine plasticity during learning and memory in vivo is unclear, but it remains a strong candidate as a factor to structurally prepare excitatory synapses for consolidation of hippocampal-dependent learning that provides evidence for a morphological basis for the synaptic deficiencies thought to underlie autism.
Various components of the multicomplex ubiquitin-proteasome system (UPS) are necessary for proper development of the brain, axon outgrowth and guidance, synapse development and plasticity.Citation61 Tight regulation of protein degradation is critical in neurodeveiopment and neurodegeneration. Glessner and colleagues reported evidence of CNVs associated with the ubiquitin pathway as a source of ASD susceptibility.Citation66 Glessner et al found that four genes (ubiquitin-protein ligase E3A [UBE3A], parkinson protein 2 [PARK2], ring finger and WD repeat domain 2 [RFWD2], F-box protein 40 [FBXO40]) were significantly enriched for CNVs only in autism, in addition to cell-adhesion molecules. Ubiquitination post-translationally modifies protein function and targets cytoplasmic polyubiquitinated proteins for 26S proteasome-mediated degradation.Citation67 Monoubiquitinated transmembrane proteins can be targeted for the lysosomal degradation or sorting for the endosomal pathway.Citation68 UBE3A, an E3 ubiquitin-protein ligase, has been extensively studied in relation to Angelman syndrome, a disorder caused by mutations or deletions of the maternal UBE3A allele and often presenting with autistic features.Citation69,Citation70 Mutations of PARK2, another ubiquitin-protein ligase, have been associated with juvenile-onset Parkinson disease, RFWD2 and FBX04 are also ubiquitin-protein ligases without previously associated disease-causing mutations. Other ubiquitin protein E3 ligases and UBE2A (E2 ubiquitin-conjugating enzyme) have been implicated in syndromic intellectual disability.Citation71
Mouse models for Angelman syndrome exhibit abnormal connectivity and synaptic development.Citation70 UPS in the Reelin-signaling cascade is relevant for proper synaptic connectivity. Reelin is a large glycoprotein that coordinates the migration of different neuronal populations in the cortex of the mammalian central nervous system.Citation72 Reelin binds to the very-low-density lipoprotein receptor (VLDLR) and the ApoE receptor 2 on target neurons.Citation73 ApoER2-deficient mice have defects in LTP because of the inability to downregulate Dabi, a cytoplasmic adaptor protein that is targeted by the UPS after Reelin signaling.Citation74 UPS mediates the intricate balance between protein synthesis and degradation to help navigate axons from extrinsic guidance cues to their target destinations. Ubiquitin is required to clear Robo from the growth cone surface and reduce the repellent effect so the growth cone can be guided across the midline.Citation68 The UPS also prevents the re -crossing with Robo upregulation and mediates the attraction to the midline by the Netrin-DCC/Fra system.Citation75,Citation76 Ubiquitination and de ubiquitination are also critical for the modification of synapse strength, which requires the insertion and removal of glutamate receptors.
Membrane excitability, synaptic vesicle maturation, and synaptic transmission
Many “synaptic” genes responsible for steps in synaptic maturation and/or neurotransmission have been identified as candidates for ASD susceptibility, including both postsynaptic (NLGN3, NLGN4, SHANK2/3, IL1RAPL1) and presynaptic proteins (NRXN1, CNTNAP2, RIMS3/NIM3).Citation77 These loci have been identified through rare yet generally recurrent clinical cases and have led to a prevailing hypothesis that autistic phenotypes are due to abnormal synaptic function and/or neural connectivity in the time window in which neuronal circuits are extensively remodeled by experience.Citation78 Underlying this hypothesis of ”synaptopathy“ is the dysfunction of excitation and inhibition in neural circuits, potentially from aberrant synaptic vesicle release,Citation79 Abnormal synaptic vesicle release would predictably alter long-term potentiation and long-term depression needed for synaptic plasticity. Several lines of evidence converge to support the hypothesis that a subgroup of autistic phenotypes may be due to abnormal synaptic vesicle maturation and release.Citation78
One study identified a Q555X mutation in synapsin 1 (SYN1), an X-linked gene encoding for a neuron-specific phosphoprotein implicated in the regulation of neurotransmitter release and synaptogenesis, in French-Canadian individuals with comorbid ASD and epilepsy.Citation80 Animal models with this mutation show impaired synaptic vesicle density and availability for the readily releasable pool. SYN3 functions in synaptogenesis and the modulation of neurotransmitter release.Citation81,Citation82
Some evidence suggests that abnormal neurotransmitter release found in autistic patients may be cell-specific and functionally alter firing patterns. Unc13a-null mice demonstrate impairment of glutamatergic synaptic vesicle maturation.Citation83 One study found three independent patients with autism that have microdeletions at NBEA and AMISYN, negative regulators of low-dense core vesicle secretion affected. Acute slices from the brain stem of Nbea knockout animals showed both reduced spontaneous excitatory and inhibitory postsynaptic currents and increased failure rates of evoked inhibitory responses, consistent with the finding that Nbea affects stimulus-release coupling, vesicle fusion, postsynaptic signal amplitude, formation or maturation of synaptic contacts.Citation84 In addition, the frequency of miniature excitatory and both the frequency and amplitudes of miniature inhibitory postsynaptic currents were severely diminished in knockout mice, indicating a perturbation of both action potential-dependent and -independent transmitter release.
