
Abstract
The oncogene protein DEK is an abundant and ubiquitous nuclear protein with implications in acute myelogenous leukemia, as translocation which results in the formation of a DEK-CAN fusion protein. In a previous study, we have identified that DEK negatively regulated peroxiredoxin 6 (Prdx 6) transcription synergistically with the p65 subunit of NF-κB. In this study, we further investigated DEK-mediated transcriptional regulation of Prdx 6 during leukemia cell differentiation. Using Chromatin Immunoprecipitation analysis and Prdx 6 reporter assays, we found that DEK operated as a negative regulator of Prdx 6 transcription during leukemia cell differentiation. DEK was highly expressed and recruited to Prdx 6 promoter along with p65 and repressed transcription after leukemia cell differentiation.
Keywords:
Introduction
The DEK protein was first identified in a specific chromosomal translocation t(6;9)(p23;q34) in acute myelogenous leukemia (AML), which results in the formation of fusion gene with the CAN nucleoporin protein NUP214 (von Lindern et al. Citation1992). The nuclear pore complex is composed of more than 30 different proteins, and CAN is one of the members in the complex. Translocated fusion gene can make the fusion protein in which part of N-terminal (1–349 amino acids) of DEK is fused with part of (813–2,090 amino acids) of CAN. In addition to these findings, recent report provides that DEK can be secreted and acts as a chemoattractant it elicits, leading to inflammation (Mor-Vaknin et al. Citation2006). Besides the immunoreactivity it elicits, DEK has long been implicated in aggressive human cancers such as hepatocellular carcinoma, glioblastoma, and melanoma (Kondoh et al. Citation1999; Grottke et al. Citation2000; Larramendy et al. Citation2002; Casas et al. Citation2003; Carro et al. Citation2006).
DEK has functional domains including DNA-binding multimerization and acidic domain (Alexiadis et al. Citation2000; Waldmann et al. Citation2002). Most of all, DEK can regulate the transcriptional activity in either activation or repression of downstream target genes through its acidic domains. For example, DEK can recruit the transcriptional corepressors such as hDaxx and HDAC2 to chromatin (Hollenbach et al. Citation2002). The promoter region of NF-κB-regulated genes, both cIAP2 and interleukin-8 (IL-8), are occupied by DEK, leading to transcriptional repression as a repressor (Sammons et al. Citation2006). DEK can interact with AP-2α and enhance the binding of AP-2α to DNA. The interaction between DEK and AP-2α leads to transcriptional activation of target gene (Campillos et al. Citation2003).
NF-κB is a transcriptional repressor with various biological functions including cell growth, differentiation, and the suppression of apoptosis. NF-κB contains five subunits – RelA (p65), c-Rel, RelB, p50, and p52 which form various hetero- and homo-dimers (Hatada et al. Citation2000). DEK interacts with p65 subunit of NF-κB complex (Sammons et al. Citation2006).
Peroxiredoxins (Prdxs) are conserved from bacteria to mammals (Wood et al. Citation2003a). There are six subgroups of Prdx enzymes in mammals (Wood et al. Citation2003b). These enzymes use redox-active cysteins to reduce peroxides and are divided into two categories of 1-Cys and 2-Cys. Prdx 6 is the member of 1-Cys Prx and has nonselenium glutathione peroxidase and phospholipase A2 activities (Chen et al. Citation2000). Prdx 6 is expressed in multiple tissues related to inhibiting of cellular oxidative stresses (Manevich et al. Citation2002). Moreover, Prdx 6-deficient mice are susceptible to oxidative stress by hyperoxia or paraquat treatment, leading to increase of mortality and lung injury (Wang et al. Citation2003; Wang et al. Citation2006). Previously, we have reported that DEK negatively regulated Prdx 6 transcription and that the NF-κB subunit p65 had a synergistic effect on this DEK-mediated repression (Kim et al. Citation2010).
In this study, we discuss the negative regulatory role of DEK in Prdx 6 transcription during leukemia cell differentiation. We found that DEK expression was repressed during leukemia cell differentiation. Additionally, DEK was recruited to the Prdx 6 promoter along with p65 and functioned as a corepressor of the target gene.
