
Abstract
Multiple genes and their interactions are involved in most human diseases. This pathway-centric view of human pathology is beginning to guide our approaches to disease research. Analytical algorithms describing human gene networks have been developed for three major tasks in disease research: (i) disease gene prioritization, (ii) disease module discovery, and (iii) stratification of complex diseases. To understand the underlying biology of human diseases, identification of disease genes and disease pathways is crucial. The functional interdependence between genes for disease progression has been identified by their connections in gene networks, which enables prediction of novel disease genes based on their connections to known disease genes. Disease modules can be identified by subnetworks that are enriched for patient-specific activated or mutated genes. Network biology also facilitates the subtyping of complex diseases such as cancer, which is a prerequisite for developing personalized medicinal therapies. In this review, we discuss network-assisted approaches in human disease research, with particular focus on the three major tasks. Network biology will provide powerful research platforms to dissect and interpret disease genomics data in the future.
Introduction
Recent revolutions in high-throughput experimental technologies have changed paradigms in human disease research. Historically, disease etiology focused on individual genes. However, recent progress in understanding the complexity of disease genetics has shifted our attention from genes to pathways. Genome-wide association study (GWAS) suggested that numerous genes were associated with complex diseases, and those disease-associated genes were close to each other in functional gene networks (Baranzini et al. Citation2009). Surveys of somatic mutations in cancer patients revealed that most genes had somatic mutations in only a few patients, but cancer signaling pathways were disrupted by mutations of some member genes in most patients (Vogelstein et al. Citation2013). These observations support the need for pathway-centric views of human pathology.
Network approaches to human disease enable in silico investigations of novel disease genes and modules through experimental analyses of the molecular networks. The principle of guilt-by-association has been a popular strategy in phenotype prediction and interpretation, which has been proven as useful with the availability of highly accurate and comprehensive molecular networks (Lee Citation2013). Network-based methods recently began to tackle another important issue in complex disease research, which is subtyping complex diseases such as cancer. We will discuss network-assisted approaches for the three major tasks in human disease research: (i) disease gene prioritization, (ii) disease module identification, and (iii) stratification of complex diseases ().
Figure 1. Network-assisted approaches for human disease research. Three major tasks are facilitated by network approaches to human disease research: (a) disease gene prioritization, (b) disease module discovery, and (c) stratification of complex diseases. (a) Networks facilitate gene prioritization by network propagation of disease information or by integrating disease-specific data with networks. DEG, differentially expressed genes; GWAS, genome-wide association study. (b) Disease module discovery uses both patient-specific information (gene expression or mutation profiles) and molecular networks, whereas conventional approaches for module identification use only network topology. (c) Network-assisted disease subtyping utilizes patient-specific gene expression or mutation data.
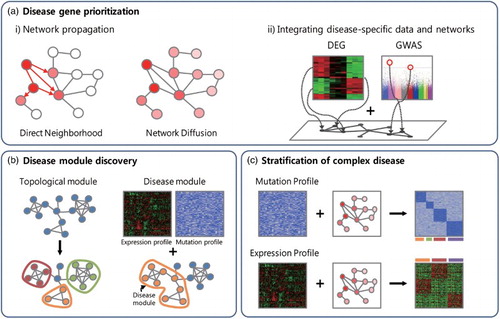
Network-assisted disease gene prioritization
An overriding task for human disease research is to establish a complete catalog of disease-associated genes. Unbiased forward and reverse genetic screens have revealed many disease-associated genes. However, these conventional genetics approaches cannot provide comprehensive catalogs because many disease-associated genes have low genetic penetrance, which result in undetectable phenotypic effects in large-scale genetic screens. By contrast, computational predictions based on genetic networks enable highly sensitive detection of disease phenotypes by critical examination of the prioritized candidate genes. Network-assisted gene prioritization can be achieved either by propagation of prior disease information throughout networks or by integrating disease-specific data within the networks.
Disease gene prioritization by network propagation
There are two conceptually distinct strategies for network propagation (Wang & Marcotte Citation2010). In the direct neighborhood strategy, prior disease information propagates only to direct neighbors. Disease information transferred from network neighbors to candidate genes is scored by simple count of the neighbors or by sum of edge weights to the neighbors, which is known as the naive Bayes (NB) algorithm. Although the direct neighborhood strategy is popular for network propagation, it has limited power if the prior disease information is scarce. This is particularly problematic for diseases with only a few known annotated genes. In this case, only a few candidate genes can be prioritized by propagated disease information from direct neighbors.