Cell signaling
Disruptions in components of signaling pathways could lead to very diffuse downstream events on two previously mentioned molecular mechanisms—transcription and translation—that effect a number of neuronal processes crucial for proper development of the nervous system. Several such genes have been identified, and of these, the tumor growth suppressors TSC1, TSC2, and phosphatase and tensin homolog (PTEN) are the most intensely investigated. Mutations in TSC1 and TSC2 cause TSC by impeding the recruitment of EIF4E downstream the mTOR pathway for cap-dependent initiation of translation.Citation85 With regard to the central nervous system, TSC is marked by the formation of cortical tubers—hamartomas within brain tissue—that result in a number of neurological manifestations including seizures, intellectual disability, and autism. Although these symptoms may arise due to disruptions of surrounding brain tissue by these cortical tubers, developing evidence suggests that TSC1 and TSC2 mutations could also have specific effects on neuron function. For example, disruptions of the TSC signaling pathway result in enlarged neurons, disrupted spine growth and morphology, and alteration of glutamatergic synapses.Citation86 The two genes also mediate axonal growthCitation87 and hippocampal mGluR-mediated long-term depression.Citation88 In addition, conditional knockout of TSC1 in GABAergic neurons of mice resulted in impaired postnatal growth, decreased numbers of interneurons, impaired neuronal migration, and a lowered seizure potential, suggesting the neurological deficits in individuals with TSC could arise from disruptions of signaling pathways in specific neuronal subtypes.Citation89 Variants in TSC1 and TSC2 may be potential susceptibility factors for autism separate from TSC. Evidence for this comes from a recent study utilizing high-throughput sequencing on nonsyndromic autistic individuals to analyze genes involved in the mGluR signaling pathway. The authors identified a number of rare, potentially disruptive single nucleotide variants in TSC1 and TSC2 never before seen in individuals with TSC.Citation90
A subset of individuals with PTEN mutations have nonsyndromic autism without the presence of tumors. A recent study suggests these individuals have mutations that preserve PTEN function, whereas PTEN hamartoma tumor-related syndrome associated mutations cause a loss of function.Citation91 PTEN is involved in dephosphorylation of the second messenger PIP3 and subsequent activation of the PI3K/Akt/mTOR signaling pathways. Like with TSC1/TSC2, mutations in PTEN could have a direct effect on neuronal function or neurodevelopment separate from indirect effects by tumor formation that results in autism pathogenesis. Conditional knockout of PTEN in adult progenitor cells of the subgranular zone of the hippocampus results in a depletion of the stem cell pool and development of hypertrophied neurons with abnormal polarity.Citation92 Furthermore, conditional deletions of PTEN to discrete neuronal subpopulations in mice result in abnormal dendrite and axonal growth.Citation93
There is also genetic evidence for impaired signaling beyond the mTOR pathway. For example, CNVs on chromosome 16 that disrupt the MAPK3 gene encoding extracellular signal-related kinase 1 (ERK1) are associated with autismCitation94,Citation95 and pinpoint disruptions of Ras/Raf/ERK1/2 signaling as a possible contributor to autism.Citation96 Upregulation of this pathway results in impaired neuronal cell migration, neurogenesis, synapse formation, and dendritic spine development.Citation97 Also, two of the recent sequencing studies already discussed implicate dual-specificity tyrosine- (Y) -phosphorylation regulated kinase 1 A (DYRK1A), Citation28,Citation30 a serine/threonine kinase involved in Down syndrome that regulates neuronal morphogenesis via cytoskeletal dynamics.Citation98 Taken together, combined in vitro and in vivo studies would suggest impairments in intracellular signaling could lead to alterations in neuronal morphology and synaptic connections. Therefore, the genetic evidence in this case highlights disruptions of activity-independent neurodevelopmental mechanisms as a contributing factor to autism, especially those of neurite outgrowth. Such deficits, in turn, could mimic the effects of epigenetic perturbations despite functioning activity-dependent processes since faulty neuronal wiring could produce an ineffective neuronal foundation for intepreting external stimuli.