Materials and methods
Plasmid constructs
For eukaryotic expression, the following pCMX plasmid constructs were used: pCMX-DEK (Ko et al. Citation2006). The proximal 5′-flanking region of the human Prdx 6 gene promoter was obtained from HeLa genomic DNA through using XhoI-HindIII site-linked primer pair (forward, 5′-CTCGAGACATTTCTCTATCGATAGGTACC-3′; reverse 5′-AAGCTTCACGTACCGGATGCCAGCTTAC-3′). The purified products were enzyme digested with XhoI and HindIII and cloned into the pGL3-basic luciferase reporter vector (Promega).
Cell culture
HL-60 and K562 cells were grown in RPMI 1640, and 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum and 0.05% penicillin-streptomycin at 37°C with 5% CO2 in humidified air. K562 and 293T cells were transfected with each construct using lipofectamine 2000 (Invitrogen) or polyethylenimine (Sigma), respectively.
Knockdown of DEK mRNA and transient transfection
We used the vector-based RNAi method to knockdown endogenous DEK and reduce the DEK expression level. pSM2c-DEK expressing DEK-specific short hairpin RNAs (nucleotides 654–674 from NM_003472), referred to as shRNA, was purchased (Open Biosystems).
RNA extraction and real-time polymerase chain reaction (PCR)
Total RNAs were prepared from cells for the reverse transcription PCR. Total RNA extraction was carried out using TRIZOL reagent (TakaRa), according to the manufacturer's instructions. Extracted total RNAs were subjected to reverse transcription PCR. The quantified cDNA was subjected to DEK, and Prdx 6 mRNA expression analysis. The following PCR primers were used: 5′-ACGGAACAGTTCTGGAATGG-3′ (forward) and 5′-TGGTGGCTCCTCTTCACTTT-3′ (reverse) (DEK), 5′-CAATACCACCGTCGGCCGCA-3′ (forward) and 5′-AAGGCTGGGGTGTGTAGCGG-3′ (reverse) (Prdx 6). Disassociation curves were generated after each PCR run to ensure that a single product of appropriate length was amplified. The mean threshold cycle (CT) and standard error (SE) were calculated from individual CT values obtained from triplicates per stage. The normalized mean CT was estimated as the ΔCT by subtracting the mean CT of β-actin. The ΔΔCT value was calculated as the difference between the control ΔCT and values obtained for each sample. The n-fold change in gene expression relative to the untreated control was calculated as 2−ΔΔC T.
Western blot analysis
Total proteins were isolated from cells using Radioimmunoprecipitation Assay buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, 1X protease inhibitor cocktail, and 1 mM EDTA), fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and transferred to nitrocellulose membranes. The membranes were probed overnight with the indicated antibodies at 4°C. The blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies (Enzo Life Sciences) and detected using an enhanced chemiluminescence (ECL) system.
Reporter assay
K562 cells were seeded in 6-well dishes and transfected by lipofectamine 2000 with pGL3-Prdx 6 and shDEK. After 24 h transfection, Hemin (30 μM) or dimethyl sulfoxide (DMSO) is treated for indicated time point. After indicated time point, cells were harvested and assayed for luciferase activity using a luciferase assay system (Promega). Each value was expressed as the mean of three replicates from a single assay, and experiments were performed at least three times.