To overcome the direct neighborhood limitation, the strategy of network diffusion was developed, which diffuses prior disease information throughout the entire network. Network diffusion allows all genes in the network to receive propagated information and become prioritized for the disease. Several algorithms have been developed for network diffusion, including random walk (RW), random walk with restart (RWR) (Woess Citation1994), PageRank (PR) (Page et al. Citation1999), and Gaussian smoothing (GS) (Zhou et al. Citation2003). Network diffusion algorithms are affected by the ratio between disease information retained in initial nodes and information transferred to neighbors after diffusion. Most diffusion algorithms utilize the following equation to determine the diffusion coefficient:
Here, a user-defined parameter α determines the amount of information to be propagated to the neighbors. Although these diffusion algorithms are based on the same propagation equation, the objective functions of each algorithm are different; RW, RWR, and PR give more weight to hub genes, whereas GS balances weights among neighbors by using a smoothing technique. Conceptually, GS finds solutions where it achieves a minimal difference between the initial and final scores of a disease gene, and between the disease score of a gene and each of its neighbors. Therefore, the GS algorithm is more robust for the number of neighbors of each gene than other network diffusion algorithms. GeneMANIA (Mostafavi et al. Citation2008) is a popular application for gene prioritization based on the GS algorithm.
Advanced network diffusion algorithms are not a governing factor for successful gene prioritization. Recently, systematic benchmarking between direct neighborhood and network diffusion has been performed (Shim et al. Citation2015). This study demonstrated that the effectiveness of each network algorithm differed for early retrieval of candidates and for prioritization of disease genes throughout the entire network. For example, in the majority of human diseases, the simple NB algorithm outperforms the advanced GS algorithm for the top 200 candidates, whereas GS outperforms NB for all ranked candidates. Considering that only a few hundred candidates generally enter subsequent experimental tests, the optimal network algorithm needs to be selected based on performance for early retrieval. This study also demonstrated that network algorithm effectiveness depends on the connectivity of disease genes in the networks. The NB algorithm is more effective for diseases whose member genes are well-connected in the gene network, whereas the GS algorithm is more effective for diseases whose member genes are disconnected in the network.
Disease gene prioritization by integrating disease-specific data with networks
Network propagation approaches require prior knowledge for the given disease. However, the amount of prior knowledge is limited for most human diseases. This lack of prior knowledge can be circumvented by using disease-specific data from high-throughput experiments such as gene expression profiling and GWAS. Differentially expressed genes (DEGs) or single nucleotide polymorphism (SNP) loci that are statistically associated with the patient population of a given disease can be integrated with molecular networks to identify novel disease genes that would not be detectable by disease-specific data or molecular networks alone.
By assuming that disease genes tend to be surrounded by network neighbors that are differentially expressed under the relevant disease conditions, Nitsch et al. (Citation2009) identified disease-causing genes that belong to network modules with significantly disrupted expression. To integrate the identified DEGs with a network, they defined the distance between two genes in a network using the Laplacian exponential diffusion kernel, and then scored candidates by aggregating the differential expression of neighbors weighted as a function of distance. This method prioritizes candidate genes by identifying their differentially expressed neighborhoods.
Molecular network information can be integrated with GWAS data by reweighting GWAS disease probability using molecular networks. For example, protein interaction network-based pathway analysis (PINBPA) (Wang et al. Citation2015) analyzes GWAS data in a network fashion. PINBPA assigns GWAS probability scores to genes using the popular tool VEGAS (Liu, Mcrae et al. Citation2010). Then, it prioritizes genes using the RWR algorithm and identifies subnetworks enriched for genes with high disease probability scores. One limitation of this approach is the reliance of GWAS data, in which only few SNPs pass stringent statistical tests for low false-positive discovery rate. To overcome the limited statistical power of GWAS, Lee et al. (Citation2011) proposed network-assisted boosting statistical signals of GWAS, which employed a functional gene network (HumanNet) constructed by integrating inferred networks from available genomic and computational data. The disease probability scores of GWAS that are reasonably high but could not pass stringent statistical tests were rescored by using GWAS scores of network neighbors. Thus, disease probability scores of genes connected to the neighbors with high GWAS score are enhanced. The effectiveness of this method was demonstrated by boosting GWAS for Crohn's disease and Type 2 diabetes.
Network-assisted disease module discovery
Many real-world networks have modular organization, including molecular networks. The modules generally represent functional units that contain functionally coherent components. For example, most cellular processes operate via pathways in which functionally related genes are interconnected to each other. A pathway-centric view of human pathology supports modular approaches for understanding disease mechanisms, and the identification of novel disease modules is a crucial task in disease research. Conventional approaches to discover modular network structures are based on network topology, in which highly interconnected subnetworks are implicated as functional system modules. Disease modules are subnetworks that are enriched for disease-specific genes or gene candidates, and they can be discovered by combining patient-specific data such as disease-specific gene expression and gene mutation data with molecular networks. Thus, disease modules can be enriched for disease-specific gene mutations or DGEs.