Postsynaptic density and cytoskeletal mechanisms
Scaffolding proteins provide multimeric protein-protein interaction domains that localize key synaptic proteins and signaling molecules to the postsynaptic terminals, enabling effective neurotransmission and synaptic plasticity necessary for normal cognitive development in the brain. From autism de novo CNV studies, some critical genes that have been identified such as SHANK2 Citation99 and SHANK3. Citation100 Many of these genes are also implicated in other neurodevelopmental disorders with potentially overlapping mechanisms such as schizophrenia.Citation46 Disrupting the function of these scaffolding proteins directly impairs the synapse organization and stabilization, and neurite outgrowth. These cellular and physiological consequences were confirmed in knockdown animal models of SHANK2, which had smaller dendritic spines and reduced AMPA receptor currents.Citation99 Dendritic morphology is intimately correlated to synaptic transmission and processing, and SHANK2 demonstrates how dysfunction of structural organization can lead to the physiological autistic phenotype of imbalanced excitatory and inhibitory currents. Current research is pursuing SHANK isoforms—one study suggests microdeletions on SHANK1 may lead to a high functioning autistic phenotype.Citation101 Initially, SHANK3 was disrupted by a de novo balanced translocation in a child with all the features of the 22q13.3 deletion syndrome and subsequent studies have confirmed SHANK3 deletions may be limited to lower functioning autism.Citation102,Citation103 The different autistic phenotypes from the various SHANK isoforms may be due to the temporal differences in recruitment into the postsynaptic density.Citation104 Recently discovered ASD candidate genes seem to center around scaffolding proteins and cell adhesion molecules, suggesting a point of convergence similar to the story unfolding for the PI3K-AKT-mTOR pathway.
Neuron cell adhesion molecules
Syndromic autism has been linked directly to mutations of genes modulating neuronal cell-adhesion molecules, which are involved in the formation, signaling, and plasticity of synaptic connections. Neuronal cell-adhesion molecules are necessary for axonal guidance and neuronal-glial interactions. Neuroligin superfamily members and numerous cell-adhesion molecules have been paths of convergence for many other complex neurodevelopmental disorders including intellectual disability and schizophrenia.Citation105 Various mutations in idiopathic autism were found: structural variations of NRXN1, Citation106 microdeletions in CNTNAP2, Citation107 R451C substitution in NLGN3, Citation108 ten mutations (2 frameshifts, 5 missense, 3 internal deletions) in NLGN4X,Citation108 and de novo CNVs in other cadherins.Citation66 NLGN3 and NLGN4 mutant mice display an autistic phenotype, and exhibit abnormal inhibitory and excitatory synaptic transmission.Citation109 These studies also support the finding that neuroligins are critical for synaptic function and transmission, not necessarily for synapse formation.Citation110
However, the role of the neuroligin-neurexin mechanism in autism remains unclear. NLGN3 and NLGN4 mutations appear to be always penetrant in males, and even female carriers with these mutations often have a phenotype, but SHANK3 point mutations are also found in the probands' nonsymptomatic relatives.Citation100 Furthermore, these mutations can lead to different phenotypes. A child with a NLGN4 microdeletion had severe autism, whereas his sibling developed Tourette syndrome.Citation111 For instance, a linkage study found a common polymorphism in CNTNAP2, another member of the presynaptic neurexin superfamily, is significantly associated with autism and a variant displays a parent-of-origin and gender pattern of inheritance,Citation112 Studying the presynaptic side of cell adhesion has been complicated, but neurexin-KO mice showed impaired neurotransmitter release and reduced NMDA-dependent synaptic responses.Citation113 Mutations in these genes may only raise the susceptibility and not always confer the disorder. These genomic studies allow us to see patterns and consider pathway interactions. For instance, ubiquitin is critical in the turnover of neuronal cell-adhesion molecules in regulating activity-dependent synaptic plasticity, which suggests another convergence point for pathways.Citation66
A brief comment on “epigenetic” mechanisms in autism
