Abstract
Polyglutamine (polyQ) diseases are caused by the expansion of polyQ repeats in the respective gene products. Intracellular accumulation of disease proteins is a hallmark of polyQ diseases; however, whether the increased aggregation of polyQ protein is the cause of diseases is still controversial. Efforts have been made to clarify the relationship between protein aggregation and pathogenesis of polyQ diseases. Especially, the roles of post-translational modifications garner attention because of their roles in regulating protein stability and aggregation. Small ubiquitin-like modifiers (SUMO) covalently attach to target proteins and affect the diverse cellular processes. PolyQ proteins such as huntingtin and ataxin-1 are covalently modified by SUMO-1 and consequently accumulate in the insoluble fraction. To study the effects of SUMO-1 on the polyQ protein ataxin-3, we examined SUMO-1 modification of ataxin-3 and tested if SUMOylation modulates ataxin-3 aggregation. Surprisingly, ataxin-3 was not covalently modified by SUMO-1; instead, SUMO-1 interacted with ataxin-3 mostly in nucleus and as a result reduced the cellular level of ataxin-3. MG132 treatment blocked the effect of SUMO-1 suggesting that SUMO-induced degradation of ataxin-3 is mediated by ubiquitin–proteasome system. Chase experiment confirmed that such effects were indeed due to the increased degradation of ataxin-3 by SUMO-1. While the aggregation of ataxin-3 was not affected by SUMO-1, the presence of polyQ-expanded ataxin-3 increased the recruitment of SUMO-1 into nuclear bodies. These results together suggest that SUMO-1 contributes to the regulation of the cellular level of ataxin-3 protein by facilitating the degradation process.
Introduction
Polyglutamine (polyQ) diseases are inherited neurodegenerative disorders caused by expansion of CAG triplet repeat in the causative genes, which produces pathogenic proteins containing expanded polyQ repeat. Expansion of polyQ repeat likely confers toxic gain-of-functions to disease proteins and promotes their misfolding and aggregation (Shao & Diamond Citation2007). Disease severity correlates with the length of polyQ repeat and the longer repeat usually leads to earlier onset of symptom and more widespread degeneration. Protein aggregation, oxidative stress, and transcriptional dysregulation are suggested as possible molecular events leading to polyQ pathogenesis; however, the precise mechanism is still elusive (Williams & Paulson Citation2008).
Post-translational modifications are rapid and reversible means of regulating dynamics and functions of proteins. Conjugation of protein modifiers, e.g., ubiquitin and ubiquitin-like proteins, are tightly regulated and provide unique control points for a variety of cellular processes (Dorval & Fraser Citation2007). Similar to ubiquitin, small ubiquitin-like modifiers (SUMO) are reversibly conjugated to specific lysine residues in target proteins. Among the isoforms of SUMO, SUMO-1, SUMO-2, and SUMO-3 are processed in vivo for covalent modification (Praefcke et al. Citation2012). SUMOylation requires a multistep pathway catalyzed by E1 (AOS1/UBA2) and E2 (Ubc9) proteins. While it is not required, E3 proteins (PIAS proteins) stimulate SUMOylation by associating Ubc9 and substrates and increase the efficiency of modification (Sarge & Park-Sarge Citation2009). Although ubiquitylated proteins are generally targeted for proteasomal degradation, consequences of SUMO modification are diverse. SUMO modification alters protein stability, modulates protein–protein interaction, or affects the subcellular localization of proteins (Verger et al. Citation2003).
Similar to ubiquitin, SUMO is detected within the neuronal inclusions observed in neurodegenerative diseases including Huntington's disease (HD) and other polyQ diseases (Dorval & Fraser Citation2007). Involvement of SUMO in polyQ pathogenesis has been suggested by a number of studies. Global impairment of SUMO pathway can increase polyQ-induced degeneration in Drosophila (Chan et al. Citation2002). Furthermore, SUMO modification exacerbates neurodegeneration caused by huntingtin (Steffan et al. Citation2004). Ataxin-1 is covalently modified by SUMO-1 at least at five different lysine residues (Riley et al. Citation2005), which is enhanced by oxidative stress (Ryu et al. Citation2010). SUMOylation decreases the aggregate propensity and toxicity of polyQ-expanded ataxin-7 (Janer et al. Citation2010). However, little is known about if SUMO modifies ataxin-3 or affects its stability and localization.