Chromatin immunoprecipitation (ChIP) analysis
ChIP analysis was performed as described in the protocols from Millipore. Briefly, HL-60 cells were differentiated by all-trans-retinoic acid (ATRA) or 12-O-tetradecanoyl-phorbol 13-acetate (TPA) and harvested 48 h later. Cells were cross-linked with 1% formaldehyde in the medium at room temperature for 10 min, followed by the addition 125 mM glycine at room temperature for 5 min, and then scraped into sodium dodecyl sulfate (SDS) lysis buffer (50 mM Tris-HCl [pH 8.1], 1% SDS, and 10 mM EDTA) with antibodies against DEK, p65, and IgG. The immunoprecipitates were recovered with protein A/G agarose beads (GenDEPOT). After reversing the cross-links, chromatin was subjected to proteinase K digestion, and the DNA was purified for PCR amplification (QIAGEN). To analyze the Prdx 6 promoter region, primer sets consisting of the promoter region (position −911 to −672 from translation start site, forward: 5′-GTTGACCTGCACACAGTAGGTCTC-3′, reverse: 5′-CCTACAGTGGAGTGGAGTGACTGCT-3′); final intron region (position 9,353 to 9,708, forward: 5′-GTCATGGCTGTAAAAGTACTGGTG-3′, reverse: 5′-CACTGGAATGGAAGTTCTATGAGGG-3′); final exon (position 9,989 to 10,347, forward: 5′-GCTTGGAGAAGAAGCTGCAGAA-3′, reverse: 5′-CTATCCCCATCCTATTGAAAGAC-3′) were used. The amplification reaction was performed under the following conditions: 45 cycles of denaturation at 94°C, annealing at 58°C, and extension at 72°C. Disassociation curves were generated after each PCR was run to ensure that a single product of appropriate length was amplified. The mean CT ± SE was calculated from individual CT values obtained from triplicate determinations per stage. The normalized mean CT was estimated as ΔCT by subtracting the mean CT of input from that of the individual region.
Statistical analysis
Data are expressed as mean ± standard deviation (SD) of three or more independent experiments. Statistically significant effects (P < 0.05) were evaluated with Microsoft Excel. Differences between groups were evaluated by one-way analysis of variance (ANOVA), followed by Student's t-test or Bonferroni's test, as appropriate.
Results and discussion
Identification of the Prdx 6 target gene during leukemia cell differentiation
A nuclear phosphoprotein, DEK, has been associated with certain human diseases including leukemia and autoimmune disorders. In the previous study, we monitored differentially expressed proteins in DEK knockdown cells using high resolution two-dimensional gel electrophoresis (2-DE) and Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF MS) to investigate the physiological role mediated by DEK (Kim et al. Citation2009). Among differential proteomic profiles in DEK knockdown HeLa cells, we have identified that Prdx 6 was up-regulated in the absence of DEK (Kim et al. Citation2009). To further investigate the possibility of Prdx 6 transcriptional regulation by DEK and their related role, we monitored expression of Prdx 6 during overexpression of DEK in 293T cells (Kim et al. Citation2010). Consistent with our previous study, real-time PCR (RT-PCR) and immunoblot analysis showed significant down-regulation of Prdx 6 expression in DEK overexpression (). To evaluate the possible role of DEK-mediated regulation of Prdx 6 during leukemia cell differentiation, we first monitored the expression levels of DEK and Prdx 6 after TPA treatment in promyelocytic HL-60 cells. First, RT-PCR analysis showed that DEK transcriptions were significantly up-regulated after leukemia cell differentiation by TPA treatments (). Up-regulation of DEK protein levels were also confirmed by immunoblots (). Consistent with previous proteomic studies, the expression levels of Prdx 6 were repressed during leukemia cell differentiation by immunoblot analysis (). These results suggest that DEK may operate as a negative regulator of Prdx 6 transcription during leukemia cell differentiation.
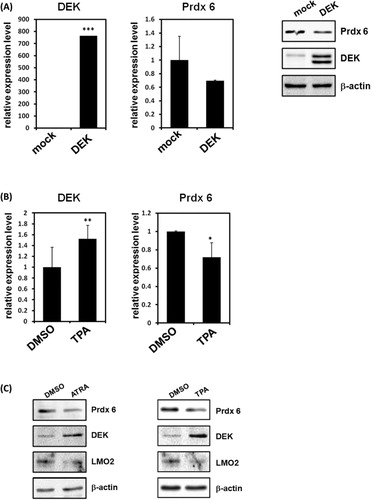
Transcriptional regulation of Prdx 6 promoter by DEK
To further investigate that DEK repressed Prdx 6 expression during leukemia cell differentiation, we next conducted a reporter assay using a Prdx 6-luc reporter system to examine DEK-mediated transcriptional regulation of Prdx 6. Initially, we checked transcription of Prdx 6 during leukemia cell differentiation. When we differentiated K562 cells using hemin treatment, transcription of Prdx 6 was significantly decreased after 24 h (). Interestingly, when DEK was knocked down by shRNA, transcription of Prdx 6 was up-regulated (). All together, these data were consistent with our previous results which showing that transcription of Prdx 6 is negatively regulated by DEK during leukemia cell differentiation.