Epigenetics is the study of heritable phenotypes caused by mechanisms other than changes in genomic sequence and that are instead frequently due to modifications of chromatin, such as methylation of DNA or various covalent histone modifications. Some authors erroneously use the term “epigenetics” to refer to effects on gene expression mediated by modification of chromatin, ie, they leave out the critical aspect requiring inheritance of these changes and the associated phenotype, or sometimes the term is used to invoke changes in chromatin mediated by environmental experience again leaving out the requirement for inheritance.Citation114 With regard to the topic of the genetics of autism, some recent studies have suggested that in some cases autism may arise due to alterations in chromatin modifications and subsequent gene expression programs, instead of due to alterations in genomic sequence.Citation115 This exciting novel hypothesis may require new methods of studying patient gene expression and also, will lead researchers to test if indeed these disease-associated chromatin modifications are heritable, ie, epigenetic, Interestingly, many of the loci that have emerged in these studies of chromatin modification are indeed genes that have been previously implicated by genetic studies, although some are novel.
Many genes may converge on only a fews steps in neurodevelopment: relevance treatment response
Treatment interventions in autism usually include a combination of psychopharmacological and behavioral/developmental/educational methods. While we are fortunate that there are choices for treatment and these treatments are effective or partially effective for a subset of patients, unfortunately there is also a large subset of patients who do not respond to current treatments.Citation116 Recent progress from autism genetics has provided some light on a path towards understanding pathophysiology mechanisms, improved diagnoses and improved treatments. One critical question is: What are the differences at the level of brain mechanism that permit some patients to respond while others do not respond? Our argument in this review is that genetics and other lines of autism research have pinpointed abnormalities in the development of circuitry in autism. Yet, there are several developmental mechanisms that when perturbed can lead to a “connectopathy.” Here we argue that there is evidence in at least some cases for morphologic abnormalities in “wiring” such as abnormal axon and/or dendritic growth and branching. In addition, genetics has provided a large amount of evidence arguing that later stages of synapse development and plasticity may be central in other patients. The large number of protein targets that have emerged in the last years provides a great deal of hope that targeting some of these mechanisms during development may provide novel treatment strategies for some patients. Mechanism-based subtyping of autism perhaps through the use of genetic testing may lead to stratified treatment paths with improved likelihood of response to given treatments.
Selected abbreviations and acronyms
| ASD | = | autism spectrum disorder |
| CNV | = | copy number variation |
| FMRP | = | fragile X mental retardation protein |
| FXS | = | fragile X syndrome |
| ID | = | intellectual disability |
| RTT | = | Rett syndrome |
| TSC | = | tuberous sclerosis |
| UPS | = | ubiquitin-proteasome system |
We would like to thank Judith Nathanson for her assistance with the illustrations. Also, we would like to thank members of the Morrow laboratory for critical reading of the manuscript. EMM has received a Career Award in Medical Science from the Burroughs Wellcome Fund and support from NIMH 1K23MH080954-05, NIGMS 5P20RR018728-09. None of the authors have a financial conflict of interest.
REFERENCES
- GanzML.The lifetime distribution of the incremental societal costs of autismArch Pediatr Adolesc Med.200716134334917404130
- Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, 14 sites. United States, 2008MMWR Surveill Summ.201261119
- AndersonDK.OtiRS.LordC.WelchK.Patterns of growth in adaptive social abilities among children with autism spectrum disordersJ Abnorm Child Psychol.2009371019103419521762
- StateMW.LevittP.The conundrums of understanding genetic risks for autism spectrum disordersNat Neurosci.2011141499150622037497
- WalshCA.MorrowEM.RubensteinJL.Autism and brain developmentCell.200813539640018984148
- TauGZ.PetersonBS.Normal development of brain circuits.Neuropsychopharmacology.20103514716819794405
- MochidaGH.Genetics and biology of microcephaly and lissencephalySemin Pediatr Neurol.20091612012619778709
- DubourgC.BendavidC.PasquierL.HenryC.OdentS.DavidV.HoloprosencephalyOrphanet J Rare Dis.20072817274816
- GleesonJG.WalshCA.Neuronal migration disorders: from genetic diseases to developmental mechanismsTrends Neurosci.20002335235910906798
- SattarS.GleesonJG.The ciliopathies in neuronal developments clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disordersDev Med Child Neurol.20115379379821679365
- SahinM.Targeted treatment trials for tuberous sclerosis and autism: no longer a dreamCurr Opin Neurobiol. In press.