To understand if ataxin-3 linked to spinocerebellar ataxia type 3 (SCA3) is covalently modified by SUMO-1, we analyzed SUMOylated forms of ataxin-3 by co-immunoprecipitation in cultured cells. In vivo interaction between SUMO-1 and ataxin-3 was also examined by immunofluorescence analysis. To investigate the effects of SUMO-1 on ataxin-3, we studied if SUMO-1 over-expression changes the cellular level of ataxin-3 or increases its accumulation in the insoluble fraction.
Materials and methods
Plasmids, cell culture, and DNA transfection
Constructs carrying HA-tagged ataxin-3 [26Q and 73Q] in pcDNA3.1/zeo vector were generated in the lab. FLAG-tagged SUMO-1 expression plasmid was provided by Prof. Chin Ha Chung (Seoul National University). BOSC 23 cells, a derivative of HEK 293T cells, were maintained in DMEM/10% FBS medium supplemented with L-glutamine, penicillin, and streptomycin at 37°C with 5% CO2. Plasmids were transfected into cells by using Lipofectamine™ 2000 reagent (Invitrogen). Unless otherwise specified, 1 µg of plasmid was used for transfection.
Immunoblot assay
After transfection with ataxin-3 with or without SUMO-1 for 48 h, cells were harvested and boiled for 5 min with 1×SDS sample buffer at 95°C. When necessary, after 24 h of transfection, cells were treated with 5 µM MG132 for 24 h to inhibit proteasomal activity. Proteins were separated by 8–10% SDS-PAGE and then transferred to PVDF membrane. After incubating with anti-HA antibody (1:1000, Sigma), anti-FLAG antibody (1:1000, Sigma), or anti-β-actin antibody (1:1000, SantaCruz) for 2 h, the membranes were incubated with peroxidase-conjugated anti-mouse IgG (1:2000, Sigma) for 1 h. The proteins were visualized by using ECL detection kit (Amersham), and images were obtained by using luminescent image analyzer (LAS-4000 mini, Fujifilm).
SUMOylation assay
After transfection with ataxin-3 and SUMO-1, cells were harvested, washed, and lysed in NP-40 lysis buffer (20 mM Tris–HCl, pH 7.5; 150 mM NaCl; 1% NP-40; 1% glycerol) supplemented with protease inhibitors (Roche) and 5 mM N-ehylmaleimide to inhibit SUMO proteases in cell lysate. After incubating 30 min on ice, the cell lysate was clarified by centrifugation at 1000×g for 10 min, and the supernatants were subjected to immunoprecipitation. To isolate HA-tagged ataxin-3, 300 µg of cell lysate was mixed with 20 µl of anti-HA-agarose beads (Sigma) at 4°C for overnight with rotation. The protein complexes were collected by centrifugation, washed with TBST buffer, and then boiled in 1×SDS sample buffer. The presence of SUMOylated forms of ataxin-3 was assayed by immunoblot analysis with anti-FLAG antibody.
Fractionation of cell lysate
To determine the relative amount of ataxin-3 in the soluble and insoluble fractions, cell lysate was subjected to a second round of centrifugation at 20,000×g for 30 min. The supernatant (‘soluble’) was collected, and the pellets (‘insoluble’) were washed with lysis buffer and solubilized by adding an equal volume of 1×SDS-PAGE sampling buffer. The relative amounts of ataxin-3 were measured by immunoblotting with anti-HA antibody. Density analysis of protein bands was carried out by using Image-J program (NIH).
Chase experiment
To analyze the direct effect of SUMO-1 on ataxin-3 degradation, 24 h after transfection with ataxin-3 and SUMO-1, cells were treated with 20 µg/ml of cycloheximide to block the synthesis of proteins. From this point (t=0), cells were collected every 8 h (for additional 24 h) and then processed for immunoblot analysis of ataxin-3 protein with anti-HA antibody.
Immuofluorescence analysis
To study protein localization, cells were seeded onto a 6-well plate with a cover glass. After 12 h, cells were transfected with ataxin-3 with or without SUMO-1 and then incubated for 24 h. The transfected cells were washed in PBS, fixed with the 3.5% paraformaldehyde, and then permeabilized with 0.1% triton X-100 for 15 min at room temperature. The cells were incubated with anti-HA antibodies (1:200) and anti-FLAG antibodies (1:200) in 1% BSA at room temperature for 2 h. After washing with PBS, cells were incubated with the secondary antibodies (1:500) in 1% BSA for 1 h. FITC-conjugated goat anti-mouse antibody (abcam) and Cy3-conjugated goat anti-rabbit antibody (Sigma) were employed. For nuclear staining, fixed/permeabilized cells were incubated with DAPI for 15 min at room temperature. Cells were observed under a laser confocal fluorescence microscope (C1+, Nikon).