Cytoscape is a popular network biology software tool that provides several plug-in applications for discovery of disease modules. One of the applications [jActiveModule (Ideker et al. Citation2002)] was developed to identify subnetworks enriched for DEGs, which are defined as active modules. This tool can be used to identify disease modules if patient-specific gene expression data are available. The algorithm applies a rigorous statistical measure for scoring network modules by calibrating the z-score against the background distribution. Other algorithms for identifying active modules are Heinz (Dittrich et al. Citation2008), CEZANNE (Ulitsky & Shamir Citation2009), and CASNet (Gaire et al. Citation2013). The algorithms HotNet (Vandin et al. Citation2011; Leiserson et al. Citation2015) and HyperModules (Leung et al. Citation2014) identify statistically mutated subnetworks among patients using local network search heuristics to detect closely connected network regions. To establish statistical significance for clinical correlations in the identified modules, HyperModules applies standard statistical tests such as log-rank test and Fisher's exact test, and corrects systematic biases across many shuffled networks.
GWAS data can be analyzed in the context of disease subnetworks. Dense module searching for GWAS (dmGWAS) (Jia et al. Citation2011) provides a dense module searching (DMS) algorithm to identify candidate subnetworks or genes for complex diseases by integrating the association signal from GWAS datasets into the human protein-protein interaction (PPI) network. The dmGWAS algorithm considers gene-centric disease association probability as the node weight and prioritizes candidate genes by DMS. Network Interface Miner for Multigenic Interactions (NIMMI) (Akula et al. Citation2011) prioritizes disease genes from GWAS using a page-rank algorithm. By combining these weights with disease association probability derived from GWAS, NIMMI produces trait-prioritized subnetworks. An excellent review on network-assisted analysis of GWAS data has been published recently (Jia & Zhao Citation2014).
Network-assisted stratification of complex diseases
Disease subtype information is crucial for successful treatment of complex diseases such as cancer. Conventionally, cancer subtypes are identified by cluster analysis of patient-specific gene expression profiles. Recent efforts to improve cancer subtyping incorporate molecular network information into analyses of gene expression or gene mutation data. Here, we introduce two network-assisted algorithms to discover cancer subtypes, which are based on non-negative matrix factorization (NMF). The advantages of NMF are superior data storage and interpretability. Due to the non-negative constraints, NMF produces a so-called “additive parts-based” representation of the data. NMF also excels in terms of factor interpretation, which also is a consequence of the non-negative constraints (Albright et al. Citation2006). The network-based stratification (NBS) method (Hofree et al. Citation2013) integrates somatic tumor mutations with a gene network. Somatic mutation profiles are extremely sparse and heterogeneous; therefore, NBS performs network smoothing and projects mutations onto a network. Subsequently, NBS clusters the smoothed profile using graph-regularized non-negative matrix factorization (GNMF).
Generation of mutation data is becoming easier with advanced high-throughput sequencing technologies, although gene expression profiles are still the most abundant data type. Network-assisted co-clustering for the identification of cancer subtypes (NCIS) (Liu, Gu et al. Citation2014) provides an algorithm that incorporates gene network information with gene expression profiles. NCIS is distinct from NBS in that it uses semi-non-negative matrix tri-factorization (SNMTF), which is a member of the matrix factorization-based clustering family. SNMTF clusters mixed-sign input data and can be used to partition a given set of patients or genes into different clusters. Here, the partition of patients into different clusters indicates potential cancer subtypes, whereas the partition of genes into different clusters potentially identifies disease-specific co-expressed gene sets. NCIS was designed to operate effectively with high-dimensional and high-throughput gene expression data.
Summary
Network-assisted disease research has steadily evolved during the past decade. During the early stages, network-assisted approaches were restricted to identification of topological subnetworks or propagation of disease probability through network information using disease-related prior knowledge. However, as high-throughput and genome-wide experimental assays have advanced, network-assisted approaches emerged as a key solution to the complexity of human disease research. Network-assisted approaches contribute to hypothesis formulation by integrating external data such as gene expression or mutation profiles with network information. Network-assisted approaches also facilitate interpretations of disease mechanisms. Some limitations and challenges remain, such as the incompleteness of molecular networks. However, it is certain that network-assisted approaches will have crucial roles in unraveling the biological complexity of human diseases in the future.
Disclosure
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Akula N, Baranova A, Seto D, Solka J, Nalls MA, Singleton A, Ferrucci L, Tanaka T, Bandinelli S, Cho YS, et al. 2011. A network-based approach to prioritize results from genome-wide association studies. Plos One. 6:e24220. doi: 10.1371/journal.pone.0024220
- Albright R, Cox J, Duling D, Langville AN, Meyer C. 2006. Algorithms, Initializations, and Convergence for The Nonnegative Matrix Factorization. Technical Report 919, NCSU Technical Report Math 81706.