- PivenJ.BaileyJ.RansonBJ.ArndtS.An MRI study of the corpus callosum in autismAm J Psychiatry.1997154105110569247388
- BoothR.WallaceGL.HappeF.Connectivity and the corpus callosum in autism spectrum conditions: insights from comparison of autism and callosal agenesisProg Brain Res.201118930331721489396
- HuttenlocherPR.Synapse elimination and plasticity in developing human cerebral cortexAm J Ment Defic.1984884884966731486
- HuttenlocherPR.de CourtenC.GareyLJ.Van der LoosH.Synaptogenesis in human visual cortex - evidence for synapse elimination during normal developmentNeurosci Lett.1982332472527162689
- KruegerDD.BearMF.Toward fulfilling the promise of molecular medicine in fragile X syndromeAnnu Rev Med.20116241142921090964
- MeguidNA.FahimC.SamiR.et al.Cognition and lobar morphology in full mutation boys with fragile X syndromeBrain Cogn.201278748422070923
- MilesJH.HaddenLL.TakahashiTN.HillmanRE.Head circumference is an independent clinical finding associated with autismAm J Med Genet.20009533935011186888
- CourchesneE.PierceK.SchumannCM.et al.Mapping early brain development in autismNeuron.20075639941317964254
- ButlerMG.DasoukiMJ.ZhouXP.et al.Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germ line PTEN tumour suppressor gene mutationsJ Med Genet.20054231832115805158
- Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006MMWR Surveill Summ.200958120
- BelichenkoPV.WrightEE.BelichenkoNP.et al.Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networksJ Comp Neurol.200951424025819296534
- GeczJ.ShoubridgeC.CorbettM.The genetic landscape of intellectual disability arising from chromosome XTrends Genet.20092530831619556021
- Bohlenvon.HalbachO.Dendritic spine abnormalities in mental retardationCell Tissue Res.201034231732321080001
- BillBR.GeschwindDH.Genetic advances in autism: heterogeneity and convergence on shared pathwaysCurr Opin Genet Dev.20091927127819477629
- SandersSJ.MurthaMT.GuptaAR.et al.De novo mutations revealed by whole-exome sequencing are strongly associated with autismNature.201248523724122495306
- NealeBM.KouY.LiuL.et al.Patterns and rates of exonic de novo mutations in autism spectrum disordersNature.201248524224522495311
- O'RoakBJ.VivesL.GirirajanS.et al.Sporadic autism exomes reveal a highly interconnected protein network of de novo mutationsNature.201248524625022495309
- O'RoakBJ.DeriziotisP.LeeC.et al.Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutationsNat Genet.20114358558921572417
- lossifovI.RonemusM.LevyD.et al.De novo gene disruptions in children on the autistic spectrumNeuron.20127428529922542183
- MartinowichK.HattoriD.WuH.et al.DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulationScience.200330289089314593184
- KorzusE.RosenfeldMG.MayfordM.CBP histone acetyltransferase activity is a critical component of memory consolidationNeuron.20044296197215207240
- GuanZ.GiustettoM.LomvardasS.et al.Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structureCell.200211148349312437922
- VoineaguI.WangX.JohnstonP.et al.Transcriptomic analysis of autistic brain reveals convergent molecular pathologyNature.201147438038421614001
- WuJl.LessardJ.OlaveIA.et al.Regulation of dendritic development by neuron-specific chromatin remodeling complexesNeuron.2007569410817920018
- KirillyD.WongJJ.LimEK.et al.Intrinsic epigenetic factors cooperate with the steroid hormone ecdysone to govern dendrite pruning in DrosophilaNeuron.2011728610021982371
- NanX.CampoyFJ.BirdA.MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatinCell.1997884714819038338
- Ben-ShacharS.ChahrourM.ThallerC.ShawCA.ZoghbiHY.Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. HumMol Genet.20091824312442
- SkenePJ.lllingworthRS.WebbS.et al.Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin stateMol Cell.20103745746820188665
- CohenS.GabelHW.HembergM.et al.Genomewide activitydependent MeCP2 phosphorylation regulates nervous system development and functionNeuron.201172728521982370
- CukierHN.RabionetR.KonidariI.et al.Novel variants identified in methyl-CpG-binding domain genes in autistic individualsNeurogenetics.20101129130319921286