Results
Interaction between ataxin-3 and SUMO-1
SUMOylated proteins usually contain the motif Ψ-K-X-D/E (Ψ is a large hydrophobic residue, K is the lysine, X is any amino acid, and D/E is an acidic residue), known as SUMO consensus sequence. These residues directly interact with Ubc9 and regulate the stability of interactions between Ubc9 and the target proteins (Gareau & Lima Citation2010). Analysis of sequence of ataxin-3 with SUMOplot™ software (http://www.abgent.com/sumoplot/) predicted that Lys166 (165VKGD168) is a probable site of SUMO modification (A). Contrary to the prediction, no bands corresponding to SUMO-1 conjugated forms were detected in the immunocomplex containing ataxin-3 (B and C). The reciprocal immunoprecipiation with anti-FLAG antibody also failed to produce clear evidence that ataxin-3 is modified by SUMO-1 under our experimental conditions (data not shown). These results raised a possibility that SUMO-1 may not conjugate to ataxin-3 and instead noncovalently interacts with this polyQ protein, as suggested previously (Shen et al. Citation2005).
Figure 1. Ataxin-3 is not covalently modified by SUMO-1. (A) A schematic representation of functional domains in ataxin-3 (top). Domain analysis was carried out using PROSITE (http://prosite.expasy.org/). Josephin domain (1–180) contains DUB activity, and UIMs (224–243, 244–263) are responsible for ubiquitin binding. QQQ denotes polyglutamine repeats. The proposed SIM sequence (IFVV) located within the ‘Josephin’ domain, indicated by shaded box, is highly conserved among mammalian and vertebrate species (bottom): bovine (Bos Taurus), chicken (Gallus gallus), rodent (Mus musculus, Rattus novergicus), swine (Sus scorfa), and human (Homo sapiens). (B) To confirm the expression, HA-ataxin-3 (26Q or 73Q) or FLAG-SUMO-1 was transfected into cells. After 48 h, cells were harvested, lysed, and analyzed by immunoblot analysis using anti-HA antibody (ataxin-3) and anti-FLAG antibody (SUMO-1), respectively. (C) To detect SUMO conjugated ataxin-3, cells were co-transfected with ataxin-3 and SUMO-1. Cell lysate was subjected to immunoprecipitation with anti-HA agarose beads followed by immunoblot with anti-HA antibody to ensure that ataxin-3 proteins were properly isolated (left) or anti-FLAG antibody to detect SUMOylated forms (right).
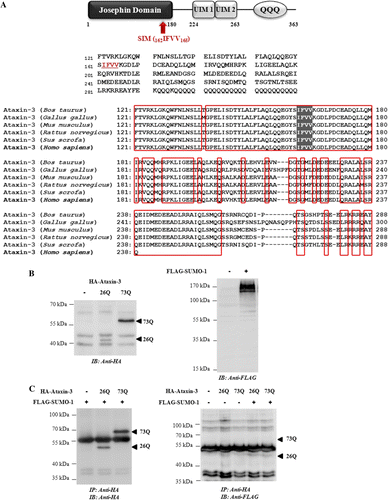
SUMO is also capable of mediating noncovalent interaction with target proteins via SUMO-interacting motif (SIM), characterized by a stretch of hydrophobic amino acids flanked by acidic residues (Gareau & Lima Citation2010). Incidentally, a sequence similar to a proposed SIM (V/I-X-V/I-V/I) was found in the coding sequence of ataxin-3 (162IFVV165), adjacent to the predicted SUMOylation site (165VKGD168) (A). Interestingly the region of ataxin-3 containing potential SIM is located within the ‘Josephin’ domain, which is highly conserved among vertebrate species, implicating that SIM is important for the normal function of ataxin-3 (A). Initially, we tried to detect monomeric SUMO-1 in the immunoprecipitates of ataxin-3 (B and C); however, due to the presence of several nonspecific bands, we failed to distinguish unconjugated SUMO-1 proteins. To prove the interaction between SUMO-1 and ataxin-3 in a different way, we then examined their localization and distribution by immunofluorescence analysis (). In the cells co-expressing ataxin-3 and SUMO-1, both proteins demonstrated intranuclear distribution pattern, and their localizations were largely overlapped. In addition to diffuse nuclear staining pattern, certain SUMO-1 proteins were present as small dot-like structures, which probably correspond to PML nuclear bodies – a known SUMO substrate (Janer et al. Citation2010). The finding that distribution of dot-like structures of SUMO-1 did not overlap with that of ataxin-3 implicates that interaction between ataxin-3 and SUMO-1 takes place outside nuclear bodies. Interestingly, polyQ-expanded ataxin-3 [73Q] increased the size of dot-like structures while SUMO-1 over-expression did not affect the aggregation of ataxin-3 (). Perhaps the polyQ-expansion in ataxin-3 affects the dynamics of SUMO pathway in nucleus and facilitates the recruitment of SUMO-1 in the nuclear bodies.