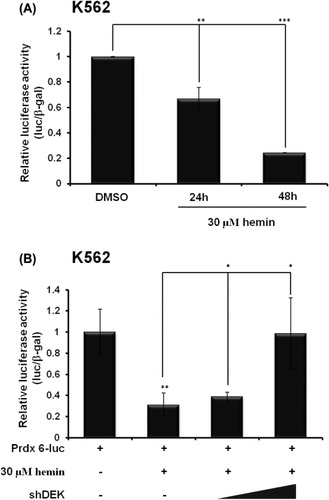
DEK is recruited to Prdx 6 promoter and represses transcription
Having established that DEK negatively regulates Prdx 6, we subsequently attempted to determine whether DEK and p65 are recruited to Prdx 6 promoter via ChIP analysis. In our previous study, we reported that NF-κB subunit p65 has a synergistic effect on DEK-mediated Prdx 6 repression (Kim et al. Citation2010). Both proteins were recruited to the Prdx 6 promoter region and regulate its transcription (Kim et al. Citation2010). We noted that there were basal levels of DEK and p65 recruitment to Prdx 6 promoter prior to TPA and ATRA treatment (). During leukemia cell differentiation by TPA and ATRA treatment, the recruitment of both DEK and p65 to the Prdx 6 promoter region was increased significantly and repressed Prdx 6 transcription (). By contrast, the Prdx 6 final intron and final exon were not occupied by DEK and p65 (). These data indicate that both DEK and p65 proteins are recruited to the Prdx 6 promoter and negatively regulate its transcription during leukemia cell differentiation.
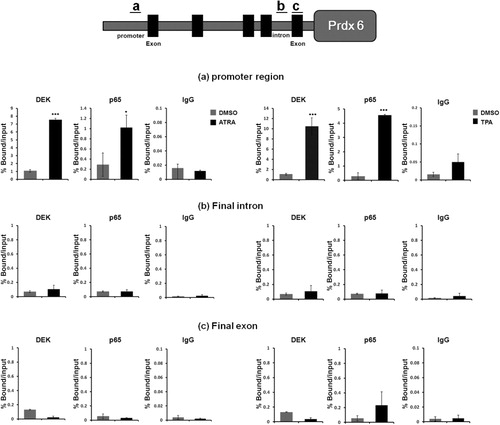
Previous reports suggested that the levels of Prdx 6 protein increase when DEK is knocked down (Kim et al. Citation2009). These reports suggest that DEK regulates Prdx 6 protein level with p65 during leukemia cell differentiation. It is possible that p65 binds preferentially to DNA and recruits DEK protein as a transcriptional repressor through protein–protein interaction.
In this study, we identified the functional role of DEK in Prdx 6 transcription during leukemia cell differentiation. DEK acted as a transcriptional repressor of Prdx 6 by interacting with the p65 subunit of NF-κB. The finding that Prdx 6 was regulated by DEK expression further links the role of DEK in regulation of Prdx 6 expression to leukemia cell differentiation. Further studies are required to elucidate the precise mechanism of DEK and p65-mediated transcriptional regulation of Prdx 6, and its biological impacts. Taken together, our results demonstrate the transcriptional regulatory role of DEK in leukemia cell differentiation through interaction with p65.
Acknowledgement
This study was supported by the Chung-Ang University Research Scholarship Grant (2013).
References
- Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. 2000. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 14:1308–1312.