- TalkowskiME.MullegamaSV.RosenfeldJA.et al.Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder.Am J Hum Genet.20118955156321981781
- TalkowskiME.RosenfeldJA.BlumenthalI.et al.Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries.Cell.201214952553722521361
- JohanssonM.GillbergC.RastamM.Autism spectrum conditions in individuals with Mobius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum diagnostic aspects.Res Dev Disabil.20103192419709852
- KleefstraT.SmidtM.BanningMJ.et al.Disruption of the gene Euchromatin Histone Methyl Transferasel (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome.J Med Genet.20054229930615805155
- MorrowEM.Genomic copy number variation in disorders of cognitive development.J Am Acad Child Adolesc Psychiatry.2010491091110420970697
- MikhailFM.LoseEJ.RobinNH.et al.Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders.Am J Med Genet A.2011155A2386239622031302
- MartinCL.DuvallJA.llkinY.et al.Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism.Am J Med Genet B Neuropsychiatr Genet.2007144B86987617503474
- ShapiroL.LoveJ.ColmanDR.Adhesion molecules in the nervous system structural insights into function and diversity.Annu Rev Neurosci.20073045147417600523
- KelleherRJ.3rdBearMF.The autistic neuron: troubled translation?Cell.200813540140618984149
- DarnellJC.Van DriescheSJ.ZhangC.et al.FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism.Cell.201114624726121784246
- HuberKM.GallagherSM.WarrenST.BearMF.Altered synaptic plasticity in a mouse model of fragile X mental retardation.Proc Natl Acad Sci USA.2002997746775012032354
- Neves-PereiraM.MullerB.MassieD.et al.Deregulation of EIF4E:a novel mechanism for autism.J Med Genet20094675976519556253
- YapCC.WincklerB.Harnessing the power of the endosome to regulate neural development.Neuron.20127444045122578496
- SchroerRJ.HoldenKR.TarpeyPS.et al.Natural history of Christiansen syndrome.Am J Med Genet A.2010152A2775278320949524
- XinhanL.MatsushitaM.NumazaM.TaguchiA.MitsuiK.KanazawaH.Na+/H+ exchanger isoform 6 (NHE6/SLC9A6) is involved in clathrin-dependent endocytosis of transferrin.Am J Physiol Cell Physiol.2011301C1431C144421881004
- OrricoA.GalliL.BuoniS.OrsiA.VonellaG.SorrentinoV.Novel PTEN mutations in neurodevelopmental disorders and macrocephaly.Clin Genet.20097519519818759867
- CastermansD.WilquetV.ParthoensE.et al.The neurobeachin gene is disrupted by a translocation in a patient with idiopathic autism .J Med Genet.20034035235612746398
- GiannandreaM.BianchiV.MignognaML.et al.Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly.Ami J Hum Genet.201086185195
- RoohiJ.TegayDH.PomeroyJC.et al.A de novo apparently balanced translocation [46,XY,t(2;9)(p13;p24)] interrupting RAB11FIP5 identifies a potential candidate gene for autism spectrum disorder.Am J Med Genet B Neuropsychiatr Genet.2008147B41141718384058
- ParkM.SalgadoJM.OstroffL.et al.Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes.Neuron.20065281783017145503
- BelC.OguievetskaiaK.PitavalC.GoutebrozeL.Faivre-SarrailhC.Axonal targeting of Caspr2 in hippocampal neurons via selective somatodendritic endocytosis.J Cell Sci.20091223403341319706678
- BurbachJP.van der ZwaagB.Contact in the genetics of autism and schizophrenia.Trends Neurosci.200932697219135727
- GirirajanS.BrkanacZ.CoeBP.et al.Relative burden of large CNVs on a range of neurodevelopmental phenotypes.PLoS Genet.20117e100233422102821
- ChapleauCA.LarimoreJL.TheibertA.Pozzo-MillerL.Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking:fundamental roles in neurodevelopmental disorders associated with mental retardation andautism. J Neurodev Disord.20091185196
- GlessnerJT.WangK.CaiG.et al.Autism genome-wide copy number variation reveals ubiquitin and neuronal genes.Nature.200945956957319404257
- JoazeiroCA.HunterT.Biochemistry. Ubiquitination - more than two to tango.Science.20002892061206211032556
- MurpheyRK.GodenschwegeTA.New roles for ubiquitin in the assembly and function of neuronal circuits.Neuron.2002365812367500