Figure 2. Ataxin-3 and SUMO-1 are associated and co-localized mostly in nucleus. After transfection of HA-ataxin-3 ([26Q] or [73Q]) with FLAG-SUMO-1, cells were incubated 24 h and fixed/permeabilized. To observe the protein localization, cells were incubated with anti-HA antibody followed by FITC-anti-IgG antibody (ataxin-3) and anti-FLAG antibody followed by Cy3-anti-IgG antibody (SUMO-1). DAPI was used for nuclear staining. Small dot-like structures in SUMO-1 panel represent SUMO-1 recruited to nuclear bodies (see text).
![Figure 2. Ataxin-3 and SUMO-1 are associated and co-localized mostly in nucleus. After transfection of HA-ataxin-3 ([26Q] or [73Q]) with FLAG-SUMO-1, cells were incubated 24 h and fixed/permeabilized. To observe the protein localization, cells were incubated with anti-HA antibody followed by FITC-anti-IgG antibody (ataxin-3) and anti-FLAG antibody followed by Cy3-anti-IgG antibody (SUMO-1). DAPI was used for nuclear staining. Small dot-like structures in SUMO-1 panel represent SUMO-1 recruited to nuclear bodies (see text).](/cms/asset/bc941523-88ae-4547-a3d2-55d9c1cc421e/tacs_a_738611_o_f0002g.jpg)
Increased degradation of ataxin-3 by SUMO-1 over-expression
SUMO also modulates stability of certain target proteins (e.g., p63α and RbAP46) and influences the processes mediated by the substrates (Ghioni et al. Citation2005; Giri et al. Citation2008). Since we observed that ataxin-3 and SUMO-1 interact and co-localize in cells, we tested if SUMO-1 over-expression affect ataxin-3 stability. The level of ataxin-3 was significantly reduced upon co-transfection with SUMO-1, and such an effect was concentration-dependent as much less ataxin-3 was present when more SUMO-1 was expressed (A). Intriguingly the effect of SUMO-1 overexpression on down-regulation was greater with normal ataxin-3 [26Q] than with the polyQ-expanded ataxin-3 [73Q] (A). These results indicate that SUMO-1 facilitates the down-regulation of ataxin-3 presumably by increasing proteolysis, and that more stable polyQ-expanded form of ataxin-3 (Matsumoto et al. Citation2004) is more resistant to the effects of SUMO-1.
Figure 3. SUMO-1 facilitates proteasomal degradation of ataxin-3. (A) BOSC cells were transfected with a fixed amount of ataxin-3 [26Q] or [73Q] (1 µg) and increasing amount of SUMO-1 (0, 1, 2, and 3 µg). After 48 h of transfection, cells were collected, lysed, and the steady-state level of ataxin-3 in cell lysate was analyzed by immunoblot with anti-HA antibody. (B) To test the effects of proteasome inhibition on ataxin-3 degradation, cells transfected with ataxin-3 [26Q] or [73Q] (1 µg) and SUMO-1 (2 µg) were treated with 5 µM MG132 for 24 h. The steady-state level of ataxin-3 was compared as described above. (C) After 24 h of transfection of ataxin-3 [26Q] with or without SUMO-1, cells were treated with 20 µg/ml of cycloheximide to block protein synthesis. From this point (t=0), cells were collected every 8 h (up to 24 h) and processed for immunoblot analysis. For loading control, β-actin was used. Gel images shown are the results from a typical experiment. Graphs are the results of image quantification from three independent experiments. Bars =SD.