- Baranzini SE, Galwey NW, Wang J, Khankhanian P, Lindberg R, Pelletier D, Wu W, Uitdehaag BM, Kappos L, GeneMSA C, et al. 2009. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Genet. 18:2078–2090. doi: 10.1093/hmg/ddp120
- Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Muller T. 2008. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics. 24:i223–i231. doi: 10.1093/bioinformatics/btn161
- Gaire RK, Smith L, Humbert P, Bailey J, Stuckey PJ, Haviv I. 2013. Discovery and analysis of consistent active sub-networks in cancers. Bmc Bioinformatics. 14(Suppl. 2):S7. doi: 10.1186/1471-2105-14-S2-S7
- Hofree M, Shen JP, Carter H, Gross A, Ideker T. 2013. Network-based stratification of tumor mutations. Nat Methods. 10:1108–1115. doi: 10.1038/nmeth.2651
- Ideker T, Ozier O, Schwikowski B, Siegel AF. 2002. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 18(Suppl. 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.S233
- Jia P, Zhao Z. 2014. Network assisted analysis to prioritize GWAS results: principles, methods and perspectives. Hum Genet. 133:125–138. doi: 10.1007/s00439-013-1377-1
- Jia P, Zheng S, Long J, Zheng W, Zhao Z. 2011. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 27:95–102. doi: 10.1093/bioinformatics/btq615
- Lee I. 2013. Network approaches to the genetic dissection of phenotypes in animals and humans. Anim Cells Syst. 17:75–79. doi: 10.1080/19768354.2013.789076
- Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM. 2011. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 21:1109–1121. doi: 10.1101/gr.118992.110
- Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. 2015. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 47:106–114. doi: 10.1038/ng.3168
- Leung A, Bader GD, Reimand J. 2014. HyperModules: identifying clinically and phenotypically significant network modules with disease mutations for biomarker discovery. Bioinformatics. 30:2230–2232. doi: 10.1093/bioinformatics/btu172
- Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Hayward NK, Montgomery GW, Visscher PM, Martin NG, et al. 2010. A Versatile gene-based test for genome-wide association studies. Am J Hum Genet. 87:139–145. doi: 10.1016/j.ajhg.2010.06.009
- Liu YY, Gu QQ, Hou JP, Han JW, Ma J. 2014. A network-assisted co-clustering algorithm to discover cancer subtypes based on gene expression. Bmc Bioinformatics. 15:37. doi: 10.1186/1471-2105-15-37
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. 2008. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9(Suppl. 1):S4. doi: 10.1186/gb-2008-9-s1-s4
- Nitsch D, Tranchevent LC, Thienpont B, Thorrez L, Van Esch H, Devriendt K, Moreau Y. 2009. Network analysis of differential expression for the identification of disease-causing genes. Plos One. 4:e5526. doi: 10.1371/journal.pone.0005526
- Page L, BS Motwani R, Winograd T. 1999. The pagerank citation ranking: bringing order to the web. Technical Report Stanford InfoLab.
- Shim JE, Hwang S, Lee I. 2015. Pathway-dependent effectiveness of network algorithms for gene prioritization. Plos One. 10:e0130589. doi: 10.1371/journal.pone.0130589
- Ulitsky I, Shamir R. 2009. Identifying functional modules using expression profiles and confidence-scored protein interactions. Bioinformatics. 25:1158–1164. doi: 10.1093/bioinformatics/btp118
- Vandin F, Upfal E, Raphael BJ. 2011. Algorithms for detecting significantly mutated pathways in cancer. J Comput Biol. 18:507–522. doi: 10.1089/cmb.2010.0265
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LAJr., Kinzler KW. 2013. Cancer genome landscapes. Science. 339:1546–1558. doi: 10.1126/science.1235122
- Wang LL, Matsushita T, Madireddy L, Mousavi P, Baranzini SE. 2015. PINBPA: Cytoscape app for network analysis of GWAS data. Bioinformatics. 31:262–264. doi: 10.1093/bioinformatics/btu644
- Wang PI, Marcotte EM. 2010. It's the machine that matters: predicting gene function and phenotype from protein networks. J Proteomics. 73:2277–2289. doi: 10.1016/j.jprot.2010.07.005
- Woess W. 1994. Random-walks on infinite-graphs and groups – a survey on selected topics. B Lond Math Soc. 26:1–60. doi: 10.1112/blms/26.1.1
- Zhou D, Bousquet O, Lal T, Weston J, Schoelkopf B. 2003. Learning with local and global consistency. Neural Information Processing Systems. 16:321–328.