- SmithSE.ZhouYD.ZhangG.JinZ.StoppelDC.AndersonMP.Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice.Sci Transi Med.20113103ra97
- MabbAM.JudsonMC.ZylkaMJ.PhilpotBD.Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes.Trends Neurosci.20113429330321592595
- NascimentoRM.OttoPA.de BrouwerAP.Vianna-MorganteAM.UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel Xlinked mental retardation syndrome.Am J Hum Genet.20067954955516909393
- FatemiSH.Reelin glycoprotein in autism and schizophrenia.Int Rev Neurobiol.20057117918716512351
- ArnaudL.BallifBA.CooperJA.Regulation of protein tyrosine kinase signaling by substrate degradation during brain development.Mol Cell Biol.2003239293930214645539
- PatrickGN.Synapse formation and plasticity:recent insights from the perspective of the ubiquitin proteasome system.Curr Opin Neurobiol.200616909416427269
- CampbellDS.HoltCE.Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation.Neuron.2001321013102611754834
- KelemanK.RajagopalanS.CleppienD.et al.Comm sorts robo to control axon guidance at the Drosophila midline.Cell.200211041542712202032
- AbrahamsBS.GeschwindDH.Connecting genes to brain in the autism spectrum disorders.Arch Neurol.20106739539920385903
- BourgeronT.A synaptic trek to autism.Curr Opin Neurobiol.20091923123419545994
- GrantynR.HennebergerC.JuttnerR.MeierJC.KirischukS.Functional hallmarks of GABAergic synapse maturation and the diverse roles of neurotrophins.Front Cell Neurosci.201151321772813
- FassioA.PatryL.CongiaS.et al.SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function.Hum Mol Genet.201120229730721441247
- KaoHT.PortonB.CzernikAJ.et al.A third member of the synapsin gene family.Proc Natl Acad Sci USA.199895466746729539796
- FengJ.ChiP.BlanpiedTA.et al.Regulation of neurotransmitter release by synapsin III.J Neurosci.2002224372438012040043
- AugustinI.RosenmundC.SudhofTC.BroseN.Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles.Nature.199940045746110440375
- VoldersK.NuytensK.CreemersJW.The autism candidate gene Neurobeachin encodes a scaffolding protein implicated in membrane trafficking and signaling.Curr Mol Med.20111120421721375492
- RichterJD.SonenbergN.Regulation of cap-dependent translation by elF4E inhibitory proteins.Nature.200543347748015690031
- TavazoieSF.AlvarezVA.RidenourDA.KwiatkowskiDJ.SabatiniBL.Regulation of neuronal morphology and function by the tumor suppressors Tsd and Tsc2.Nat Neurosci.200581727173416286931
- ChoiYJ.Di NardoA.KramvisI.et al.Tuberous sclerosis complex proteins control axon formation.Genes Dev.2008222485249518794346
- BateupHS.TakasakiKT.SaulnierJL.DenefrioCL.SabatiniBL.Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function.J Neurosci.201131886288921677170
- FuC.CawthonB.ClinkscalesW.BruceA.WinzenburgerP.EssKC.GABAergic interneuron development and function is modulated by the tsc1 gene.Cereb Cortex.2012222111211922021912
- KelleherRJ.GeigenmullerU.HovhannisyanH.et al.High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism.PLoS One.20127e3500322558107
- Rodriguez-EscuderoI.OliverMD.Andres-PonsA.MolinaM.CidVJ.PulidoR.A comprehensive functional analysis of PTEN mutations:implications in tumor- and autism-related syndromes.Hum Mol Genet.2011204132414221828076
- AmiriA.ChoW.ZhouJ.et al.Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis.J Neurosci.2012325880589022539849
- KwonCH.LuikartBW.PowellCM.et al.Pten regulates neuronal arborization and social interaction in mice.Neuron.20065037738816675393
- KumarRA.KaraMohamedS.SudiJ.et al.Recurrent 16p11.2 microdeletions in autism.Hum Mol Genet.20081762863818156158
- WeissLA.ShenY.KornJM.et al.Association between microdeletion and microduplication at 16p11.2 and autism.N Engl J Med.200835866767518184952
- ZouH.YuY.SheikhAM.et al.Association of upregulated Ras/Raf/ERK1/2 signaling with autism.Genes Brain Behav.20111061562421595826
- YangK.CaoF.SheikhAM.et al.Up-regulation of Ras/Raf/ERK1/2 signaling impairs cultured neuronal cell migration, neurogenesis, synapse formation, and dendritic spine development.Brain Struct Funct. In press.