![Figure 3. SUMO-1 facilitates proteasomal degradation of ataxin-3. (A) BOSC cells were transfected with a fixed amount of ataxin-3 [26Q] or [73Q] (1 µg) and increasing amount of SUMO-1 (0, 1, 2, and 3 µg). After 48 h of transfection, cells were collected, lysed, and the steady-state level of ataxin-3 in cell lysate was analyzed by immunoblot with anti-HA antibody. (B) To test the effects of proteasome inhibition on ataxin-3 degradation, cells transfected with ataxin-3 [26Q] or [73Q] (1 µg) and SUMO-1 (2 µg) were treated with 5 µM MG132 for 24 h. The steady-state level of ataxin-3 was compared as described above. (C) After 24 h of transfection of ataxin-3 [26Q] with or without SUMO-1, cells were treated with 20 µg/ml of cycloheximide to block protein synthesis. From this point (t=0), cells were collected every 8 h (up to 24 h) and processed for immunoblot analysis. For loading control, β-actin was used. Gel images shown are the results from a typical experiment. Graphs are the results of image quantification from three independent experiments. Bars =SD.](/cms/asset/8d72098a-493a-4419-b651-d776c4b07c3c/tacs_a_738611_o_f0003g.jpg)
To determine which proteolytic pathway is responsible for SUMO-induced down-regulation of ataxin-3, we treated cells co-expressing ataxin-3 and SUMO-1 with the proteasome-specific inhibitor MG132 and then examined the level of ataxin-3. As shown in B, MG132 treatment reversed the effect of SUMO-1 and increased the steady-state level of ataxin-3. Again, the effect of MG132 was more prominent with ataxin-3[26Q] confirming the resistance of polyQ-expanded form of ataxin-3 to proteasomal degradation. These results suggest that SUMO-1 could facilitate ataxin-3 degradation by ubiquitin–proteasome system.
To verify these results, we carried out chase experiments to examine if SUMO-1 over-expression changes the half-life of ataxin-3. Ataxin-3 [26Q] protein was relatively stable and showed a half-life of approximately 18 h. By contrast, when co-expressed with SUMO-1, the half-life of ataxin-3 greatly reduced from 18 h to 6 h (C). Therefore, the effect of SUMO-1 on the down-regulation of ataxin-3 is indeed due to the increased turnover of this polyQ protein by UPS.
SUMO-1 did not cause the accumulation of ataxin-3 in insoluble fraction
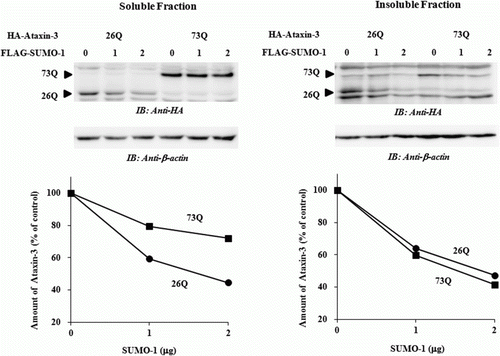
SUMOylation of proteins can stabilize and increase aggregation of certain neurodegenerative proteins (Ryu et al. Citation2010; Kim et al. Citation2011). Although SUMO-1 interacts with ataxin-3 noncovalently and accelerates its degradation, we could not rule out a possibility that the effects of SUMO-1 is, at least partially, due to the increased partitioning of aggregation-prone ataxin-3 into the insoluble fraction. To clarify this, we fractionated the cell lysate and analyzed the relative amount of ataxin-3 in the soluble and insoluble fractions. In both fractions, the level of ataxin-3 was decreased by SUMO-1 overexpression in a concentration-dependent manner confirming that SUMO-1 causes destabilization of ataxin-3 without increasing aggregation (). Surprisingly, while normal ataxin-3 [26Q] was degraded at similar rates in both fractions, polyQ-expanded ataxin-3 [73Q] in the soluble fraction was much slowly degraded than that in the insoluble fraction. These findings suggest that the reduced degradation of ataxin-3 [73Q] is not due to the propensity to aggregate but to the inherent resistance to the proteolysis.
Figure 4. SUMO-1 does not affect the aggregation of ataxin-3. After co-expression of HA-tagged ataxin-3 (1 µg) with increasing amount of FLAG-tagged SUMO-1 (0, 1, and 2 µg), cells were lysed and fractionated as described in Materials and methods section. Relative amounts of ataxin-3 in the soluble and insoluble fractions were measured by immunoblot analysis. For loading control, β-actin was used. Gel images and graphs shown are the results from a typical experiment.
Discussion
Ataxin-3 (MJD1) is a multifunctional protein; it contains a deubiquitinating (DUB) enzyme activity that resides in the Josephin domain (A) and functions as a transcriptional co-repressor (Reina et al. Citation2010). Ataxin-3 plays a role in the ER-associated degradation through modulating protein extraction from the ER mediated by p97 (Zhong & Pittman Citation2006), which mediates the shuttling of misfolded proteins onto 26S proteasomes (Choi & Lee Citation2010). Ubiquitin-interacting motifs (UIMs) present in ataxin-3 (A) are important for both ubiquitin binding and DUB activities and required for the aggresome formation (Reina et al. Citation2010). Ataxin-3 is now established as a crucial component of protein quality control process mediated by ubiquitin–proteasome system (UPS).