- Campillos M, Garcia MA, Valdivieso F, Vazquez J. 2003. Transcriptional activation by AP-2alpha is modulated by the oncogene DEK. Nucleic Acids Res. 31:1571–1575. 10.1093/nar/gkg247
- Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, Muller H. 2006. DEK expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 5:1202–1207. 10.4161/cc.5.11.2801
- Casas S, Nagy B, Elonen E, Aventin A, Larramendy ML, Sierra J, Ruutu T, Knuutila S. 2003. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leuk Lymphoma. 44:1935–1941. 10.1080/1042819031000119299
- Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 2000. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem. 275:28421–28427. 10.1074/jbc.M005073200
- Grottke C, Mantwill K, Dietel M, Schadendorf D, Lage H. 2000. Identification of differentially expressed genes in human melanoma cells with acquired resistance to various antineoplastic drugs. Int J Cancer. 88:535–546. 10.1002/1097-0215(20001115)88:4%3C535::AID-IJC4%3E3.0.CO;2-V
- Hatada EN, Krappmann D, Scheidereit C. 2000. NF-kappaB and the innate immune response. Curr Opin Immunol. 12:52–58. 10.1016/S0952-7915(99)00050-3
- Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. 2002. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J Cell Sci. 115:3319–3330.
- Kim DW, Chae JI, Kim JY, Pak JH, Koo DB, Bahk YY, Seo SB. 2009. Proteomic analysis of apoptosis related proteins regulated by proto-oncogene protein DEK. J Cell Biochem. 106:1048–1059. 10.1002/jcb.22083
- Kim DW, Kim JY, Choi S, Rhee S, Hahn Y, Seo SB. 2010. Transcriptional regulation of 1-cys peroxiredoxin by the proto-oncogene protein DEK. Mol Med Rep. 3:877–881.
- Ko SI, Lee IS, Kim JY, Kim SM, Kim DW, Lee KS, Woo KM, Baek JH, Choo JK, Seo SB. 2006. Regulation of histone acetyltransferase activity of p300 and PCAF by proto-oncogene protein DEK. FEBS Lett. 580:3217–3222. 10.1016/j.febslet.2006.04.081
- Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, Goseki N, Matsubara O, Takenaka K, Shichita M, et al. 1999. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 59:4990–4996.
- Larramendy ML, Niini T, Elonen E, Nagy B, Ollila J, Vihinen M, Knuutila S. 2002. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica. 87:569–577.
- Manevich Y, Sweitzer T, Pak JH, Feinstein SI, Muzykantov V, Fisher AB. 2002. 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc Natl Acad Sci USA. 99:11599–11604. 10.1073/pnas.182384499
- Mor-Vaknin N, Punturieri A, Sitwala K, Faulkner N, Legendre M, Khodadoust MS, Kappes F, Ruth JH, Koch A, Glass D, et al. 2006. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol Cell Biol. 26:9484–9496. 10.1128/MCB.01030-06
- Sammons M, Wan SS, Vogel NL, Mientjes EJ, Grosveld G, Ashburner BP. 2006. Negative regulation of the RelA/p65 transactivation function by the product of the DEK proto-oncogene. J Biol Chem. 281:26802–26812. 10.1074/jbc.M600915200
- von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, Grosveld G. 1992. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 12:1687–1697.
- Waldmann T, Eckerich C, Baack M, Gruss C. 2002. The ubiquitous chromatin protein DEK alters the structure of DNA by introducing positive supercoils. J Biol Chem. 277:24988–24994. 10.1074/jbc.M204045200
- Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, Brown A, Lerner CP, Paigen B. 2003. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J Biol Chem. 278:25179–25190. 10.1074/jbc.M302706200
- Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. 2006. Peroxiredoxin 6 gene-targeted mice show increased lung injury with paraquat-induced oxidative stress. Antioxid Redox Signal. 8:229–237. 10.1089/ars.2006.8.229
- Wood ZA, Poole LB, Karplus PA. 2003a. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 300:650–653. 10.1126/science.1080405
- Wood ZA, Schroder E, Robin Harris J, Poole LB. 2003b. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 28:32–40. 10.1016/S0968-0004(02)00003-8