- Martinez de LagranM.Benavides-PiccioneR.Ballesteros-YanezI.et al.DyrkIA influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons.Cereb Cortex. In press.
- BerkelS.MarshallCR.WeissB.et al.Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation.Nat Genet.20104248949120473310
- DurandCM.BetancurC.BoeckersTM.et al.Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders.Nat Genet.200739252717173049
- SatoD.LionelAC.LeblondCS.et al.SHANK1 deletions in males with autism spectrum disorder.Am J Hum Genet.20129087988722503632
- SykesNH.TomaC.WilsonN.et al.Copy number variation and association analysis of SHANK3 as a candidate gene for autism in the IMGSAC collection.Eur J Hum Genet.2009171347135319384346
- BonagliaMC.GiordaR.ManiE.et al.Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome.J Med Genet.20064382282816284256
- GrabruckerAM.KnightMJ.ProepperC.et al.Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation.EMBO J.20113056958121217644
- ChoiYB.LiHL.KassabovSR.et al.Neurexin-neuroligin transsynaptic interaction mediates learning-related synaptic remodeling and long-term facilitation in aplysia.Neuron.20117046848121555073
- MarshallCR.NoorA.VincentJB.et al.Structural variation of chromosomes in autism spectrum disorder.Am J Hum Genet.20088247748818252227
- AlarconM.AbrahamsBS.StoneJL.et al.Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene.Am J Hum Genet.20088215015918179893
- JamainS.QuachH.BetancurC.et al.Mutations of theX-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism.Nat Genet.200334272912669065
- SudhofTC.RothmanJE.Membrane fusion:grappling with SNARE and SM proteins.Science.200932347447719164740
- VaroqueauxF.AramuniG.RawsonRL.et al.Neuroligins determine synapse maturation and function.Neuron.20065174175416982420
- Lawson-YuenA.SaldivarJS.SommerS.PickerJ.Familial deletion within NLGN4 associated with autism and Tourette syndrome.Eur J Hum Genet.20081661461818231125
- ArkingDE.CutlerDJ.BruneCW.et al.A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism.Am J Hum Genet.20088216016418179894
- KattenstrothG.TantalakiE.SudhofTC.GottmannK.MisslerM.Postsynaptic N-methyl-D-aspartate receptor function requires alpha-neurex-ins.Proc Natl Acad Sci U S A.20041012607261214983056
- JiangY.LangleyB.LubinFD.et al.Epigenetics in the nervous system.J Neurosci.200828117531175919005036
- ShulhaHP.CheungI.WhittleC.et al.Epigenetic signatures of autism:trimethylated H3K4 landscapes in prefrontal neurons.Arch Gen Psychiatry.20126931432422065254
- AndersonDK.LordC.RisiS.et al.Patterns of growth in verbal abilities among children with autism spectrum disorder.J Consult Clin Psychol.20077559460417663613
- ystronI.BlakemoreC.RakicP.Development of the human cerebral cortex: Boulder Committee revisited.Nat Rev Neurosci.2008911012218209730
- StilesJ.JerniganTL.The basics of brain development.Neuropsychol Rev.20102032734821042938
- VolpeJJ.Neurology of the Newborn.5th ed. Philadelphia, PA: Saunders2008