Ataxin-3 itself is also modified by ubiquitin and degraded by 26S proteasomes. Interestingly, in vivo and in vitro degradation of ataxin-3 is affected by its own DUB activity (Todi et al. Citation2007). Conversely, the ubiquitination of ataxin-3 is shown to enhance its DUB activity, which does not require the presence of UIM (Todi et al. Citation2009). Ubiquitination of ataxin-3 is changed when proteasome function is impaired or under stress conditions and ubiquitinated ataxin-3 is usually rapidly de-ubiquitinated by DUBs associated with proteasomes (Todi et al. Citation2009). It is evident that the DUB activity and ataxin-3 degradation are tightly regulated by UPS. Possibly polyQ expansion in ataxin-3 disrupts such precisely controlled mechanism and may lead to neuronal death.
SUMO proteins, similar to ubiquitin, serve as modifiers that exert the effects through covalent modification of substrates (Gareau & Lima Citation2010). SUMO modifies a number of proteins involved in neurodegenerative diseases such as ataxin-1, ataxin-7, huntingtin, α-synucelin, and SOD1 (Sarge & Park-Sarge Citation2009). However, the consequences of SUMOylation are quite diverse. SUMOylation of huntingtin, ataxin-7, and α-synuclein reduces their aggregation and toxicity (Steffan et al. Citation2004; Janer et al. Citation2010; Krumova et al. Citation2011). By contrast, SUMOylation increases aggregation of ataxin-1 and SOD-1, presumably by stabilizing the proteins (Fei et al. Citation2006; Ryu et al. Citation2010). The finding that SUMO-1 accelerates ataxin-3 degradation without affecting aggregation suggests that SUMO-1 plays a distinct role in regulating the stability of ataxin-3.
Initially SUMOylation of proteins was considered to be functionally unrelated to the ubiquitylation. Analysis of SUMO conjugation sites for certain proteins (e.g., PCNA), however, revealed that SUMO can compete with ubiquitin for the same lysine residues and thereby stabilize the substrates. Accumulating evidence indicates that SUMO system is tightly interconnected to UPS. SUMOylation can prevent proteasomal degradation of phosducin by inhibiting ubiquitin conjugation (Klenk et al. Citation2006). Moreover, the SUMO-1 over-expression increases the stability of Rb-associated protein 46 and suppresses cell growth (Giri et al. Citation2008). By contrast, SUMO-1 destabilizes ΔNα isoform of p63 and thus regulates its transcriptional activity (Ghioni et al. Citation2005). Recent studies have unraveled the presence of a novel family of RING-finger ubiquitin ligases (E3), called as SUMO-targeted ubiquitin ligases (STUbL), which recognize SUMOylated proteins and promote ubiquitin-mediated degradation. Importantly STUbL proteins contain functional SIMs that allow binding with SUMO or SUMOylated proteins (Perry et al. Citation2008).
While most of neurodegenerative proteins are conjugated with SUMO, ataxin-3 does not appear to be modified by SUMO-1 in vivo, although it is possible that SUMOylated forms of ataxin-3 are difficult to detect, especially since SUMO over-expression can lead to the rapid degradation of ataxin-3. To determine whether SUMO-1 is conjugated to ataxin-3, we probably need to use different strategies. For example, co-expression of Ubc9 to enhance in vivo SUMOylation or application of stress conditions that enhance SUMOylation (Bossis & Melchior Citation2006) should be helpful. The observation that MG132 reversed the effect of SUMO-1 over-expression (C) also suggests that SUMOylated form of ataxin-3 is rapidly degraded by UPS, and the inhibition of proteasome is needed to stabilize the labile species of SUMO-conjugated ataxin-3. Use of SUMO-1 mutant lacking C-terminal glycine residues may also provide more direct evidence to understand whether ataxin-3 is modified by SUMO-1.
Noncovalent effects of SUMO are also well documented. Noncovalent interaction of SUMO and Ubc9 leads to the down-regulation of dynamin-mediated endocytosis of transferrin (Mishra et al. Citation2004). Generation of Aβ peptide from amyloid precursor protein in cultured cells is enhanced by SUMO-3, and the effects are independent of covalent attachment (Dorval et al. Citation2007). Moreover, noncovalent association of SUMO-1 with parkin modulates not only the protein level but also the enzymatic activity and intracellular localization (Um & Chung Citation2006). Except for ataxin-3 and parkin, evidence that SUMO-1 can promote turnover of proteins in a noncovalent manner is hard to find, and the underlying mechanism is largely unknown. As mentioned above, STUbL might be involved in this process. Possibly ataxin-3 is recognized by STUbL via SIM and then subjected to proteasomal degradation. Since other E3 ligases also mediate ubiquitin-dependent degradation of ataxin-3, it is necessary to test if STUbL can cooperate with other E3 ligase when SUMO-1 is over-expressed. Since we have only studied the effects of SUMO-1 on ataxin-3 degradation, it would be interesting to examine if SUMO-1 also modulates the DUB activity of ataxin-3 or interaction with other factors such as p97. It is also necessary to determine whether ataxin-3 is conjugated with SUMO-2/3 proteins.
In conclusion, we here present the evidence that SUMO-1 associates with ataxin-3 noncovalently and facilitates its turnover. Such effects are not due to the propensity of ataxin-3 to aggregate but rather due to the increased proteasomal degradation. Future studies will tell us more about the exact role of SUMO-1 in the proteasomal degradation of ataxin-3 and help us understand if SUMO pathway is functionally linked to the pathogenesis of SCA3.
Acknowledgements
We thank Prof. Chin Ha Chung for providing SUMO-1 constructs. This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grant number 2010-0006080).
References
- Bossis , G and Melchior , F. 2006 . Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes . Mol. Cell. , 21 : 349 – 357 . doi: 10.1016/j.molcel.2005.12.019
- Chan , HY , Warrick , JM , Andriola , I , Merry , D and Bonini , NM. 2002 . Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila . Hum. Mol. Genet. , 11 : 2895 – 2904 . doi: 10.1093/hmg/11.23.2895
- Choi , JS and Lee , DH. 2010 . CHIP promotes the degradation of mutant SOD1 by reducing its interaction with VCP and S6/S6' subunits of 26S proteasome . Ani. Cells Sys. , 14 : 1 – 10 . doi: 10.1080/19768351003765145
- Dorval , V and Fraser , PE. 2007 . SUMO on the road to neurodegeneration . Biochim. Biophys. Acta. , 1773 : 694 – 706 . doi: 10.1016/j.bbamcr.2007.03.017
- Dorval , V , Mazzella , MJ , Mathews , PM , Hay , RT and Fraser , PE. 2007 . Modulation of Aβ generation by small ubiquitin-like modifiers does not require conjugation to target proteins . Biochem. J. , 404 : 309 – 316 . doi: 10.1042/BJ20061451
- Fei , E , Jia , N , Yan , M , Ying , Z , Sun , Q , Wang , H , Zhang , T , Ma , X , Ding , H Yao , X . 2006 . SUMO-1 modification increases human SOD1 stability and aggregation . Biochem. Biophys. Res. Commun. , 347 : 406 – 412 . doi: 10.1016/j.bbrc.2006.06.092
- Gareau , JR and Lima , CD. 2010 . The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition . Nat. Rev. Mol. Cell. Biol. , 11 : 861 – 871 . doi: 10.1038/nrm3011
- Ghioni , P , D'Alessandra , Y , Mansueto , G , Jaffray , E , Hay , RT , La Mantia , G and Guerrini , L. 2005 . The protein stability and transcriptional activity of p63α are regulated by SUMO-1 conjugation . Cell Cycle. , 4 : 183 – 190 . doi: 10.4161/cc.4.1.1359
- Giri , R , Yeh , H-H , Wu , C-H and Liu , H-S. 2008 . SUMO-1 overexpression increases RbAP46 protein stability and suppresses cell growth . Anticancer Res. , 28 : 3749 – 3756 .
- Janer , A , Werner , A , Takahashi-Fujigasaki , J , Daret , A , Fujigasaki , H , Takada , K , Duyckaerts , C , Brice , A , Dejean , A and Sittler , A. 2010 . SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7 . Hum. Mol. Genet. , 19 : 181 – 195 . doi: 10.1093/hmg/ddp478
- Kim , YM , Jang , WH , Quezado , MM , Oh , Y , Chung , KC , Junn , E and Mouradian , MM. 2011 . Proteasome inhibition induces α-synuclein SUMOylation and aggregate formation . J. Neurol. Sci. , 307 : 157 – 161 . doi: 10.1016/j.jns.2011.04.015
- Klenk , C , Humrich , J , Quitterer , U and Lohse , MJ. 2006 . SUMO-1 controls the protein stability and the biological function of phosducin . J. Biol. Chem. , 281 : 8357 – 8364 . doi: 10.1074/jbc.M513703200
- Krumova , P , Meulmeester , E , Garrido , M , Tirard , M , Hsiao , H-H , Bossis , G , Urlaub , H , Zweckstetter , M , Kugler , S Melchior , F . 2011 . SUMOylation inhibits α-synuclein aggregation and toxicity . J. Cell Biol. , 194 : 49 – 60 . doi: 10.1083/jcb.201010117
- Matsumoto , M , Yada , M , Hatakeyama , S , Ishimoto , H , Tanimura , T , Tsuji , S , Kakizuka , A , Kitagawa , M and Nakayama , KI. 2004 . Molecular clearance of ataxin-3 is regulated by a mammalian E4 . EMBO J. , 23 : 659 – 669 . doi: 10.1038/sj.emboj.7600081
- Mishra , RK , Jatiani , SS , Kumar , A , Simhadri , VR , Hosur , RV and Mittal , R. 2004 . Dynamin interacts with members of the SUMOylation machinery . J. Biol. Chem. , 279 : 31445 – 31454 . doi: 10.1074/jbc.M402911200
- Perry , JJ , Tainer , JA and Boddy , MN. 2008 . A SIM-ultaneous role for SUMO and ubiquitin . Trends Biochem. Sci. , 33 : 201 – 208 . doi: 10.1016/j.tibs.2008.02.001
- Praefcke , GJK , Hoffman , K and Dohmen , RJ. 2012 . SUMO playing tag with ubiquitin . Trends in Biochem. Sci. , 37 : 23 – 31 . doi: 10.1016/j.tibs.2011.09.002
- Reina , CP , Zhong , X and Pittman , RN. 2010 . Proteotoxic stress increases nuclear localization of ataxin-3 . Hum. Mol. Genet. , 19 : 235 – 249 . doi: 10.1093/hmg/ddp482
- Riley , BE , Zoghbi , HY and Orr , HT. 2005 . SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal . J. Biol. Chem. , 280 : 21942 – 21948 . doi: 10.1074/jbc.M501677200
- Ryu , J , Cho , S , Park , BC and Lee , DH. 2010 . Oxidative stress-enhanced SUMOylation and aggregation of ataxin-1: implication of JNK pathway . Biochem. Biophys. Res. Commun. , 393 : 280 – 285 . doi: 10.1016/j.bbrc.2010.01.122
- Sarge , KD and Park-Sarge , OK. 2009 . SUMOylation and human disease pathogenesis . Trends in Biochem. Sci. , 34 : 200 – 205 . doi: 10.1016/j.tibs.2009.01.004
- Shao , J and Diamond , MI. 2007 . Polyglutamine diseases: emerging concepts in pathogenesis and therapy . Hum. Mol. Genet. , 16 : R115 – R123 . doi: 10.1093/hmg/ddm213
- Shen , L , Tang , JG , Tang , BS , Jiang , H , Zhao , GH , Xia , K , Zhang , YH , Cai , F , Tan , LM and Pan , Q. 2005 . Research on screening and identification of proteins interacting with ataxin-3 . Chin. J. Med. Genet. , 22 : 242 – 247 .
- Steffan , JS , Agrawal , N , Pallos , J , Rockabrand , E , Trotman , LC , Slepko , N , Illes , K , Luka-csovich , T , Zhu , YZ Cattaneo , E . 2004 . SUMO modification of Huntingtin and Huntington's disease pathology . Science. , 304 : 100 – 104 . doi: 10.1126/science.1092194
- Todi , SV , Laco , MN , Winborn , BJ , Travis , SM , Wen , HM and Paulson , HL. 2007 . Cellular turnover of the polyglutamine disease protein ataxin-3 is regulated by its catalytic activity . J. Biol. Chem. , 282 : 29348 – 29358 . doi: 10.1074/jbc.M704126200
- Todi , SV , Winborn , BJ , Scaglione , KM , Blount , JR , Travis , SM and Paulson , HL. 2009 . Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3 . EMBO J. , 28 : 372 – 382 . doi: 10.1038/emboj.2008.289
- Um , JW and Chung , KC. 2006 . Functional modulation of Parkin through physical interaction with SUMO-1 . J. Neurol. Res. , 84 : 1543 – 1554 . doi: 10.1002/jnr.21041
- Verger , A , Perdomo , J and Crossley , M. 2003 . Modification with SUMO. A role in transcriptional regulation . EMBO Rep. , 4 : 137 – 142 . doi: 10.1038/sj.embor.embor738
- Williams , AJ and Paulson , HL. 2008 . Polyglutamine neurodegeneration: protein misfolding revisited . Trends in Neurosci. , 31 : 521 – 528 . doi: 10.1016/j.tins.2008.07.004
- Zhong , X and Pittman , RN. 2006 . Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates . Hum. Mol. Genet. , 15 : 2409 – 2420 . doi: 10.1093/hmg/ddl164